TESE DE DOUTORADO
Conversão catalítica de clorometano em
hidrocarbonetos
Leopoldo Oswaldo Alcázar Rojas
Orientador: Prof. Dr. João Fernandes de Souza
Natal / RN Março / 2012Julho
Departamento de Engenharia Química
Leopoldo Oswaldo Alcázar Rojas
Conversão catalítica de clorometano em
hidrocarbonetos
Tese apresentada ao Programa de Pós-Graduação em
Engenharia Química - PPGEQ, da Universidade
Federal do Rio Grande do Norte - UFRN, como
parte dos requisitos necessários para a obtenção do
título de Doutor em Engenharia Química sob a
orientação do Prof. Dr. João Fernandes de Sousa.
Natal / RN
hidrocarbonetos. Tese de Doutorado, UFRN, Programa de Pós-Graduação em Engenharia
Química.
Orientador: Prof. Dr. João Fernandes de Sousa
________________________________________________________________________
RESUMO:
Sílica alumina amorfa e modificada por impregnação incipiente de precursores de
ferro, níquel, zinco e cromo foram sintetizados na forma de óxido e reduzidos, tendo sido
posteriormente avaliados como catalisadores na conversão de clorometano em
hidrocarbonetos. Propriedades texturais e técnicas dinâmicas em temperatura programada
foram usadas para a determinação das propriedades ácidas dos materiais. Um modelo
termodinâmico para representar a capacidade de adsorção e dessorção de clorometano foi
avaliado. Dois tipos de reações foram abordados. No primeiro, o clorometano foi convertido
cataliticamente a hidrocarbonetos (T = 300 – 450 oC e m = 300 mg) em um reator de leito fixo
catalítico com controle de fluxo, pressão e vazão mássica e, no segundo, com o suporte puro
(T = 300 oC e m = 250 mg) foi estudado a reação de desativação do catalisador pelo coque em
um micro –adsorvedor com monitoramento gravimétrico. O teor de metal no suporte (2,5 %)
e o percentual de clorometano na mistura (10 %) foram mantidos constantes durante os
experimentos. Com os resultados obtidos foram avaliadas a conversão e a seletividade dos
produtos gasosos, ou seja, H2, CH4, C3 e C4 bem como a energia da etapa de dessorção (75,2
KJ/mol para o Ni/Al2O3-SiO2 e 684 KJ/mol para o catalisador Zn/Al2O3-SiO2), considerando
a taxa de adsorção do gás em função da temperatura. A presença do metal no suporte mostrou
uma importância significativa em termos de atividade na reação de condensação do
clorometano. Os catalisadores na forma de óxido apresentaram melhor desempenho para
obtenção de hidrocarbonetos. É possível destacar o ZnO/Al2O3-SiO2 que , em termos de
constituinte gasoso, produziu apenas C3 (Máx. de 83 %) e C4 (Máx. de 63%),
respectivamente, na temperatura de 450 oC e 20 horas de reação,. O gás hidrogênio foi
formado exclusivamente com os catalisadores FeO/Al2O3-SiO2 (Máx. de 15 %, T = 550 oC e
tempo de reação de 5,6 h) e Ni/SiO2-Al2O3 (Máx. 75 %, T = 400 oC e tempo de reação de 21,6
h). Todos os catalisadores produziram o gás metano (10 à 92 %), salvo os do tipo Ni/Al2O3
e o sequencial, onde a etapa de dessorção é a competidora. Com as equações do balanço de
massa baseadas no mecanismo proposto foram determinados duas constantes cinéticas (a
primeira de valor 8,01·10-4 min-1, relacionada à etapa da formação de hidrocarbonetos e a
segunda, 1,46·10-1 min-1, ao coque depositado no sítio do material), três constantes de
equilíbrio (a global 0,003, relativa ao clorometano 0,417 bar-1, e a de formação de
hidrocarbonetos 2,266 bar-1) e o do perfil do fator de atividade (1,516). Em termos de ajustes,
o modelo representou um comportamento satisfatório em relação ao mecanismo proposto.
Palavras-chave:
Clorometano, sílica-alumina, modelo de desativação, reator de leito fixo, hidrocarbonetos,
Amorphous silica-alumina and modified by incipient impregnation of iron, nickel, zinc
and chromium were synthetized in oxide and metal state and evaluated as catalysts for the
chloromethane conversion reaction. With known techniques their textural properties were
determined and dynamics techniques in programmed temperature were used to find the acid
properties of the materials. A thermodynamic model was used to determine the adsorption and
desorption capacity of chloromethane. Two types of reactions were studied. Firstly the
chloromethane was catalytically converted to hydrocarbons (T = 300 – 450 oC e m = 300 mg)
in a fixed bed reactor with controlled pressure and flow. Secondly the deactivation of the
unmodified support was studied (at 300 °C and m=250 g) in a micro-adsorver provided of
gravimetric monitoring. The metal content (2,5%) and the chloromethane percent of the
reagent mixture (10% chloromethane in nitrogen) were fixed for all the tests. From the results
the chloromethane conversion and selectivity of the gaseous products (H2, CH4, C3 and C4)
were determined as well as the energy of desorption (75,2 KJ/mol for Ni/Al2O3-SiO2 to 684
KJ/mol for the Zn/Al2O3-SiO2 catalyst) considering the desorption rate as a temperature
function. The presence of a metal on the support showed to have an important significance in
the chloromethane condensation. The oxide class catalyst presented a better performance
toward the production of hydrocarbons. Especial mention to the ZnO/Al2O3-SiO2 that, in a gas
phase basis, produced C3 83 % max. and C4 63% max., respectively, in the temperature of 450
o
C and 20 hours on stream. Hydrogen was produced exclusively in the FeO/Al2O3-SiO2
catalysts (15 % max., T = 550 oC and 5,6 h on stream) and Ni/SiO2-Al2O3 (75 % max., T =
400 oC and 21,6 h on stream). All the catalysts produced methane (10 à 92 %), except for
Ni/Al2O3-SiO2 and CrO/Al2O3-SiO2. In the deactivation study two models were proposed:
The parallel model, where the product production competes with coke formation; and the
sequential model, where the coke formation competes with the product desorption dessorption
step. With the mass balance equations and the mechanism proposed six parameters were
determined. Two kinetic parameters: the hydrocarbon formation constant, 8,46 10-4 min-1, the
coke formation, 1,46 10-1 min-1; three thermodynamic constants (the global, 0,003, the
chloromethane adsorption 0,417 bar-1, the hydrocarbon adsorption 2,266 bar-1), and the
activity exponent of the coke formation (1,516). The model was reasonable well fitted and
presented a satisfactory behavior in relation with the proposed mechanism.
AGRADECIMENTOS
À minha esposa Elizabeth e filhas Liliana, Blanca Virginia e Ellen, pelo apoio, compreensão e amor recebido.
Aos meus pais Oswaldo e Nilda que ainda distante sempre senti sua confiança e apoio, aos meus irmãos (Martha, Virginia, Hermann e Alonso) e respectivas famílias pelo constante carinho e incentivo.
A esperança se baseia em sonhos, em
imaginação e na coragem daqueles que se
atrevem em converter os sonhos em realidade.
“Hope lies in dreams, in imagination, and in the
courage of those who dare to make dreams into
reality”.
1 Introdução 2
1.1 Hipóteses do Trabalho ... 5
2 Revisão da Literatura 7 2.1 Indústria Petroquímica... 7
2.2 Gás Natural ... 8
2.3 Conversão de gás natural ... 9
2.4 Conversão direta de metano ... 11
2.4.1 Pirólise térmica de metano ... 11
2.4.2 Pirólise catalítica de metano ... 13
2.4.3 Homologação de metano em duas etapas (acoplamento de metano em baixas temperaturas) ... 14
2.4.4 Oxidação acoplativa de metano... 15
2.4.5 Oxidação parcial de metano a metanol e formaldeído ... 17
2.4.6 Processos indiretos ... 20
2.5 Conversão de clorometano ... 22
2.6 Catálise metálica... 23
2.7 Catálise ácida... 25
2.8 Desativação de catalisadores ... 26
3 Metodologia 36 3.1 Síntese dos catalisadores ... 36
3.2 Caracterização dos catalisadores ... 37
3.3 Sistema Reacional ... 39
3.3.1 Síntese dos catalisadores óxidos... 40
3.3.2 Síntese dos catalisadores reduzidos... 41
3.4 Desativação de catalisadores ... 41
4 Resultados e discussão 46 4.1 Caracterização dos catalisadores ... 46
4.2.2 Atividade catalítica da sílica alumina modificada com Ni ... 61
4.2.3 Atividade catalítica da sílica alumina modificada com Zn... 63
4.2.4 Atividade catalítica da sílica alumina modificada com Cr ... 65
4.3 Mecanismo de Desativação do catalisador... 67
4.4 Resolução do Modelo ... 80
5 Conclusões e futuros trabalhos 85 5.1 Conclusões... 85
5.2 Sugestões para futuros trabalhos ... 86
Referências bibliográficas 88
LISTA DE FI GURAS
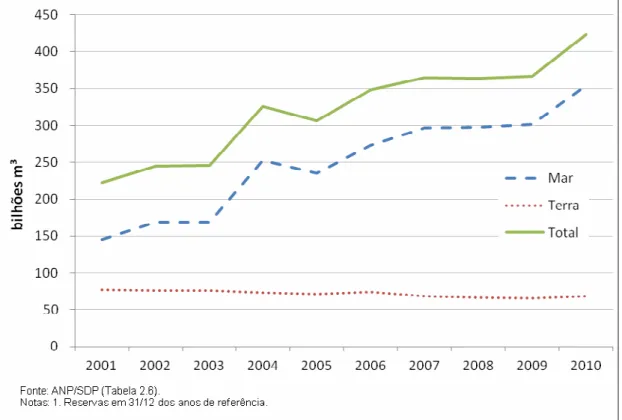
Figura 1-1: Evolução das reservas de gás natural em terra, mar e total - 2001-2010 (ANP,
2011)... 2
Figura 2-1: Cadeia Petroquímica (modificada de BRASKEM, 2011). ... 7
Figura 2-2: Produtos da cadeia petroquímica( BRASKEM, 2011). ... 8
Figura 2-3: Rotas de aproveitamento do Gás Natural ( ANDERSON, 1989). ... 10
Figura 2-4: Energia de formação dos hidrocarbonetos em função da temperatura ( HOLMEN, OLSVIK e OKSTAD, 1995) ... 12
Figura 2-5: Produção de acetileno da pirólise de metano ( HOLMEN, ROKSTAD e SOLBAKKEN, 1976)... 12
Figura 2-6: Mecanismo proposto para a reação de acoplamento oxidativo de metano sob La2O3 a 1073 K. ( SIMON, 2004) ... 16
Figura 2-7: Oxidação parcial de metano (TABATA, TENG, et al., 2002) ... 18
Figura 2-8: Mecanismo de reação da ativação de metano para bisulfato de metila com complexo de Pt (PERIANA, 1998). ... 20
Figura 2-9: Comparativo da distribuição de produtos na reação de conversão de MeX (X= Cl, Br) e de MeOH para hidrocarbonetos (OLSBYE, SAURE, et al., 2011). ... 21
Figura 2-10: Mecanismo de ativação do CH3Cl proposta por Olah (OLAH e ARPADR, 2003)... 23
Figura 2-11: Comportamento catalítico na série periódica (SINFELT, 1991)... 24
Figura 2-12: Efeito de envenenamento dos catalisadores de metanação pela adição de H2S. . 28
Figura 2-13: BARTHOLOMEV, 2001 Processo de desativação do catalisador metálico suportado: encapsulação e bloqueio de poros (). ... 29
Figura 2-14: Formação de coque sob catalisador de Ni suportado (BARTHOLOMEW, 2001). ... 30
Figura 2-15: Mecanismo de formação de coque a partir de hidrocarbonetos. ... 31
Figura 2-16: Taxas de sinterização do Pt/Al2O3 nos processos de oxidação e redução. ( FORZATTI e LIETTI, 1999). ... 32
Figura 3-1: Sistema reacional ... 40
modificado com adição de metais. ... 47
Figura 4-2: Determinação da acidez do suporte puro e modificado... 50
Figura 4-3: Representação da curva vulcano para a energia de ativação da dessorção de clorometano sob sílica alumina. ... 53
Figura 4-4: TPD de clorometano com sílica pura e óxidos dos metais ... 54
Figura 4-5: TPD de clororometano com sílica pura e metais na forma reduzida... 55
Figura 4-6: Desempenho catalítico do suporte puro de Sílica Alumina (SiAl)... 57
Figura 4-7: Desempenho catalítico da SiAl modificada com Fe na forma inicial de óxido... 60
Figura 4-8: Desempenho catalítico da SiAl modificada com Fe... 61
Figura 4-9: Desempenho catalítico da SiAl modificada com Ni na forma inicial de óxido... 62
Figura 4-10: Desempenho catalítico da SiAl modificada com Ni na forma incial de metal. ... 63
Figura 4-11: Desempenho catalítico da SiAl modificada com Zn na forma inicial de óxido .. 64
Figura 4-12: Desempenho catalítico da SiAl modificada com Zn na forma inicial de metal .. 65
Figura 4-13: Desempenho catalítico da SiAl modificada com Cr na forma inicial de óxido... 66
Figura 4-14: Desempenho catalítico da SiAl modificada com Cr reduzido... 67
Figura 4-15: Esquema reacional com formação de coque. (a) No mecanismo paralelo, (b) no mecanismo sequencial. ... 68
LISTA DE T ABEL AS
Tabela 2-1 Desempenho catalítico de catalisadores de Re e Mo suportados em HZM-5
(WANG, TAO, et al., 1993)... 14
Tabela 2-3 Desempenho catalítico para a reação acoplativa de metano ( MLECSKO e BAERNS, 1995). ... 15
Tabela 2-4 Resultados para oxidação parcial de metano em diferentes catalisadores OTSUKA e WANG, 2001. ... 19
Tabela 2-5 Exemplos de venenos para catalisadores comerciais ( FORZATTI e LIETTI, 1999) ... 27
Tabela 3-1 – Reagentes usados na síntese dos catalisadores... 36
Tabela 4-1 Conteúdo de metal impregnado no suporte (massa de metal / massa de suporte). 46 Tabela 4-2- Resultados das analises texturais para o suporte puro e modificado na forma óxido. ... 46
Tabela 4-3 Resumo dos resultados de ensaios dinâmicos em temperatura programada. ... 52
Tabela 4-4 Reações elementares com as respectivas velocidades de reação e constantes de equilíbrio envolvendo os dois modelos. ... 69
Tabela 4-5 Expressão para a concentração de sítios... 69
Tabela 4-6 Resumo de expressões cinéticas para a velocidade de reação superficial limitante. ... 72
Tabela 4-7. Balanços de massa... 73
Tabela 4-8 Expressões dos balanços para o mecanismo de coqueamento em paralelo. ... 73
Tabela 4-9 Expressões dos balanços para o mecanismo de coqueamento sequencial. ... 74
Tabela 4-10 Equações simplificadas de balance de massa considerando excesso de A. ... 75
Tabela 4-11 Parâmetros do modelo de desativação para o mecanismo paralelo... 80
SIGLA DEFINIÇÃO
bbl Barril
Boe Barris de óleo equivalente
CTGAS-ER Centro de Tecnologias do Gás e Energias Renováveis
GLP Gás liquefeito de petróleo
GTL Gas to liquid
LPG Laboratório de Processamento de Gás
Tm3 Trilhões de metros cúbicos
Bm3 Bilhões de metros cúbicos
TPD Dessorção à temperatura programada
TPR Redução à temperatura programada
UPGN Unidade de processamento de gás natural
FCC Craqueo catalítico fluidizado
TCD Detector de condutividade térmica
LETRAS AL FABÉT ICAS
SIMBOLO DESCRIÇÃO UNIDADE -
SI
∆H° Entalpía de reação kJ/mol
∆Gf° Energia livre de Gibbs de formação kJ/mol
A clorometano
A* clorometano adsorvido
ai parâmetro auxiliar (i = 1,..6)
B o grupo dos hidrocarbonetos (C1, C2 e C3),
formados no sítio catalítico
irreversivelmente no catalisador, chamado
de coque.
Ci concentração do composto i na fase gasosa
(bar)
bar
Ci* concentração do composto i adsorvido
(mol/g)
mol/g
Cl concetração de sítios livres (mol/g) mol/g
CT concentração total de sítios (mol/g) mol/g
Edes energia de ativação por mol de adsorbato kJ/mol
K constante de equilíbrio da reação
k°des constante pré-exponencial min-1
KA constante de equilíbrio de adsorção de A bar-1
KB constante de equilíbrio de adsorção de B bar-1
ki constante cinética direta da reação i min-1
k-i constante cinética inversa da reação i min-1
l sítio livre do catalisador
n ordem da taxa de dessorção.
Nc número de compostos, o balanço do
composto “i” no sistema reacional será:
Ne : fluxo molar na entrada e na saída (mol/h)
rads: taxa de dessorção mol h-1 m-2
Si seletividade a carbono convertido para a
espécie “i” produzida
LETRAS GREGAS
SIMBOLO DESCRIÇÃO UNIDADE - SI
β taxa de aquecimento (K/min)
Γdes densidade superficial do adsorbato
adsorvido
mol m-2
θ fração de cobrimento
ϑi número de carbonos do composto “i”
produzido
νi,j coeficiente estequiométrico do
composto “i” na reação “j”.
:-:-::-:::-:::::-::::::::-:::::::::::::-:::::::::::::::::::::-::::::::::::::::::::::::::::::::::-
CAPÍTULO I
INTRODUÇÃO
1
Introdução
A evolução das reservas mundiais de gás natural tiveram um continuo crescimento,
sendo que nos últimos nove anos estas reservas foram incrementadas de 168,44 trilhões de
m3, em finais de 20010 até 187,01 trilhões de m3 no final de 2010 (ANP, 2011). As maiores
reservas mundiais de gás natural encontram-se no Oriente Medio e paises da ex União
Soviética, sendo que juntos detentam o 70% do total de reservas. O país com maiores reservas
de gás natural é a Russia com 63,08 trilhões de metros cúbicos (Tm3) de gás natural (GN),
seguido de Irã e Catar com reservas da ordem de 29,61 e 25,32 Tm3. Na visão regional,
América Central e do Sul detem 8,1 Tm3 em reservas de gás natural, sendo que Venezuela
possui 70 % destas reservas seguida da Bolivia e Trinidade e Tobago com 8,8 e 5,5 % das
reservas. O Brasil possui 4,64 % das reservas da região que equivale a 0,42 Tm3, ainda que
estas reservas devam sofrer grandes incrementos caso confirmadas as estimativas das
descobertas na região do pré-sal ( HYPERLINK \l "ANP11" ANP, 2011 ). A evolução destas
reservas é apresentada na Figura 1-1.
A produção mundial de gás natural foi aproximadamente 3 Tm³ em 2010, ou seja,
7,31% maior que a produção do 2009. O Brasil terminou produzindo 14,4 bilhões de metros
cúbicos (Bm³), recuperando a queda de 2009, reflexo da crise de 2008, e aumentando a
produção de 2008 de 13,7 Bm3, o que significa um aumento consistente da produção de gás
natural no país. Segundo fontes da ANP (2011), no mês de setembro de 2010 a produção do
GN foi de 63,92 milhões de metros cúbicos por dia (Mm3/dia) dos quais após a reinjeção,
queima e o uso interno nas unidades de processamento resultou num total de 36,72 Mm3/dia
disponíveis, o que representa um incremento médio de 10% com respeito ao ano de 2009.
Atualmente, a oferta não acompanha a procura e isso se deve ao sistema termoelétrico do país
que deve servir como pulmão de energia para atender os picos de demanda ou eventualmente
uma perda da produção de base (no caso de diminuição da produção das hidroelétricas). Isso
faz com que se mantenha uma estrutura física e disponibilidade de gás para uma eventual
entrada em produção do sistema termoelétrico. Como não se tem armazenamento de grandes
volumes de gás, existem contratos de fornecimento de gás, principalmente em alguns setores
industriais, onde seu suprimento é diminuído ou suspenso quando da demanda de gás para as
termoelétricas. Isto está sendo contornado pela Petrobrás com a instalação de pontos de
recebimento e vaporização de gás natural liquefeito (GNL) comprado no mercado
internacional. A perspectiva é promissora para o gás natural, pois com o funcionamento de
novos campos como os de Santos e Espírito Santo se prevê um aumento significativo da
produção.
Estima-se que, a produção de GNL no pré-sal possa alcançar 190 a 200 Mm3/dia, um
pouco mais de três vezes o volume de produção atual. Assim, o mercado interno estará
equilibrado, tendo ainda um excedente de gás que poderá ser destinado ao mercado de
exportação que ainda é pouco regulado, apesar de não está sendo considerado como
commodity. Isto implica em ter que fechar contratos para garantir o mercado do gás. Inicialmente, o gás do pré-sal seria exportado para Estados Unidos, mas esta possibilidade se
viu afetada pelas novas descobertas de gás de xisto que aumentou a disponibilidade interna de
gás nesse país. Assim, o Brasil deverá investir em outros mercados com alguns países da
Europa que procuram alternativas ao gás da Rússia ( OLIVEIRA, 2010). Também deve-se
considerar um aumento na disponibilidade do gás pela diminuição da queima em tochas para
cumprir com recomendações ambientais. Tudo isto leva a procurar outras rotas para monetizar
as reservas, apontando à busca de alternativas para agregar valor a este gás.
Um dos problemas no consumo de gás natural é sua baixa densidade energética (em
transporte ou armazenagem. Uma alternativa é a de liquefazer o gás aumentando em 600
vezes sua densidade normal. O processo é relativamente custoso e precisa de cuidados para
sua manipulação adequada. Um outro processo consiste na transformação química de modo a
aumentar a cadeia de carbonos (hidrocarbonetos mais pesados) e ter um combustível líquido
(como a gasolina ou diesel) ou um outro facilmente liquidificável (como propano, butano,
dimetil éter).
Um processo comercialmente usado é a síntese de combustível via a rota da
transformação do gás de síntese (mistura de CO e H2) a altas pressões e temperaturas entre
200 a 350ºC conhecida como síntese de Fischer Tropsch. Dependendo das condições de
operação, tipo de reator usado e o catalisador empregado, a distribuição de produtos obtida
pode ter uma tendência a hidrocarbonetos de baixo (gasolina), médio (diesel) ou alto (ceras e
especialidades) peso molecular. Um dos grandes problemas econômicos que esta tecnologia
apresenta é a produção do gás de síntese ( WILHELM, SIMBECK, et al., 2000). A rota de
produção tem uma tecnologia madura e se têm várias fontes para a obtenção desse gás. No
caso do gás natural, este processo, conhecido como reforma de gás natural, se realiza com
uma mistura de metano e água sob catalisadores metálicos em base de Ni-Zn-Cr-Cu. Como
esta reação é altamente endotérmica a empresa Haldor Topsoe, líder na tecnologia de reforma,
introduziu uma seção de queima parcial de gás natural na parte superior do reator para
fornecer a energia necessária na reação. Ainda com este avanço tecnológico este processo
continua sendo custoso devido à unidade de fracionamento de ar para a obtenção de oxigênio
puro. Assim, a produção do gás de síntese é uma etapa cara que pode chegar a representar o
50% dos custos (operacional e de investimento) totais do projeto.
Para evitar a rota de obtenção do gás de síntese, uma alternativa consiste em ativar o
gás natural com ar (fonte de oxigênio) e cloreto de hidrogênio de modo a realizar uma
oxicloração do gás natural e obter seletivamente o cloreto de metila (ou clorometano). Esta
reação é exotérmica e se realiza em condições suaves, (baixa pressão e temperatura). Por sua
vez o cloreto de metila em determinadas condições reage produzindo hidrocarbonetos de
maior valor agregado. O ácido clorídrico é liberado nesta segunda etapa e reciclado para o
primeiro processo. Desta forma, esta é uma rota alternativa à síntese de Fischer Tropsch onde
que as condições de operação são muito menos rigorosas. Uma das desvantagens deste
processo está na manipulação de ácido clorídrico que é altamente corrosivo. Este problema
Assim, neste trabalho foi realizada a síntese e caracterização de catalisadores e
avaliação do desempenho para a reação de conversão de clorometano em hidrocarbonetos de
maior peso molecular.
O material escolhido para ser usado como catalisador foi a sílica alumina (SiAl)
extrapolando a idéia de paralelismo entre a conversão de metanol e clorometano, visto que
existem trabalhos na literatura usando este suporte para a conversão de metanol para
hidrocarbonetos ( ESPINOZA, 1986, ESPINOZA, STANDER e MANDERSLOOT, 1983,
SEDRAN, COMELLI e FÍGOLI, 1984, COMELLI e FÍGOLI, 1991).
O presente trabalho encontra-se estruturado em quatro capítulos. No primeiro tem-se
uma revisão da literatura abordando o estado da arte e revisão bibliográfica. No segundo
capítulo, é apresentada a metodologia experimental e os materiais e métodos empregados. Os
resultados e discussões são apresentados no capítulo terceiro e por último é apresentado um
modelo de desativação que representa a deposição do coque no catalisador. Para finalizar
estão enumeradas as conclusões e recomendações para futuros trabalhos.
1.1
Hipóteses do Trabalho
A conversão de clorometano em hidrocarbonetos é feito mediante um mecanismo
heterolítico (iônico).
A sílica alumina é um suporte estável térmica e quimicamente com propriedades
ácidas e texturais (distribuição de poros e área superficial) adequadas para que a reação seja
realizada.
À semelhança da reação de conversão de metanol, a sílica alumina deverá ser ativa
:-:-::-:::-:::::-::::::::-:::::::::::::-:::::::::::::::::::::-::::::::::::::::::::::::::::::::::-
CAPÍTULO II
REVISÃO BIBLIOGRÁFICA
2
Revisão da Literatura
Neste capítulo, são abordados os aspectos teóricos relacionados com a revisão bibliográfica
que tangem os assuntos referentes ao presente trabalho visando facilitar a compreensão das
atividades abordadas.
2.1
Indústria Petroquímica
A indústria petroquímica organiza-se em produtos de primeira, segunda e terceira
geração dependendo da fase de transformação dos insumos petroquímicos (Figura 2-1). Esta
indústria transforma subprodutos do petróleo, principalmente nafta e gás natural, em produtos
de consumo final e insumos para diversas indústrias ( BRASKEM, 2011).
BRASKEM, 2011
Figura 2-1: Cadeia Petroquímica (modificada de BRASKEM, 2011).
Os principais produtos de cada fase da cadeia petroquímica são apresentados na Figura
2-2. No Brasil, a principal fonte de matéria-prima da petroquímica é a nafta de petróleo e em
menor quantidade o gás natural, basicamente o etano. Pelas características de ter uma cadeia
interligada a indústria petroquímica se desenvolve em base de arranjos industriais chamados
de pólos. Estes pólos se localizam junto a refinarias ou fontes de gás natural. Grandes plantas
plantas também produzem as resinas (produtos de segunda geração). A terceira fase
normalmente esta distribuída em muitas empresas que produzem o bem de consumo.
Figura 2-2: Produtos da cadeia petroquímica( BRASKEM, 2011).
A primeira geração de produtos está baseada em três tipos principais de
intermediários: as olefinas na faixa de C2-C4, os aromáticos na faixa de C6-C8 e o gás de
síntese (H2 + CO) ( IWASAKI, REINNIOKANENB, et al., 1998). O gás natural é uma
matéria-prima mais barata que a nafta e dele podem-se obter as olefinas leves (principalmente
o eteno), sendo que a nafta fornece tanto as olefinas (por craqueamento) como os compostos
aromáticos da primeira geração.
2.2
Gás Natural
O gás natural não processado é uma mistura de hidrocarbonetos leves, composto
principalmente de metano (até 98%) e de alguns hidrocarbonetos de maior peso molecular (C2
a C6), acompanhados de outros componentes não hidrocarbônicos. Os constituintes não
hidrocarbônicos variam de acordo com o local de origem. Os mais comuns são o gás
Alguns reservatórios de gás natural contêm grande quantidade de hélio, viabilizando a
comercialização deste último.
O gás natural pode ser classificado em gás não associado ou “livre” e gás associado. É
classificado como não associado quando produzido em poços com pouco petróleo. Quando a
sua produção é realizada em poços com predominância de óleo, onde pode estar dissolvido no
petróleo ou acumulado na forma de uma capa de gás, ele é classificado como gás associado.
Neste caso, a sua produção está “associada” à produção do petróleo ( THOMAS, 2001).
O gás natural, antes de ser disponibilizado para uso é submetido a tratamento para a
remoção de H2S, de umidade e demais impurezas. Além disto, os componentes mais pesados,
como o propano e o butano são separados da corrente de gás natural para serem usados como
combustíveis e/ou insumos petroquímicos. Assim, etano, pode ser separado e empregado para
a síntese do etileno. Propano e butano são recuperados e comercializados como gás liquefeito
de petróleo (GLP).
2.3
Conversão de gás natural
As grandes reservas de gás natural fazem do metano um importante material para a
indústria química bem como o etano e o propano. Os processos industriais de obtenção destes
compostos acontecem por meio do craqueamento catalítico seguido do processo de
deshidrogenação para a obtenção das respectivas olefinas.
Ainda que o metano seja uma excelente fonte de materia prima o seu maior uso é
como combustível tanto para uso doméstico como industrial. Entretanto, enormes reservas de
gás natural encontram-se distantes do mercado de consumo. Assim, como o caso do gás do
pré-sal uma opção interessante é converter o gás para líquidos (GTL) de modo a aumentar a
sua densidade energética para poder ser transportado e armazenado com maior facilidade. Na
Figura 2-3: Rotas de aproveitamento do Gás Natural ( ANDERSON, 1989).
O metano pode ser convertido por duas rotas, uma via gás de síntese ou diretamente
para hidrocarbonetos C2 ou metanol. Atualmente os processos comerciais para a conversão de
gás natural em grande escala passam pela produção de gás de síntese, ainda que na Figura 2-3
se mostram outras rotas alternativas. A reforma a vapor é um processo predominante na
produção de gás de síntese.
(1)
Uma rota alternativa para produzir gás de síntese é a oxidação parcial de metano que
apresenta um gás de síntese com relação H2:CO de 2,0. A combinação de ambas as reações dá
a reforma auto-térmica com uma relação H2:CO que dependerá da composição da carga sendo
ajustada dependendo do uso do que terá o gás de síntese.
(2)
As indústrias têm produzido em grande escala gás de síntese a partir da síntese de
amônia e metanol. Combustíveis sintéticos também podem ser produzidos a partir de gás de
síntese (Figura 2-3) via reação de Fischer-Tropsch (GTL). Nos últimos anos muito esforço
tem sido dado para melhorar a tecnologia GTL. Varias plantas de demonstração foram
O grande problema da tecnologia GTL está na produção de gás de síntese que pode
representar 50% ou mais do custo de capital ( WILHELM, SIMBECK, et al., 2001). Assim, é
de interesse o desenvolvimento de outra rota que não passe pela produção de gás de síntese o
qual será o foco desta revisão cuja visão geral consiste em apresentar uma rota direta de
conversão de metano.
2.4
Conversão direta de metano
Metano é uma molécula bastante estável, com um ponto de fusão em -182.5 °C e
ponto de ebulição de - 161.5 °C, devido ás ligações C–H serem muito fortes (425 kJ/mol) e
devido a ausência de grupos funcionais, momento dipolar magnético e assimetria o que
dificulta o ataque químico. Para poder quebrar a ligação C-H se precisa de altas temperaturas
e/ou a ajuda de agentes oxidantes. Nestes processos oxidativos a catálises tem um papel
importante para a conversão de metano.
Apesar dos enormes esforços em desenvolver a rota direta de conversão de gás natural,
até o momento não se tem um processo bem definido. Um dos motivos são as restrições
cinéticas e termodinâmicas que devem ser considerados. É necessário temperaturas elevadas
para ativar o metano e nessas condições as reações de radicais livres na fase gasosa são
preponderantes. A força da ligação C-H no metano é maior que a força de seus produtos,
significando que os produtos serão mais reativos que o metano. Desta forma o desafio está
mais na seletividade que na reatividade. Algumas soluções em nível de catálises e engenharia
das reações foram propostas, as quais são descritas a seguir.
2.4.1 Pirólise térmica de metano
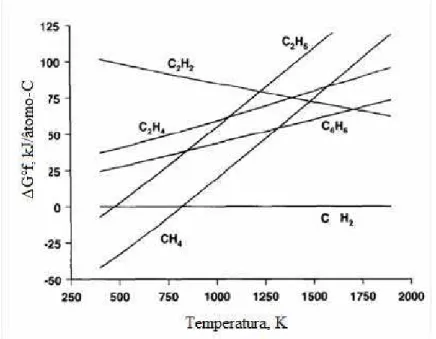
Cálculos no equilíbrio ( HOLMEN, OLSVIK e OKSTAD, 1995) indicam que a
composição dos produtos na pirólise térmica de metano contém etileno, acetileno, benzeno e
hidrogênio como produtos principais sempre que a reação não acontece até a formação de
coque. A Figura 2-4 apresenta o comportamento do equilíbrio termodinâmico como resposta a
Figura 2-4: Energia de formação dos hidrocarbonetos em função da temperatura ( HOLMEN, OLSVIK e OKSTAD, 1995)
∆G°f, kJ/átomo-C
Acetileno e hidrogênio são os principais produtos (Figura 2-5). A formação de
carbono pode ser controlada diminuindo o tempo de residência e a pressão parcial de metano,
diluindo a alimentação preferencialmente com hidrogênio. O resfriamento rápido da reação é
também importante. Altas taxas de produção de acetileno (aprox. 90%) podem ser obtidas a
temperaturas extremas (maiores de 2000 K) e tempos de contato baixos (<10-2s).
Figura 2-5: Produção de acetileno da pirólise de metano ( HOLMEN, ROKSTAD e SOLBAKKEN, 1976).
O mecanismo de reação está baseado num complexo sistema de reações de radicais
livres na fase gasosa. A reação global pode ser descrita como a deshidrogenação em altas
2 CH4 → C2H6 + H2 → C2H4 + H2 → C2H2 + H2 → 2C + H2 (3)
Para esta reação acontecer em condições comerciais é necessário de uma engenharia
adequada. Alguns exemplos de operação industrial são o arco elétrico da Huls, (Huels,
DuPont), técnicas regenerativas (Wulff process), combustão incompleta com O2 (BASF,
SBA, Tsutsumi) e misturas com gases de combustão. A diferença fundamental entre estas
tecnologias é a forma como se fornece e se remove o calor ao reator.
Algumas unidades têm sido propostas e inclusive testadas em escala piloto. O
processo dispensa o uso do gás de síntese e não produz cera. O metano é convertido a
acetileno numa câmara de craqueamento. A temperatura é suficientemente alta para converter
o metano e produzir acetileno e hidrogênio. Vapor de água é adicionado para diminuir a
formação de coque e por questões de segurança o acetileno é hidrogenado a etileno. O etileno
pode ser oligomerizado para obter produtos mais pesados. Se a oligomerização for conduzida
a baixas pressões se obtém gasolina de aviação e incrementando a pressão óleo diesel. Após o
reator, a corrente é estabilizada, separando os produtos líquidos e reciclando os gasosos.
O Instituto Frances do Petróleo (IFP) também construiu e operou uma planta piloto de
pirólise de metano a 1473 K. Usando 50% de hidrogênio como diluente o IFP reportou uma
seletividade a acetileno e etileno de 32 e 23%, respectivamente, com uma conversão de 31%.
Entretanto o processo apresenta seletividades de 15% a benzeno e 18% a coque.
2.4.2 Pirólise catalítica de metano
Metano pode ser convertido a benzeno, tolueno, naftaleno e hidrogênio
(desidroaromatização) a temperaturas menores que as usadas na decomposição térmica,
porém as conversões estão limitadas pela termodinâmica. Para temperaturas de 700 °C e 1
atm a conversão de metano é de 12%, sendo que quase a metade é convertido a benzeno e
metade a naftaleno. Na temperatura de 800 °C a conversão de equilíbrio é de 24%.
Vários catalisadores bifuncionais promovem esta reação. Os catalisadores são
típicamente zeólitas e a atividade do metal segue a tendência Mo > W > Fe > V > Cr.
O catalisador de Mo/HZSM-5 é o mais estudado desde o trabalho pioneiro de
WANG, TAO, et al., 1993. Alguns resultados típicos deste estudo são apresentados a seguir
Tabela 2-1 Desempenho catalítico de catalisadores de Re e Mo suportados em HZM-5 (WANG, TAO, et al., 1993)
Seletividade (%) Catalisador Tempo
(h)
Conver. de CH4 (%)
C2 Benzeno Naftaleno Coque
5% Re 2 7 4 48 11 33
2% MoO3 1 6 4 50 n.a. 43
2% Mo 2 9 3 57 15 15
4% Mo 2 10 2 65 18 3
O mecanismo pode acontecer em duas etapas, convertendo o metano a etileno (ou
acetileno) com carbeto de molibdênio seguido da conversão de etileno (ou acetileno) para
aromáticos nos sítios ácidos da HZSM-5. A desativação do catalisador ocorre pela deposição
de coque e compostos pesados que se formam na superfície externa do catalisador bloqueando
os poros. Várias técnicas foram testadas para melhorar a estabilidade do catalisador como
tratamento com vapor, adição de CO/C02 na alimentação, inertização dos sítios ácidos externo
da zeólita por moléculas organosilano. Também foi levantado o fato que com uma boa
escolha do precursor de Co a estabilidade aumenta. Uma unidade piloto foi construída para
demonstrar a tecnologia.
Uma tecnologia usada para melhorar o sistema consiste em retirar o hidrogênio
formado de modo a deslocar o equilíbrio para os produtos. Isso é conseguido com um agente
oxidante do hidrogênio ou com uma membrana de hidrogênio. Com o catalisador de
Mo/HZSM-5 ( KINAGE, OHNISHIi e ICHIKAAWA, 2003) uma melhora significativa da
conversão de metano e a formação de benzeno, tolueno, naftaleno e hidrogênio foram
observados.
2.4.3 Homologação de metano em duas etapas (acoplamento de metano em
baixas temperaturas)
Neste processo o metano se descompõe numa primeira etapa seguida pela
hidrogenação das espécies carbonáceas para obter o C2. Como este processo tem restrições
termodinâmicas devido ao metano ser mais estável que o C2 a reação se faz em duas
temperaturas. A primeira etapa, a decomposição de metano é realizada a 700 K sob
catalisadores de Ru, Rh e na segunda, a hidrogenação acontece a 373 K ( KOERTS, DEELEN
e VAN SANTEN, 1992). Os produtos apresentam uma distribuição de espécies do tipo
e a amorfa. A fase carbônica é a responsável pela formação dos hidrocarbonetos, entretanto a
fase amorfa se apresenta como inativa.
Alguns aspectos fundamentais foram estudados onde a quimissorção de metano sob
Ru apresenta espécies CH e CCH2 a 400 e 700 K. A hidrogenação de CCH2 leva à formação
de etano, entretanto, as espécies CH podem ser hidrogenadas a metano ou transformadas a
CCH2.
2.4.4 Oxidação acoplativa de metano
Na reação acoplativa o metano e oxigênio reagem num catalisador sólido para formar
etileno e acetileno. Desde o trabalho de KELLER, 1982 muitas pesquisas foram
desenvolvidas. O grande problema desta reação é que ao ativar a ligação C-H do metano, os
sítios catalíticos são ativados promovendo a ativação das ligações C-H do etano e etileno
resultando na formação de CO2 por combustão. A taxa de produção de C2 está limitada a 25%
com uma seletividade a C2 de 80%. Um dos melhores catalisadores reportados foram o
SrO/La2O3 e Mn/Na2WO4/SiO2. Não é evidenciado um melhoramento na taxa de produção de
C2. Existem trabalhos ( SU, YING e GREEN, 2003) que impõem um limite teórico de 30% na
pressão atmosférica. Alguns resultados são apresentados na Tabela 2-2 a seguir.
Tabela 2-2 Desempenho catalítico para a reação acoplativa de metano ( MLECSKO e BAERNS, 1995).
C2 (%)
Catalisador T (K) XCH4 (%)
Seletividade Produção
Li/MgO 1013 37,8 50,3 19,0
BaF2/Y2O3 1023 36,1 62,1 22,4
Rb2WO4/SiO2 1123 32,0 78,0 25,0
La2O3–CeO2 1048 22,3 66,0 14,7
Na2WO4/SiO2 1123 44,0 52,0 22,9
A oxidação acoplativa de metano é uma reação exotérmica e a formação de óxidos de
carbono aumenta o calor gerado além de reduzir a seletividade, de modo que a remoção deste
calor é um problema de engenharia a ser levado em conta.
A reação transcorre num mecanismo heterogêneo e homogêneo. O metano é ativado no
catalisador para formar radicais metila. Estes radicais ao passar á fase gasosa reagem para
O mecanismo representado na Figura 2-6 descreve duas rotas distintas, uma leva à formação
de oxigenados e a outra a hidrocarbonetos. Em altas conversões, uma terceira rota, a
decomposição do etileno ganha importância. Estudos com marcadores isotópicos mostram
que em baixas conversões o CO2 origina-se do metano, enquanto o etileno surge em altas
conversões ( LUNSFORD, 2000).
Figura 2-6: Mecanismo proposto para a reação de acoplamento oxidativo de metano sob La2O3 a 1073 K. ( SIMON, 2004)
Os desafios da reação de acoplamento oxidativo são encontrar um catalisador que
ative simultaneamente o metano e iniba a reação de etano. Algumas propostas incluem o uso
de membranas onde os gases metano e oxigênio são separados. O uso de reatores cíclicos
onde o oxigênio é fornecido pelo catalisador também foi proposto por GREISH, GLUKHOV,
et al., 2010.Um exemplo é observado nas reações oxidativas do metano utilizando materiais do tipo perovsquita ( TAN, 2006). No reator, o oxigênio é alimentado em um lado da
membrana enquanto o metano do lado oposto. Outra aproximação é a proposta por
problema desta tecnologia é a necessidade de membranas e/ou adsorvedores para retirar
seletivamente o etileno. Para a zeólita 5A foi usado um processo de TSA (temperature swing
adsorption), adsorvendo a 30 °C e dessorvendo a 400 °C incrementando assim a taxa de produção de etileno. Numa variação do processo de reciclo, o etileno produzido passa por um
segundo reator onde reage a 500 °C sob o catalisador de Ga/HZSM-5 para produzir
aromáticos ( LUNSFORD, 2000). A presença de etileno incrementa a produção de aromáticos
visto que este é um intermediário da reação de aromatização.
O uso de CO2 substituindo o O2 foi também proposto MLECSKO e BAERNS, 1995.
Vários catalisadores foram usados, porém, ainda a produção de etano é baixa.
2.4.5 Oxidação parcial de metano a metanol e formaldeído
A oxidação seletiva de metano para metanol é uma rota muito atrativa que poderia
inovar a forma de utilização do metano. A reação básica é:
(4)
Muitas pesquisas têm sido feitas com esta reação onde são propostos vários caminhos
possíveis:
• Rota de alta temperatura em fase homogênea baseada em radicais livres, TABATA, TENG, et al., 2002;
• Rota catalítica de baixa temperatura, OTSUKA e WANG, 2001;
• Rota homogênea em solução PERIANA, TAUBE, et al., 1993;
• Rota de catálises enzimática LABINGER, 1995.
Para as reações em fase gasosa com radicais livres em altas pressões, a presença do
catalisador não parece ter algum efeito. Alguns resultados obtidos por TABATA, TENG, et
Figura 2-7: Oxidação parcial de metano (TABATA, TENG, et al., 2002)
A pressão tem um efeito pronunciado na seletividade da oxidação parcial de metano.
Nas condições onde o oxigênio foi consumido completamente um aumento de pressão leva a
uma diminuição na formação de CO e um aumento à seletividade a metanol. Na literatura se
menciona que se a reação está sendo feita entre 430 – 470 °C seria necessário uma pressão de
50 bar (ZHAN, HE, et al., 2008). Os resultados mostram uma seletividade de 30 - 40% para
uma conversão de 5-10% em temperaturas entre 350 a 400 °C e pressões entre 30 a 60 bar.
Dados teóricos e experimentais apontam baixas taxas de produção de metanol. Reciclando o
metano não convertido é um esquema proposto para obter uma possível rota GTL.
Em pressões baixas, o catalisador tem um efeito importante nas taxas de produção de
metanol. Embora maiores pressões favoreçam a síntese de metanol, a combustão a altas
pressões é controlada pela fase gasosa. Na revisão de Otsuka e Wang (2001Tabela 2-3
Resultados para oxidação parcial de metano em diferentes catalisadores OTSUKA e WANG,
2001.) para a conversão direta de metano para oxigenados, são apresentados resultados de
vários autores, Tabela 2-3. Os resultados foram obtidos a temperaturas acima de 500 °C,
sendo que as conversões de metano foram baixas e o único produto oxigenado observado foi o
formaldeído. Quando a reação acontece com um catalisador de MoO3/SiO2 a uma temperatura
de 600 °C com excesso de vapor de água, têm-se uma conversão de metano de
aproximadamente 25% e altas seletividades a metanol e formaldeído. Este aumento na
Tabela 2-3 Resultados para oxidação parcial de metano em diferentes catalisadores OTSUKA e WANG, 2001.
Seletividade (%) Catalisador Temperatura
(°C)
Conversão de CH4 (%) CH
3OH HCHO
SiO2 620,0 4,8 0,0 24,0
Mo/SiO2 650,0 5,2 0,0 32,0
V2O5/SiO2 650,0 13,5 0,0 25,3
MoSnP/SiO2 500,0 7,2 0,0 64,8
Tem sido reportado que com a adição de 1% de NO na alimentação a produção de
oxigenados (metanol e formaldeído) foi aumentada em 16% com catalisadores de V2O5/SiO2
a 650 °C, indicando que este efeito é representativo de um mecanismo misto
heterogêneo-homogêneo ( BARBERO, ALVARES, et al., 2002).
As espécies isoladas de molibdênio sob a sílica têm a melhor atividade e seletividade
para a conversão direta de metano para formaldeído. Por enquanto não se encontrou um
catalisador capaz de produzir diretamente metanol de metano. A razão parece ser que na
temperatura de decomposição do metano, o metanol formado seria descomposto a formadeído
e óxidos de carbono (COx). Assim o desafio é obter catalisadores que a baixas temperaturas
ainda possam ativar a ligação C-H do metano.
A ativação de metano em baixas temperaturas está sendo estudada em reações
homogêneas desde o primeiro trabalho de PERIANA, TAUBE, et al., 1993, os quais
avaliaram a atividade de um catalisador constituído de um complexo de Hg com ácido
sulfúrico concentrado:
(5)
O bisulfato pode se hidrolisar a metanol, na forma:
(6)
Um complexo de platino II também foi usado como catalisador ao invés do complexo
de mercúrio, obtendo-se uma conversão de 90% e uma seletividade a bisulfato de metila de
sulfúrico, o que é um processo custoso. Um esquema do mecanismo proposto por (PERIANA,
1998) é apresentado a seguir na Figura 2-8..
Figura 2-8: Mecanismo de reação da ativação de metano para bisulfato de metila com complexo de Pt (PERIANA, 1998).
2.4.6 Processos indiretos
Com o aumento do preço do petróleo as rotas de transformação do gás natural
(principalmente o metano como maior constituinte) ganharam interesse. Assim, ganha
importância o trabalho de OLAH (1983) onde reportou uma interessante rota de três etapas
para converter gás natural em olefinas. Neste processo o metano é inicialmente halogenado
seletivamente para obter um composto monohalogenado sob um catalisador ácido suportado
ou metálico com platina. Na segunda etapa a metila halogenada é transformada a metanol via
uma reação de hidrólise catalítica e finalmente o metanol é convertido a hidrocarbonetos pelo
processo MTG sob o catalisador de HZM-5. No entanto LERSCH e BANDERMANN, 1991;
MURRAY, CHANG e HAW, 1993 e JUAMAIN e SU, 2002, apresentaram estudos onde
mostraram que este processo pode ser melhorado convertendo diretamente o halo metano
processo proposto por Olah pode ser reduzido a duas etapas, representado pelas seguintes
equações:
(7)
(8)
Na primeira etapa o metano passa por um processo de oxicloração seletiva a
clorometano e na segunda etapa o clorometano é convertido a hidrocarbonetos, num processo
de oligomerização. O ácido clorídrico liberado na segunda etapa pode ser reciclado para ser
usado novamente na etapa de oxicloração.
Por outro lado outros autores, ( OLSBYE, SAUREe, et al., 2011) ressaltam o
paralelismo entre as reações de metanol para hidrocarbonetos (MTH) e de halometanos para
Figura 2-9: Comparativo da distribuição de produtos na reação de conversão de MeX (X= Cl, Br) e de MeOH para hidrocarbonetos (OLSBYE, SAURE, et al., 2011).
Na Figura 2-9 é feita uma comparação da conversão do metanol e clorometano e a
distribuição dos produtos obtidos. Esta comparação é feita quando se usa a zeólita ZSM-5 ou
o SAPO (silicoalumino-fosfato) como catalisadores, tanto para a conversão de metanol como
a conversão de clorometano. A similaridade na distribuição de produtos de ambas as reações
para os materiais sugere um mecanismo de reação similar ( WEI, ZHANG, et al., 2005;
SVELLE, ARAVINTHAN, et al., 2006).
Na reação de oxicloração numerosos catalisadores foram propostos. Os principais se
baseiam em sais de cobre com certos promotores de potássio e lantânio que servem como
redutor do ponto de fusão e estabilização do cobre no catalisador, respectivamente.
Por outro lado, na reação de conversão de clorometano foram testados muitos
catalisadores entre estruturados e amorfos, com propriedades ácidas e básicas, com e sem
centros metálicos. Uma revisão dos catalisadores usados na conversão de clorometano pode
2.5
Conversão de clorometano
A reação de conversão do clorometano para hidrocarbonetos é uma reação heterolítica
sendo o cloro o elemento altamente eletrofílico (OLAH e ARPADR, 2003). Para alterar a
ligação carbono cloro (C-Cl) é necessário de um doador de elétrons (sítio ácido de Lewis) que
debilite a ligação, levando eventualmente a sua incisão. Assim, após a adsorção e cisão do
clorometano no sitio ácido, o cloro ficará adsorvido no sítio, liberando um grupo metila. Os
grupos metilas reagem, via deshidrogenação, para formar um novo radical na superfície
(principalmente a metálica), e o hidrogênio liberado é transportado pela superfície (spillover)
reagindo ao encontrar o cloro adsorvido para formar o ácido clorídrico que eventualmente
será dessorvido. A Figura 2-10 representa este mecanismo de forma simplificada. As espécies
dentro do círculo representa a superfície catalítica bem como as espécies nela adsorvidas. As
espécies gasosas são adsorvidas e dessorvidas segundo a círculo externo. O movimento do
hidrogênio está representado no círculo interno.
CH3Cl
CH3---Cl
CH3 CH2=CH2
sítio ácido
HCl CH3CH3
CH3Cl
H H H
Figura 2-10: Mecanismo de ativação do CH3Cl proposta por OLAH e ARPAR (2003).
Este mecanismo será descrito em maior detalhe no item a seguir, mas o que se pode
depreender é que existem dois mecanismos que devem atuar paralelamente, um nos sítios
bifuncionais, como descrito na patente de (OLAH, 1983) onde a função ácida do catalisador
permite a adsorção e remoção do cloro da molécula de clorometano e a função metálica
realiza a condensação dos grupos metilas via uma reação de deshidrogenação. De modo a
entender melhor estes mecanismos, a seguir é apresentado um resumo dos aspectos teóricos
da catálise metálica e da catálise ácida de modo a se ter um melhor entendimento dos
diferentes catalisadores propostos na literatura.
2.6
Catálise metálica
Na catálise metálica as reações de hidrogenação e dehidrogenação são as mais
importantes. A maior parte dos catalisadores metálicos são metais de transição, portanto os
orbitais d têm um papel preponderante neste mecanismo (GATES, KATZER e SCHULTZ,
1979).
Os metais são arranjos de átomos ordenados tridimensionalmente. Ainda que as
propriedades do seio destes arranjos sejam importantes para entender o comportamento
metálico, deve se considerar que a catálise é um fenômeno de superfície sendo justamente a
superfície do metal onde as interações metal adsorbato acontecem. Estes átomos superficiais
têm deficiência de átomos vizinhos, conferindo à superfície diferentes características
eletrônicas comparadas com os átomos interiores. O grande número de átomos e a
proximidade deles fazem que os orbitais dos átomos se superponham. Assim, as faixas de
energia dos orbitais s, p e d terminam se superpondo formando uma banda energética continua
que contem os elétrons de enlace. O elétron de máxima energia define o nível de Fermi. Por
baixo dele todos os orbitais estão ocupados e por acima dele desocupados. A energia que
apresenta os orbitais dos átomos superficiais encontra-se acima do nível de Fermi. Nos
átomos superficiais os orbitais tipo d não ocupados ficam livres em direção normal à
superfície, sendo altamente reativos. O fenômeno de adsorção depende da interação destes
orbitais superficiais com os orbitais moleculares da molécula no adsorbato. Conforme
aumenta o número atômico numa fila da tabela periódica, tem-se um aumento da quantidade
de orbitais superficiais que interagem com os orbitais moleculares do adsorbato, aumentando
assim a interação e, portanto, a força de adsorção (GATES, KATZER e SCHULTZ, 1979).
A relação entre a série periódica, o calor de adsorção e a atividade catalítica para a
Figura 2-11: Comportamento catalítico na série periódica (SINFELT, 1991).
Como é possível observar, a atividade catalítica passa por um máximo o qual é
atribuído à força de interação superfície adsorbato ao longo da série. No extremo esquerdo a
interação é fraca conseqüentemente uma fraca adsorção, sem conseguir segurar o adsorbato o
tempo suficiente para acontecer a reação. Por outro lado no extremo direito esta interação é
muito forte dificultando a reação (pois a interação do adsorvente com a superfície é maior que
a afinidade com os reagentes) mantendo o adsorbato adsorvido. Entre estes dois efeitos
encontra-se o termo ótimo que origina o máximo na atividade catalítica. Mudando o adsorbato
e as condições da reação a interação muda. Os reagentes adsorvidos apresentam uma baixa
energia de ativação que faz que a reação possa acontecer. Assim se pode dizer resumir que na
catálise metálica os orbitais d superficiais tem alta afinidade pelos elétrons formando uma
ligação com o adsorbato de forma que dependendo do metal esta ligação pode ser muito fraca
o forte e no caso ótimo será o suficientemente forte para quimissorver o reagente para reagir e
o suficientemente fraco para permitir os produtos se dessorverem.
2.7
Catálise ácida
Segundo SANTILLI e GATES, 2008, as reações ácido-base se caracterizam por
serem cíclicas onde ocorre transferência de hidrogênio. Quando o grupo doador é hidratado e
se dissocia, o íon (H3O)+ é formado tornando-se uma espécie catalítica ativa. Este tipo de
catálise é chamada de “catálise ácida específica”. Por outro lado quando o ácido não se
complexo e mais comum nos processos catalíticos. Tipicamente os óxidos metálicos têm a
capacidade de serem doadores de prótons através dos grupos OH- (ácidos Bronsted) e
aceptores de par de elétrons (ácido Lewis). Ainda que se possa perceber uma relação entre a
atividade catalítica e a acidez do catalisador, a medida da acidez não da informação que
permita entender o mecanismo envolvido ou a natureza dos sítios.
As reações superficiais onde há transferência de carga, transporte de próton ou elétron,
são definidas como reações do tipo ácido-base. Em geral um óxido pode ser considerado
como ácido ou base a depender de sua capacidade de doar ou receber elétrons (definição de
Lewis) ou de receber e doar prótons (definição de Bronsted). Assim, de acordo a definição de
Lewis, um ácido é aquele que tem a capacidade de receber um par de elétrons e uma base é
um composto que pode doar (ou transferir) um par de elétrons. A acidez de um íon metálico
aumenta com sua carga (Na+, Ca+2, Y+3, Th4+). Por outro lado a capacidade de uma superfície
catalítica de transferir um próton define a acidez do catalisador segundo a definição de
Bronsted. Uma forma de medir a acidez é adsorvendo uma base fraca de modo a medir os
sítios ácidos existentes e na dessorção uma rampa linear de temperatura se medirá a força do
ácido. A força ácida classifica os catalisadores em catalisadores ácidos e superácidos quando
a acidez destes é superior à força ácida do ácido sulfúrico (SOMORJAI, 1994).
Uma das reações mais importantes dos catalisadores ácidos com os hidrocarbonetos é
a formação de íons carbônios. Para a reação com olefinas se tem:
(9)
Estes íons carbônios são muito instáveis. Os íons de carbono terciários são mais ativos que os
secundários e estes mais ativos que os primários. Entre as principais reações que transcorrem
via estes intermediários podem ser mencionadas as reações de incisão da ligação
carbono-carbono, de isomerização e de formação de ligação carbono-carbono (alquilação). Na
indústria de refino de petróleo estas reações são feitas com zeólitas, que são particularmente
ativas para converter as olefinas e cicloparafinas em parafinas e aromáticos, respectivamente.
Um dos maiores problemas do uso de catalisadores sólidos é a perda de atividade que
estes apresentam ao longo do tempo. Este é um processo físico e químico e normalmente
inevitável, mas tem algumas técnicas que podem minimizar os seus efeitos.
Entre os principais fatores que levam à desativação do catalisado, é possível citar:
• Envenenamento
• Coqueamento ou bloqueio
• Sinterização ou transformação de fases
Apresentaremos uma breve descrição destes tipos para seguidamente apresentar os aspectos
cinéticos da desativação de catalisadores.
Envenenamento é um processo químico onde certas impurezas da alimentação são
fortemente adsorvidas nos sítios ativos do catalisador, alguns exemplos são apresentados na
Tabela 2-4Tabela 2-1. A adsorção de uma base num sítio ácido é um exemplo de
envenenamento. O envenenamento do sítio pode ser um processo geométrico onde o
adsorbato cobre o sítio ou mediante um efeito elétrico onde este campo elétrico distorce a
afinidade de adsorção de outros adsorbatos. Os venenos podem inclusive reagir quimicamente
com o sítio formando novos produtos (reconstrução) de modo que o desempenho do
catalisador é mudado.
Para distinguir entre um processo de alta interação e irreversível de outro de pouca
interação e reversível,tem-se a diferença entre veneno e inibidor, respectivamente. Quando o
veneno atua em todos os sítios se diz que é não seletivo, sendo que a atividade do catalisador
cai linearmente com a quantidade de veneno adsorvido. No caso de venenos seletivos, por
exemplo, em sítios ácidos, o veneno atacará primeiramente os sitos de maior (ou menor)
acidez, mudando assim a atividade do catalisador de acordo à função específica de cada tipo
de sítio. Alguns catalisadores atacados com venenos podem ser regenerados de maneira
reversível, que é o caso do COx e H20 na superfície do catalisador de amônia. Para regenerar
só é preciso retirar esses compostos da alimentação e logo regenerar a superfície do
catalisador passando hidrogênio. Mais quando a concentração superficial é alta estes venenos
reagem com o sólido formando produtos irreversíveis como a carbonila de ferro. Em caso de
reações insensíveis à estrutura, a perda de atividade não muda necessariamente a seletividade,
mas em catalisadores multifuncionais ou reações sensíveis à estrutura a diminuição na
Tabela 2-4 Exemplos de venenos para catalisadores comerciais ( FORZATTI e LIETTI, 1999)
Processo Catalisador Veneno
Síntese de amônia Fe
CO, CO2, H2O, C2H2, S, Bi, Se, Te, P
Reforma de vapor Ni/Al2O3 H2S, As, HCl
Síntese de Metano, Shift de baixa T Cu H2S, AsH3, PH3, HCl
Craqueo Catalítico SiO2±Al2O3, zeolitas
Bases orgânicas, NH3, Na, metais pesados
Hidrogenação de CO Ni, Co, Fe H2S, COS, As, HCl
Oxidação V2O5 As
Convertidores automotivos
(oxidação de CO e HC, redução de NO) Pt, Pd Pb, P, Zn
Oxidação de metanol para formaldeído Ag Fe, Ni, carbonilas
Etileno a óxido de etileno Ag C2H2
Muitos outros processos
óxidos dos Metais de
transição Pb, Hg, As, Zn
O efeito do veneno pode ser visto quando se tem uma alta acumulação deste na
superfície do catalisador, mas em alguns casos este efeito é dramático na presença de traços
do veneno. Este é o caso do H2S nos catalisadores de Fe, Ni, Co e Ru para a reação de
metanação de CO. A reação cai em 4 ordens de magnitude quando a concentração de ácido
sulfídrico varia de 15 a 100 ppb (partes por bilhão), como é mostrado na Figura 2-12. Assim,
o envenenamento é um processo que deve ser evitado limpando a carga, colocando
armadilhas para os venenos e procurando novas formulações resistentes a estes venenos.
Nas reações que envolvem hidrocarbonetos, geralmente surgem reações secundárias
na superfície do catalisador que leva à deposição e acumulação de resíduos carbonáceos.
Estes compostos podem chegar a acumular entre 15 a 20% do peso do catalisador. Estes
compostos terminam cobrindo os sítios ativos do catalisador e/ou bloqueando os poros de
Figura 2-12: Efeito de envenenamento dos catalisadores de metanação pela adição de H2S .
( BARTHOLOMEW, 2001)
Menon ( EDWIN e MENON, 1991) classificaram as reações do tipo coque sensível e
coque insensível. Nas reações tipo coque-insensível a atividade do catalisador diminui devido
à deposição do sólido e bloqueio dos sítios. Já no coque-sensível o sólido depositado tem
precursores ativos que na presença de produtos (ex. hidrogênio) reagem e são eliminados.
Exemplos de reações com coque-sensíveis são o craqueo catalítico (FCC) e a hidrogenólise;
De outro lado, exemplos de reações coque insensíveis são a síntese de Fischer-Tropsch, a
síntese de metanol, e a reação de reformado. Assim, os efeitos da deposição de coque não só
depende da quantidade mas também da localização e estrutura do carvão. A estrutura do
coque depende do mecanismo de formação que está associado ao catalisador. Em geral o
coque formado sob catalisadores metálicos é distinto que aquele proveniente de óxidos ou
sulfetos. Assim se tem os seguintes mecanismo de coque:
Um esquema da desposição de coque é apresentado na Figura 2-13. A desativação do
catalisador pode se dar por deposição do coque nos sítios em mono ou míltiplas camadas,
bloqueio na entrada dos poros impedindo o acesso dos reagentes aos centros ativos ao interior
do poro. Um outro mecanismo é a deposição e crescimento do carvão no interior do poro
aumentando os esforços e provocando a quebra da partícula.
Figura 2-13: BARTHOLOMEW, 2001 Processo de desativação do catalisador metálico suportado: encapsulação e bloqueio de poros ().
Mesmo tendo o CO u um hidrocarboneto como precursor, o mecanismo de formação
do coque é difererente. O CO tem uma adsorção dissociativa,na forma:
a
CO→Cα +O , (10)
O monóxido de carbono se dissocia em carbono atômico, Cα, que se deposita na
superfície e oxigênio atômico Oa que é adsorvido, reage segundo é apresenta na Figura 2-14,
de três maneiras: a primeira formando um carvão polimérico (Cβ) que se deposita sob o
metal, a segunda reagindo com o metal e a terceira formado metano através de uma reação
Figura 2-14: Formação de coque sob catalisador de Ni suportado (BARTHOLOMEW, 2001).
Por outro lado quando o coque se forma a partir de hidrocarbonetos o mecanismo
proposto por BARTHOLOMEW, 2001, pode ser visto na Figura 2-15.
No caso da formação de coque a partir de hidrocarbonetos, como na reforma a vapor,
três tipos de carbono podem se identificar:
- O CnHz, coque de encapsulamento formado pela polimerização lenta sob Ni, em temperatura
< 500 °C;
- Carvão filamentoso ou “whisker”, formado por difusão do carvão no Ni seguida da
separação do Ni da superfície;
- Carvão pirolítico proveniente do craqueo de compostos pesados a temperatura acima de 600
°C.
Estes mecanismos são representados na Figura 2-15