Os princípios observados para essas máquinas foram generalizados em postulados, conhecidos como 1ª e 2ª leis da termodinâmica. A base da Termodinâmica é a observação experimental, seus postulados não possuem comprovação matemática (SMITH, VAN NESS & ABBOTT, 2000). Iremos nos concentrar principalmente nas Leis da Termodinâmica – a Primeira Lei da Termodinâmica e a Segunda Lei da Termodinâmica.
A primeira lei da termodinâmica estuda as diferentes formas de energia (cinética, potencial, interna) e é utilizada no cálculo das necessidades de calor e trabalho para processos físicos e químicos. A segunda lei da termodinâmica diz respeito à possibilidade de ocorrência de determinado fenômeno, ou seja, mostra se uma transformação é possível ou não. Processos industriais importantes requerem a separação de componentes em misturas fluidas de operações unitárias.
Por exemplo, destilação, cujo objetivo é separar os componentes de uma mistura líquida por diferenças na sua volatilidade; absorção, que visa extrair um componente de uma mistura gasosa dissolvendo-o em um líquido; extração, cujo objetivo é retirar um componente de uma mistura líquida e transferi-lo para outro líquido; e lixiviação, na qual um componente é extraído de uma mistura sólida utilizando um solvente líquido.
Conceitos e Definições
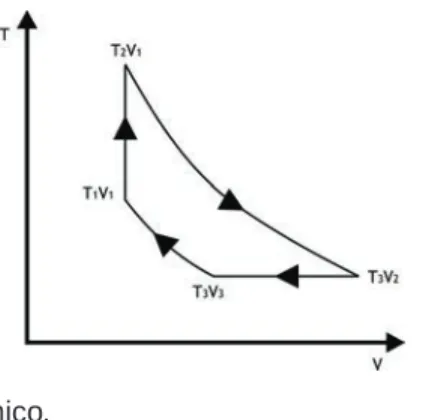
Quando o sistema muda de um estado de equilíbrio para outro, o sistema passa por um conjunto de estados denominado processo. Por exemplo, na Figura 1.3, se todas as massas m no pistão forem retiradas repentinamente, o pistão sobe rapidamente, causando um processo de desequilíbrio no gás (sistema). A temperatura pode ser definida como uma medida da agitação molecular de um sistema e expressa seu potencial de transferência de energia na forma de calor.
Os estados líquidos ao longo dos segmentos a-b, e-f e ij (Figura 2.3) são chamados de líquido subresfriado (o líquido tem uma temperatura inferior à temperatura de saturação a uma certa pressão) ou líquido comprimido (a pressão é maior que a certa pressão de saturação na temperatura correspondente ). A discussão acima é bem resumida pelo diagrama de fases (Figura 2.5), que mostra como as fases sólida, líquida e vapor podem coexistir em equilíbrio. Se o processo de pressão constante for realizado a uma pressão abaixo do ponto triplo (AB na Figura 2.5), a fase sólida passa diretamente para o vapor.
Os parâmetros "a" e "b" das equações de Van der Waals e Redlich-Kwong podem ser avaliados ajustando os dados p-v-T disponíveis ou, se não estiverem disponíveis, estimados a partir das propriedades críticas (Tc e pc).

Leis da Termodinâmica e suas Aplicações
Todas estas questões podem ser respondidas através da primeira lei da termodinâmica ou balanço energético. Calor e trabalho são formas de energia em trânsito onde um sistema interage com outro sistema ou com seu entorno. A equação (3.5) afirma que a variação da energia total em um sistema com massa constante é igual ao balanço de energia transportada para o sistema na forma de calor (Q) e rede (W = WS + WpV).
No sistema SI, a unidade de energia é o Joule (J), porém, outras unidades ainda estão em uso, como a caloria (cal), pé x libra-força (ft x lbf) e a unidade térmica britânica. (Btu). O sinal ∑ é alimentado no balanço de energia para contabilizar todas as entradas e saídas do volume de controle. A Equação (3.18) representa o Balanço Energético para processos em estado estacionário com uma entrada e uma saída.
As formas mais comuns de trabalho calculadas no balanço de energia são trabalho de expansão/contração, trabalho axial e trabalho de fluxo.

Entropia
A primeira lei da termodinâmica estabelece a conservação da energia e não impõe restrições à direção do calor e dos fluxos de trabalho em uma determinada transformação. É impossível para qualquer sistema funcionar de tal maneira que o único resultado seja a transferência de energia na forma de calor de um corpo mais frio para um corpo mais quente. A segunda lei não proíbe a produção de trabalho a partir do calor, mas estabelece um limite para a porção de calor que pode ser convertida em trabalho em qualquer processo cíclico.
A afirmação de Kelvin-Planck refere-se às máquinas térmicas e estabelece que é impossível construir uma máquina térmica que opere num ciclo em que recebe uma certa quantidade de calor de um corpo de alta temperatura (TH) e produz uma quantidade igual de calor. quantidade de calor, quantidade de trabalho (Figura 4.3). Refrigeradores ou bombas de calor são dispositivos construídos para transferir calor de uma fonte de baixa temperatura para uma fonte de alta temperatura à custa da energia transferida para o ciclo na forma de trabalho. Um frigorífico ou uma bomba de calor podem ser utilizados para os seguintes fins:
Nesta etapa, o vapor é resfriado e condensado com rejeição de calor em um nível de temperatura mais elevado. É impossível construir uma máquina funcionando entre dois reservatórios de calor que seja mais eficiente do que uma máquina de Carnot funcionando entre os dois reservatórios. Todas as máquinas de Carnot que funcionam entre dois reservatórios de calor nas mesmas temperaturas têm a mesma eficiência térmica.
A Equação (4.14) mostra que em motores térmicos reversíveis a razão entre as quantidades de calor pode ser substituída pela razão das temperaturas absolutas dos dois dissipadores de calor. A eficiência dada pela equação (4.14) é a maior eficiência que uma máquina térmica operando entre dois dissipadores de calor pode ter nas temperaturas TF e TQ. Exemplo 4.3 O compartimento de alimentos de um refrigerador é mantido a 4°C removendo calor a uma taxa de 360 kJ/min.
Ou seja, 3 kJ de calor são removidos do espaço de refrigeração para cada kJ de trabalho realizado. Um sistema fechado não envolve fluxo de massa através da fronteira, e a mudança de entropia é devida à transferência de entropia, que acompanha a transferência de calor, e a geração de entropia dentro do sistema devido a irreversibilidades.

Termodinâmica do Equilíbrio de Fases
A equação (5.35) é conhecida como equação da soma e permite o cálculo das propriedades de uma mistura a partir das propriedades parciais de seus componentes. A equação de Gibbs-Duhem é obtida pela diferenciação da equação de Gibbs-Duhem (Equação (5.37)) que impõe a condição de que a fração molar de qualquer fase pode variar com T, p e xi. A integral da equação (5.65) pode ser avaliada a partir de dados p-v-T ou de EDE (ver p.
A Equação (5.71) diz que para que processos reais ocorram espontaneamente, quando o sistema está em T e p constantes, a energia livre de Gibbs deve diminuir. A Equação (5.77) aplicada a problemas de equilíbrio de fases requer o uso de modelos que expressam G e µι como funções de T, p, ni. Dados necessários para utilização da Equação (5.84): i) Os valores de Zi para calcular φisat podem vir de uma EDE, experimentos ou correções gerais;. ii) vli é normalmente igual ao valor do líquido saturado;. iii) psati pode ser calculado a partir da equação de Antoine.
A definição da fugacidade de uma substância em uma mistura ou solução é derivada da equação (5.60), adaptada para misturas não ideais, substituindo a pressão parcial (yip) por uma quantidade chamada de fugacidade de uma espécie. A equação (5.89) pode ser usada quando dados de mistura estão disponíveis, como volume molar (vm) ou fator de compressibilidade (Zm), com os quais os volumes parciais molares vi ou Zi podem ser calculados. Partindo da equação (5.58), gigi =gi9i+RTln( )yi , se substituirmos gi9i (energia livre de Gibbs molar de uma substância "i" no estado de gás ideal) por gi (energia livre de Gibbs molar de uma substância "i" , que faz parte de uma mistura, dado que se encontra nas mesmas condições de T e p e do estado de agregação da mistura), podemos aplicá-lo a soluções reais - líquidas, sólidas ou gasosas.
Todas as outras propriedades termodinâmicas de uma solução ideal podem ser obtidas na Equação (5.102). A energia livre de Gibbs em excesso molar, gE, é particularmente importante devido à sua dependência de variáveis diretamente mensuráveis (T, p e composição) e, conhecendo gE, calcula-se vE (Eq. (5.124)), hE (Eq. (5.125) )) e lnlnγi(Equação (5.126)). A Equação (5.144) é truncada por conveniência e, a partir da Equação (5.126), temos as respectivas equações para lnlnγi.
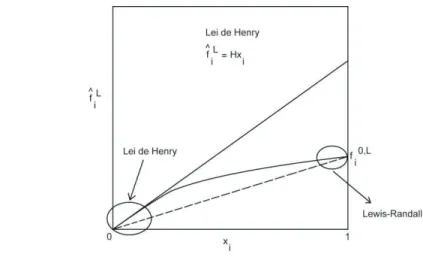
Na Figura 5.1, a linha tracejada representa o comportamento da fugacidade com composição em solução ideal (Equação (5.161)). A Equação (5.173) mostra que o coeficiente de atividade de um composto pouco solúvel em água é o inverso de sua fração molar em água na saturação. O coeficiente de fugacidade para uma espécie "i" em solução (φi) pode ser estimado a partir de EDEs ou correlações generalizadas.
Para uma espécie “i” numa mistura de gases ideais, a fugacidade é igual à pressão parcial de “i” na mistura fi = =pi y pi.
Equilíbrio Químico
A seguir, apresentaremos os critérios utilizados para prever o equilíbrio das reações químicas e como utilizá-los para determinar a constante de equilíbrio de uma reação química. Por fim, mostraremos as relações entre a constante de equilíbrio e a composição das espécies químicas presentes no meio reacional, expressas, para os casos mais simples, em termos de frações molares. A termodinâmica nos dá informações sobre o calor da reação e a conversão máxima possível, ou seja, a conversão de equilíbrio.
É assim fundamental estudar o equilíbrio das reações químicas e ter ferramentas adequadas para calcular as conversões de equilíbrio. A vantagem de definir ξ consiste no fato de que desta forma o número de variáveis do problema é reduzido: conhecendo ξ, integrando a equação (6.4), podemos calcular o número de mols de cada componente e seus componentes. fração molar (yi ou xi, dependendo da fase, vapor ou líquido). Os coeficientes νi e Xi são números adimensionais. Portanto, tanto a equação (6.4) quanto a equação (6.7) exigem que a variável ξ seja expressa em moles e, portanto, ξ é uma variável estendida.
O critério de equilíbrio para sistemas reacionários baseia-se na Segunda Lei da Termodinâmica; especificamente, o princípio do aumento da entropia. Para uma reação química ocorrendo em um reator adiabático (Q=0), a Equação (6.9) se reduz a dSsis≥0, ou seja, a reação prossegue na direção de entropia crescente (Figura 6.1). Para um sistema reacional com transferência de calor, a Equação (6.9) não é prática, pois precisamos conhecer a transferência de calor entre o sistema e a vizinhança.
De acordo com a equação (5.71), uma reação química a T e p constantes prossegue na direção de diminuir a energia livre de Gibbs (figura 6.2). Embora este critério de equilíbrio seja dado para sistemas fechados, não se limita apenas a este caso, uma vez que as propriedades termodinâmicas são independentes do caminho seguido. A partir da equação (5.71), podemos escrever uma expressão para G como uma função de ξ e procurar o valor de ξ que minimize G, ou podemos diferenciar a expressão, igualá-la a zero e resolver para ξ.
Porém, é comum simplificar a equação (6.28), tendo em mente que as reações geralmente ocorrem à pressão atmosférica e, portanto, o desvio da idealidade dos gases é normalmente pequeno. Sabendo que γi = f (T, xi) e que xi pode ser relacionado com ξ, a equação (6.32) pode ser resolvida numericamente para obter as composições de equilíbrio.