Alzheimer’s Disease Amyloid-
b
Nicole Schonrock1*, Yazi D. Ke1, David Humphreys2, Matthias Staufenbiel3, Lars M. Ittner1, Thomas Preiss2,4, Ju¨rgen Go¨tz1*
1Alzheimer’s and Parkinson’s Disease Laboratory, Brain and Mind Research Institute, University of Sydney, Sydney, New South Wales, Australia,2Molecular Genetics Division, Victor Chang Cardiac Research Institute (VCCRI), Darlinghurst, Sydney, New South Wales, Australia, 3Novartis Institute for BioMedical Research, Basel, Switzerland,4School of Biotechnology and Biomolecular Sciences and St. Vincent’s Clinical School, University of New South Wales, Sydney, New South Wales, Australia
Abstract
Normal brain development and function depends on microRNA (miRNA) networks to fine tune the balance between the transcriptome and proteome of the cell. These small non-coding RNA regulators are highly enriched in brain where they play key roles in neuronal development, plasticity and disease. In neurodegenerative disorders such as Alzheimer’s disease (AD), brain miRNA profiles are altered; thus miRNA dysfunction could be both a cause and a consequence of disease. Our study dissects the complexity of human AD pathology, and addresses the hypothesis that amyloid-b(Ab) itself, a known causative factor of AD, causes neuronal miRNA deregulation, which could contribute to the pathomechanisms of AD. We used sensitive TaqMan low density miRNA arrays (TLDA) on murine primary hippocampal cultures to show that about half of all miRNAs tested were down-regulated in response to Abpeptides. Time-course assays of neuronal Abtreatments show that Ab is in fact a powerful regulator of miRNA levels as the response of certain mature miRNAs is extremely rapid. Bioinformatic analysis predicts that the deregulated miRNAs are likely to affect target genes present in prominent neuronal pathways known to be disrupted in AD. Remarkably, we also found that the miRNA deregulation in hippocampal cultures was paralleled invivoby a deregulation in the hippocampus of Ab42-depositing APP23 mice, at the onset of Abplaque formation. In addition, the miRNA deregulation in hippocampal cultures and APP23 hippocampus overlaps with those obtained in human AD studies. Taken together, our findings suggest that neuronal miRNA deregulation in response to an insult by Abmay be an important factor contributing to the cascade of events leading to AD.
Citation:Schonrock N, Ke YD, Humphreys D, Staufenbiel M, Ittner LM, et al. (2010) Neuronal MicroRNA Deregulation in Response to Alzheimer’s Disease Amyloid-b. PLoS ONE 5(6): e11070. doi:10.1371/journal.pone.0011070
Editor:Mel B. Feany, Brigham and Women’s Hospital, Harvard Medical School, United States of America
ReceivedFebruary 23, 2010;AcceptedMay 21, 2010;PublishedJune 11, 2010
Copyright:ß2010 Schonrock et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding:N.S. is supported by the Human Frontier Science Program. L.I. is supported by the National Health and Medical Research Council (NHMRC) and the Australian Research Council (ARC), and J.G. is supported by grants from the University of Sydney, the National Health and Medical Research Council (NHMRC), the Australian Research Council (ARC), and the J.O. & J.R. Wicking Trust. Postgraduate scholarship support has been provided by the Wenkart Foundation, GlaxoSmithKline and Alzheimer’s Australia. Novartis (via Matthias Staufenbiel) provided the APP23 mouse strain. All experiments were performed in Sydney, and Novartis did not fund any aspect of this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests:The authors have a purely academic collaboration with Matthias Staufenbiel of Novartis. There is no commercial interest attached to this work with him. All PLoS ONE policies regarding the sharing of data and materials will be adhered to.
* E-mail: nschonrock@med.usyd.edu.au (NS); jgoetz@med.usyd.edu.au (JG)
Introduction
Alzheimer’s disease (AD) is a prominent neurodegenerative disorder characterized by progressive loss of memory and other cognitive functions. Histopathologically, AD is characterized by neurofibrillary tangles (NFTs) consisting of the microtubule-associated protein tau and neuritic plaques composed of amyloid-b (Ab). Ab is a naturally occurring, predominantly 40 amino acid long polypeptide (Ab40) derived from the larger amyloid precursor protein (APP) [1]. Increases in the proportion of the longer, more neurotoxic form, Ab42, result in the formation of higher order aggregates and subsequently, plaque deposition. In familial AD (FAD), the increases in Ab42 are caused by aberrant processing of APP due to mutations in either theAPPgene itself or in genes that encode subunits of the APP processing machinery. In addition,APPpromoter polymorphisms [2], gene duplications [3] or trisomy 21 [4] can cause increased APP expression levels, resulting in elevated Ab42. While increased Ablevels characterize
AD pathology, the precise mechanism(s) and signaling cascades it uses to cause cellular toxicity and cell death are not fully understood [5,6].
sequence-specific manner. Indeed, profiling of postmortem human AD brain has verified that significant changes in miRNA expression occur in several brain regions [10]. This includes miRNAs that regulate genes such asAPPitself, andBACE1, that encodes an enzyme involved in APP processing [12,13,14]. However, whether the deregulated miRNAs are a cause or a consequence of disease, and what triggers miRNA dysfunction in AD is unknown. We therefore explored the hypothesis that Ab
itself causes neuronal miRNA deregulation which could contribute to the pathology associated with AD. To remove the complexity inherently associated with human studies, we used mature murine primary hippocampal cultures to determine the effects of Ab
specifically on neuronal miRNAs.
Sensitive TaqMan low density miRNA arrays (TLDA) revealed that 47% of all miRNAs tested were down-regulated in response to Ab42. This response may be extremely rapid and bioinformatic analysis predicts that the deregulated miRNAs are likely to affect target genes present in prominent neuronal pathways disrupted in AD. Remarkably, when we analyzed hippocampi of APP23 mice at the onset of Abplaque formation, we found a similar miRNA deregulation as in our invitromodel. These findings support the notion that an insult by Ab peptides causes a considerable neuronal miRNA deregulation that may be an important factor in the pathocascade of events leading to AD.
Materials and Methods
Ethics Statement
All animal experiments were approved by the Animal Ethics Committee (AEC) of the University of Sydney under AEC approval numbers K00/1-2009/3/4914 and K00/1-2009/3/ 4915.
Cell culture and Abtreatments
Primary hippocampal neurons were prepared from 16.5-day-old embryonic C57BL/6 mice (E16.5) as described [15]. 600,000 cells were plated per dish and cultivated in Neurobasal medium supplemented with 1% (v/v) B27 supplements (Gibco) and 0.25% (v/v) 200 mM L-glutamine (Gibco) to minimize growth of astrocytes and microglia. Synthetic Ab42 peptides (Bachem, Germany) dissolved in PBS were aged by incubation at 37uC for 24 h with shaking at 1000 rpm to allow fibril formation [16]. We applied a protocol as described which used a range of biophysical methods to determine the fibrillar nature of our preparation [17,18]. At 23 days in vitro (DIV) cells were treated for 0, 1, 6, 15 or 24 hours with either 5mM aged Ab42 or a mock treatment
containing PBS. Following treatments, cells were washed once in PBS and lysed by resuspension in QIAzol reagent (Qiagen). Cell lysates were stored in QIAzol at 280uC until further use. Experiments were performed in triplicate.
Transgenic mouse strain
APP23 mice expressing human APP751 cDNA containing the Swedish double mutation (K651M and N652L) were used for this study [8]. Brains from mice at different ages (ranging from two to thirteen months) were harvested following cervical dislocation and the hippocampus was isolated, snap frozen and stored at280uC until use. Hippocampi were not pooled and analysis was performed on individual animals. Non-transgenic littermates were used as controls.
RNA extraction and microarray analysis
RNA was extracted from mouse primary hippocampal neurons and dissected hippocampi using the miRNeasy Kit (Qiagen)
according to the manufacturer’s instructions. RNA quantity was routinely assessed on a NanoDrop 1000 spectrophotometer (Thermo Scientific). For microarray analysis, RNA quality was determined on a Bioanalyser 2100 (Agilent) and only RNA samples with an RNA integrity number (RIN) between 8 and 10 were used. Megaplex profiling using rodent TaqMan Low Density miRNA Arrays (TLDA) (Applied Biosystems) was used to assay the expression of 380 miRNAs as described by the manufacturer. Briefly, 100 ng of total RNA obtained from Ab42- or control-treated primary hippocampal cells was used in the megaplex reverse transcription (RT) reaction containing about 450 miRNA-specific RT primers provided by the manufacturer. No prior miRNA preamplification step was needed. The RT product was mixed with 2X TaqMan Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems) and loaded onto the TLDA containing the 48-plex PCR reaction mix. TLDAs were run on a 7900HT Thermocycler (Applied Biosystems) using Sequence Detection Systems (SDS) software version 2.3. A single TLDA was used per Ab- or control-treated sample. Manual inspection of all amplification plots was performed and miRNAs were excluded from the analysis if: Ctvalues were too high (above 35, indicating a miRNA expression too low for accurate detection), if amplification was not achieved in all six samples, or if very high variation was found. Data analysis was performed using SDS RQ manager v1.2 (Applied Biosystems) which utilizes the delta-delta CT method [19]. The endogenous small nucleolar control RNA, snoRNA234, was used for normalization. Significance was calculated using the student’s T-test.
Quantitative real-time PCR
Individual TaqMan assays (Applied Biosystems) were used to analyse the expression of the following mature mouse miRNAs: 181c, 9, 20b, 21, 30c, 148b, miR-361, miR-409-3p and Let-7i. 10 ng of total RNA was used in the RT reaction and the transcribed cDNA was then used for subsequent PCR amplification using TaqMan 2X Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems) as described by the manufacturer. Assays were run on an Mx3000P thermocycler (Stratagene) as follows: 95uC for 10 min, and 40 cycles at 95uC for 151s followed by 60uC for 1 min. To avoid any miRNA degradation, RNA extractions, reverse transcription reactions and real-time runs were all performed on the same day. Mouse snoRNA135 expression was assayed for normalization. All reactions were performed in triplicate, and relative miRNA expression was normalized against endogenous controls using the comparative delta-delta CT method calculated using MxPRO Software V4.0 (Stratagene).
In-situ hybridisation and immunohistochemistry
Ketamine/xylazine (Troy Laboratories, Australia) -anaesthe-tized mice were perfused with 20 ml PBS. Brain tissue was dissected and postfixed over night at 4uC in 4% paraformaldehyde (Sigma, Australia). Tissue embedding in paraffin was done in a Shandon Excelsior tissue processor (Thermo, USA). In-situ hybridization was performed as described [20]. Briefly, 15mm
antidigox-igenin antibody (Sigma), and developed in NBT/BCIP solution (Sigma).
For immunohistochemistry, antigen retrieval of 5mm sections of APP23 and WT brain was performed in 10 mM citrate buffer, pH 5.8 in a RHS-1 microwave vacuum histoprocessor (Milestone, USA) at 120uC. For standardization, all stainings were carried out in Sequenza racks (Thermo, USA). Sections were blocked with PBS containing 3% heat inactivated goat serum and 5% BSA for 1 hour at room temperature followed by incubation at 4uC over night with the primary antibody 6E10 (1:1000, Signet, USA), which is reactive against amino acid residues 1-16 of Ab. Antibody staining was visualized using the AP-ABC Elite Kit (Vector, USA).
Immunocytochemistry
Coverslips containing mouse primary hippocampal neurons grown for 24 DIV were fixed with 4% paraformaldehyde in 80 mM PIPES, 1 mM MgCl2, and 1 mM EGTA, pH 6.8. Cells were permeabilized with 0.1% Triton in phosphate-buffered saline and stained with primary antibodies to rabbitb3 tubulin (1:400, Covance, USA). Antibody staining was visualized using Alexa labeled secondary antibodies (Molecular Probes, USA). Pictures were taken with a BX51 fluorescence microscope equipped with a DP70 CCD color camera (Olympus, USA).
Pathway enrichment analysis of deregulated miRNAs
TargetScanMouse v5.1 [21] was used to generate a list of potential target genes for each of our significantly deregulated miRNAs found from the microarray analysis. Due to miRNA sequence similarities between family members, TargetScan mostly predicts target genes for miRNA families rather than for individual family members. We performed an enrichment analysis of target gene lists predicted for all significantly deregulated miRNAs using the DAVID (Database for Annotation, Visualization and Interrogated Discovery) bioinfor-matics database [22,23]. Gene lists were uploaded into DAVID and enrichment analysis was performed by comparing each set of genes to all available biological pathways provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG) [24]. A cut-off P-value of 0.01 was used to show KEGG Pathways likely to be affected by predicted targets of deregulated miRNAs.
Results
Neuronal miRNA expression changes upon exposure to amyloid-b
To determine whether neuronal miRNAs are deregulated in response to Abwe used a cell culture model of murine primary hippocampal neurons [15]. Neurons were matured in vitro and neuronalb3 tubulin staining showed that they had developed dense axonal networks indicative of fully differentiated and healthy neurons (Fig 1A). At 23 DIV, triplicate cultures were exposed for 24 hours to either 5mM of aged Ab42 preparations or a PBS
control. Under these conditions, no evidence of Abtoxicity or cell death was observed. Propidium iodide (PI) uptake was negligible in Ab-treated neurons indicative of viable cultures and b3 tubulin staining of Ab-treated cells showed no neuronal fragmentation, an early sign of degeneration (data not shown). Total RNA was then isolated and expression of 381 miRNAs analyzed by qRT-PCR using rodent TaqMan Low Density miRNA Arrays (TLDA) (Applied Biosystems). Careful manual inspection of all amplification plots excluded miRNAs which did not amplify in all six samples, had very high variation, or had Ctvalues above 35 indicating that their expression was too low for accurate analysis. Relative miRNA expression was normalized against the endogenous control snoRNA234 using the comparative delta-delta CT method as
calculated using SDS RQ manager v1.2 software. For comparison, normalization was also performed using two other endogenous controls, snoRNA135 and 18S, included in each reverse transcrip-tion (RT) primer pool and the same end result was achieved (data not shown). 230 miRNAs (60%) were reliably detected on the array (Supplementary Table S1) of which 35% were considered unchanged as their expression levels varied only up to 15% from untreated controls. Interestingly, Ab42 induced a considerable down-regulation of miRNAs with 47% showing decreased levels compared to untreated controls (Fig 1B). A much smaller fraction (18%) of miRNAs was up-regulated in response to Ab42. When applying a cut-off P-value of ,0.05, twenty miRNAs showed a significant down-regulation, of up to four fold, with the exception of miR-376b whose expression seemed to be strongly induced (2.6 fold) by Abtreatment (Fig 1C). Amongst the strongest significantly down-regulated miRNAs were 409-3p, 361, 20b, 21, 181c and 148b. miRNAs 700, 146a, 365, 30c and 301 showed a moderate decrease, while miRNAs 9, 664, 187, 125b, 433, 137, 30b, Let-7i and Let-7g had a mild but significant down-regulation in response to Ab. One advantage of using TaqMan assays is that their design, with three levels of specificity per miRNA, allows differentiation between mature miRNAs differing in only a few nucleotides. Despite the fact that the Let-7 family consists of eight members varying only by one to two nucleotides, only Let-7i, Let-7g and miR-98 were down-regulated by Ab, whilst the others remained mostly unchanged. In addition, specific members of the miR-30 family (30c and 30b) were also significantly down-regulated in response to Ab. Thus, treatment of mouse primary neurons with Ab42 does indeed evoke a strong change in miRNA profiles with a substantial portion of miRNAs being down-regulated.
Neuronal miRNA response to amyloid-boccurs rapidly
Due to the low amounts of total RNA obtained from primary neuronal cells we chose to validate our microarray results by quantitative PCR using individual TaqMan assays on independent preparations of Ab42-treated primary cultures. Eight significantly down-regulated miRNAs from the array data were selected for further validation and analysis. This selection was based on the fact that these miRNAs produced the most significant fold-changes in our study. In addition, an interesting overlap between human studies and ours was observed (miR-9, 181c, 30c, 148b, 20b and Let-7i) (Table 1) and therefore it was of great interest to validate and analyze these miRNAs in particular [13,25]. miRNA expression was thus assayed in independent 24 DIV cultures treated with 5mM aged Ab42. As Abis known to cause cellular
toxicity and cell death [26], a time course of 1, 6 and 15 hour treatments was used to exclude any effects due to apoptosis and to gauge the rapidity of the miRNA response evoked by Ab. Similar to our TLDA microarray results, all of the eight tested miRNAs showed a significant down-regulation upon exposure to Ab at some stage during the Ab time-course treatment compared to untreated controls (Fig 2). Interestingly, Abcaused an extremely rapid neuronal response of distinct mature miRNA sequences with miR-9, 181c, 409-3p and 361 responding even after a one hour Ab treatment. Expression of miRNAs 148b, 21, 20b and Let-7i was more variable and therefore no conclusions can be made concerning the rapidity of their response to Ab. Thus our analysis not only validates the microarray data but further shows that Ab42 is a powerful regulator of certain mature miRNA sequences.
Deregulated miRNAs may affect targets in key pathways altered in AD
Figure 1. Deregulated miRNAs in mouse primary hippocampal cells treated with Ab42. A. Mouse primary hippocampal neurons grown for 24 DIV (days in vitro) stained with neuronalb3 tubulin showing dense axonal networks indicative of healthy mature neurons. Scale bar = 25 um.B. Neuronal miRNA response to Abtreatment. Overview of directional miRNA changes after Abtreatment. miRNAs altered by 15% compared to untreated controls were considered unchanged.C. Summary of significantly deregulated microRNAs in primary hippocampal cells with or without Ab42 treatment (n = 3) analyzed by rodent TLDA. miRNA expression levels can be gauged using average (Ave) Ctvalues. T-test P-value significance: **P,0.01, *P,0.05.
doi:10.1371/journal.pone.0011070.g001
Table 1.Overlap in miRNA changes between Ab42-treated murine hippocampal neurons and APP23 hippocampus found in this study and sporadic human AD brain.
Mouse Neurons APP23 Hippocampus Human AD Brain
miRNA FC P-value FC P-value FC P-value Brain Region Reference
miR-9 0.76 0.01146 0.54 0.00000002 0.71 0.0053 Anterior temporal cortex [14]
0.38 (–1.39*) 0.0231 Hippocampus, Braak 5,6 [26]
miR-181c 0.35 0.01771 0.66 0.00006444 0.71 0.0018 Anterior temporal cortex [14]
0.1 (–3.30*) 0.0015 Parietal lobe cortex [51]
let-7i 0.75 0.02112 0.74 0.00000481 0.88 0.0283 Anterior temporal cortex [14]
miR-30c 0.74 0.04302 0.72 0.00008285 0.3 (–1.76*) 0.0194 Hippocampus, Braak 3,4 [26]
miR-148b 0.38 0.03563 0.76 0.00139502 0.21 (–2.24*) 0.0062 Parietal lobe cortex [51]
miR-20b 0.28 0.0188 0.80 0.25607460 0.28 (–1.84*) 0.0085 Parietal lobe cortex [51]
Deregulated miRNA fold changes (FC) and significance (P-value) found in our study on Ab-treated mouse primary neurons and APP23 hippocampus compared to those found in human AD profiling experiments on various brain regions (using the Braak staging). * FC values represented in the original reference from some human studies were in log form.
hence changes in miRNA expression can have profound effects on biological systems [21]. To increase the likelihood of identifying biological processes most relevant to the miRNAs deregulated by Ab we performed a gene ontology (GO) enrichment analysis on predicted target genes. Of the available prediction algorithms such as miRBase, PicTar, miRanda, PITA and TargetScan, all of which use site conservation as a prediction criterion, the latter was shown to result in the most accurate predictions upon target validation [27,28]. Therefore, we used TargetScanMouse v5.1 to generate lists of predicted target genes regulated by our miRNAs of interest. Lists of target genes can be found at http://www.
targetscan.org/mmu_50/. To extract biological meaning associ-ated with these large gene lists we used the bioinformatics database DAVID. Pathway enrichment analysis was performed by comparing each list of target genes to all available biological pathways provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG) [24]. Encouragingly, many pathways associat-ed with brain function were enrichassociat-ed in the pathway prassociat-ediction analysis (Table 2). Axon guidancewas among the most significant pathways to be affected by the predicted target genes and was the top prediction for miR-9, miR-30 and miR-20. These three miRNAs that are down-regulated by Abpotentially target a total
Figure 2. miRNA expression pattern in response to Abtime course in murine primary hippocampal neurons assessed by real-time PCR using TaqMan assays.Independent primary neuronal preparations treated with Ab42 for 1, 6 or 15 hours were assayed for miRNA expression relative to untreated controls. T-test P-value significance: ***P,0.001, **P,0.01, *P,0.05. Expression was normalized to snoRNA135.
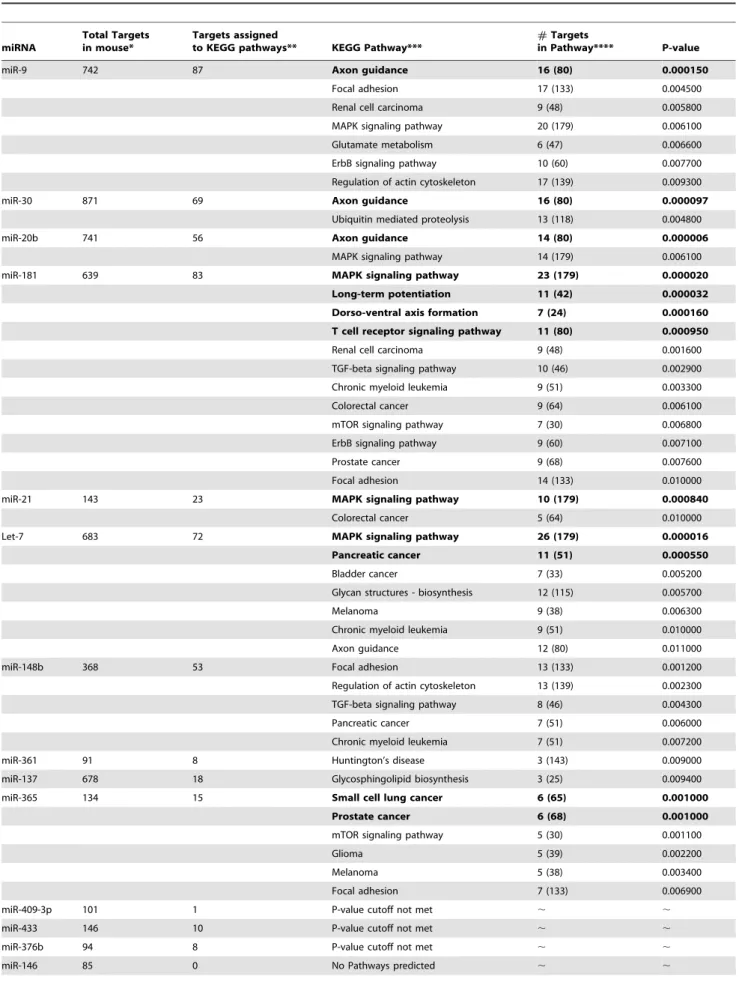
Table 2.Pathway enrichment analysis for deregulated miRNAs in mouse primary hippocampal neurons after Abtreatment.
miRNA
Total Targets in mouse*
Targets assigned
to KEGG pathways** KEGG Pathway***
#Targets
in Pathway**** P-value
miR-9 742 87 Axon guidance 16 (80) 0.000150
Focal adhesion 17 (133) 0.004500
Renal cell carcinoma 9 (48) 0.005800
MAPK signaling pathway 20 (179) 0.006100
Glutamate metabolism 6 (47) 0.006600
ErbB signaling pathway 10 (60) 0.007700
Regulation of actin cytoskeleton 17 (139) 0.009300
miR-30 871 69 Axon guidance 16 (80) 0.000097
Ubiquitin mediated proteolysis 13 (118) 0.004800
miR-20b 741 56 Axon guidance 14 (80) 0.000006
MAPK signaling pathway 14 (179) 0.006100
miR-181 639 83 MAPK signaling pathway 23 (179) 0.000020
Long-term potentiation 11 (42) 0.000032
Dorso-ventral axis formation 7 (24) 0.000160
T cell receptor signaling pathway 11 (80) 0.000950
Renal cell carcinoma 9 (48) 0.001600
TGF-beta signaling pathway 10 (46) 0.002900
Chronic myeloid leukemia 9 (51) 0.003300
Colorectal cancer 9 (64) 0.006100
mTOR signaling pathway 7 (30) 0.006800
ErbB signaling pathway 9 (60) 0.007100
Prostate cancer 9 (68) 0.007600
Focal adhesion 14 (133) 0.010000
miR-21 143 23 MAPK signaling pathway 10 (179) 0.000840
Colorectal cancer 5 (64) 0.010000
Let-7 683 72 MAPK signaling pathway 26 (179) 0.000016
Pancreatic cancer 11 (51) 0.000550
Bladder cancer 7 (33) 0.005200
Glycan structures - biosynthesis 12 (115) 0.005700
Melanoma 9 (38) 0.006300
Chronic myeloid leukemia 9 (51) 0.010000
Axon guidance 12 (80) 0.011000
miR-148b 368 53 Focal adhesion 13 (133) 0.001200
Regulation of actin cytoskeleton 13 (139) 0.002300
TGF-beta signaling pathway 8 (46) 0.004300
Pancreatic cancer 7 (51) 0.006000
Chronic myeloid leukemia 7 (51) 0.007200
miR-361 91 8 Huntington’s disease 3 (143) 0.009000
miR-137 678 18 Glycosphingolipid biosynthesis 3 (25) 0.009400
miR-365 134 15 Small cell lung cancer 6 (65) 0.001000
Prostate cancer 6 (68) 0.001000
mTOR signaling pathway 5 (30) 0.001100
Glioma 5 (39) 0.002200
Melanoma 5 (38) 0.003400
Focal adhesion 7 (133) 0.006900
miR-409-3p 101 1 P-value cutoff not met , ,
miR-433 146 10 P-value cutoff not met , ,
miR-376b 94 8 P-value cutoff not met , ,
of 36 out of 80 genes present in theaxon guidancepathway, eight genes (DPYSL2, DPYSL5, EPHA7, NFAT5, NFATC3, NTNG1, PPP3R1andSEMA6D) may be targeted by two out of the three miRNAs, andsrGAP3is predicted to be co-regulated by all three miRNAs. The other major pathway highlighted by this enrich-ment analysis is themitogen-activated protein kinase (MAPK) signaling pathway. The MAPK cascade represents a prototypic signal transduction system through which extracellular stimuli are transduced. Three of the down-regulated miRNAs (miR-181, miR-21 and Let-7) have this pathway as their top candidate with p-values of less than 0.001. These three miRNAs alone are predicted to affect a total of 48 genes in the MAPK signaling pathway (containing 179 genes in total) with eight genes (BRAF, FASl, MAP3K1, MAP4K4, TAOK1, TGFBR1, RASGRP1, RASA2
andNLK) co-targeted by two miRNAs andACVR1C, encoding a serine/threonine protein kinase, predicted to be regulated by all three. In addition, categories such as ErbB signaling and TGFb signaling pathwaysare targeted by several miRNAs as are pathways involved in apoptosis such asubiquitin mediated proteolysis. Pathways essential for correct neuronal function such asglutamate metabolism,
long term potentiation and regulation of the actin cytoskeleton are all enriched in our analysis and they are known to be disrupted in AD.
miRNA down-regulation is paralleled in the APP23 mouse model
To determine whether miRNA deregulation in response to Ab
also occurs in vivo we chose to analyze the well characterized APP23 mouse model [8]. The hippocampus was selected for analysis not only because the human APP mutant transgene is highly expressed in this brain region (Fig 3A) but it is also highly vulnerable to Ab and it is where neuropathological changes are initiated early in AD [29]. The hippocampus also has a prominent role in learning and memory and its malfunction results in early memory loss and gradual decline of other cognitive functions characteristic of AD. Immunohistochemistry using the antibody 6E10 shows that Ab is present at high levels in hippocampi of APP23 mice (Fig 3B). This was confirmed by ELISA analysis showing that both Ab40 and Ab42 are greatly overproduced in APP23 brain compared to WT (data not shown). In contrast to the published studies in humans which mostly show miRNA profiles at the end-stage of disease, we chose to analyze APP23 mice at various ages ranging from two month-old animals (pre-symptom-atic, plaque-free stage), seven month-old (cognitive deficits apparent and onset of plaque deposition) and thirteen month-old
mice (established AD pathology) [30,31]. Utilizing their highly quantitative nature, individual TaqMan assays were used to analyze miRNA expression in hippocampi of four APP23 mice
versusfour wild-type littermate controls. In juvenile two month-old APP23 mice most of the miRNAs tested showed no change in expression levels compared to littermate controls. However, miR-409-3p, Let-7i and miR-30c already exhibited a significant down-regulation while miR-148b was the only up-regulated miRNA in hippocampus at this age (Fig 3C). As mice approach 6-7 months of age they reach the critical period of Ab-plaque formation and it was here that a significant down-regulation was seen in nearly all miRNAs tested including the previously up-regulated miR-148b (Fig 3D). As mice continue to age only miRNAs 9, 409-3p and 21 maintain the significant down-regulation exhibited in seven month-old animals whilst the remaining miRNAs tested resume expression patterns similar to WT mice (Fig 3E). Thus the miRNA down-regulation found in Ab42-treated hippocampal neurons was paralleled invivoin hippocampi of APP23 mice, at the onset of plaque formation. Altogether, our studies reveal that Ab is not only a powerful regulator of miRNAs invitro, but also invivo.
Discussion
An important role for miRNAs has been shown in development and specifically in brain, where more distinct miRNAs are expressed than in any other tissue [32]; with evidence increasing for their implication in neurodegenerative disease [10,11]. miRNA profiles are known to be altered in several regions of the AD brain, however what is cause or consequence of the disease is unknown [12,13,25,33,34,35]. Our study dissects the complexity of early human AD pathogenesis, in the absence of a post mortem delay, and provides the first miRNA profiling analysis addressing the contribution of Ab, a known causative factor of AD, to the de-regulation of neuronal miRNA expression. We used both invitro
and invivomodels to show that Abevokes a substantial change in neuronal miRNA profiles that is not only predominantly down-regulated, but can occur rapidly, within a few hours of Ab
treatment. Several of these deregulated miRNAs overlap with those found in human AD studies and potentially affect important biological pathways essential for proper brain function relevant to AD.
miRNAs seem to provide essential neuroprotective functions, as decreases result in neurodegeneration [36,37,38]. Importantly, this paradigm is reiterated in our study where Ab predominantly causes a down-regulation of miRNAs in hippocampal neurons. We
miRNA
Total Targets in mouse*
Targets assigned
to KEGG pathways** KEGG Pathway***
#Targets
in Pathway**** P-value
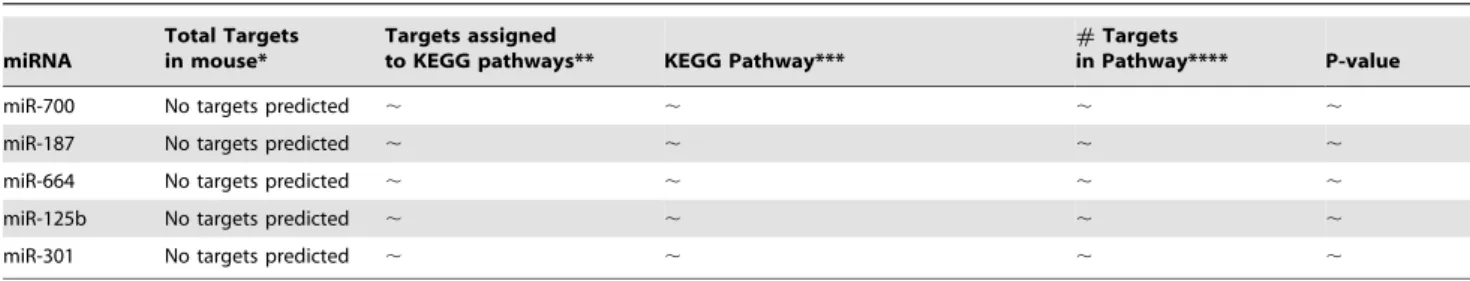
miR-700 No targets predicted , , , ,
miR-187 No targets predicted , , , ,
miR-664 No targets predicted , , , ,
miR-125b No targets predicted , , , ,
miR-301 No targets predicted , , , ,
*The total number of target genes predicted to be regulated by individual miRNAs was calculated using TargetScanMouse v5.1. Note that TargetScan gives lists of predicted targets for miRNA families and not for individual family members due to sequence similarity. **Gene lists were uploaded into DAVID bioinformatic database and the number of targets recognized by DAVID assigned to known KEGG pathways is given. ***Enrichment analysis showing KEGG Pathways likely to be affected by predicted targets of deregulated miRNAs are indicated using a cut-off P-value of 0.01. The most significant pathways with a P-value of 0.001 are shown in bold. ****For each pathway predicted to be affected the number of miRNA target genes in that pathway is indicated followed by the total number of genes in that pathway in brackets.
also found that this down-regulation of mature miRNAs is extremely rapid for several miRNAs tested and is paralleled in
vivoin the hippocampus of the APP23 AD mouse model at the onset of plaque formation.
Although changes in miRNA levels have been linked to several disease states such as Parkinson’s disease [36], Huntington’s disease [39,40], schizophrenia [41] and Down’s syndrome [42], the mechanisms responsible for stabilized or reduced miRNA expression have remained largely elusive. The biological effects of miRNAs are coordinated by the abundance of mature miRNA molecules and accumulation of a specific miRNA depends on the rates of transcription, processing and decay. There are several possible mechanisms that would explain the rapid down-regulation of certain mature miRNAs observed in Ab-treated neurons:
1) Substantial nucleotide substitutions, additions and deletions have been detected in animal miRNAs [43,44]. Although the sensitivity and specificity of TaqMan assays is far greater than for spotted arrays, each assay is highly specific for a predefined, mature miRNA sequence. Thus, any changes in sequence or length such as uridylation or adenylation of 39 ends will interfere with TaqMan detection and may result in a down-regulated read-out [45,46]. 2) In brain, miRNA turn-over may be rapid, and Abmay interfere with the multiple steps involved in the production of mature, ,22 nt
miRNAs [47]. That the stability of mature miRNAs varies considerably was shown for the highly abundant, hepatocyte-specific microRNA miR-122 (T1/2.24 hrs) [48], while several
brain-enriched miRNAs, such as miR-9, 125b, 146a, 132 and 183 exhibit short half-lives ranging from 1 to 3.5 hrs [35]. The decay rates for miR-9 are comparable in human brain tissue (T1/2= 48 min) and
neuronal cells in culture (T1/2= 42 min), highlighting the validity of
the invitromodel used by us. 3) Abmay induce a rapid activation of miRNA-specific nucleases such as XRN-2, a 59to 39exonuclease [49]. However, whether XRN-2 or other unknown nucleases are
up-regulated in AD remains to be determined. 4) Incorporation into Argonaute-containing protein complexes protects mature miRNAs from exonucleolytic pathways [50]. Target availability also affects miRNA release from Argonaute, which may be triggered by Ab, resulting in subsequent degradation [49].
5) Finally, miRNAs are not only required during development but are essential to maintain function of the adult brain, e.g. at the synapse [51]. Ab-induced miRNA regulation may involve alterations in cellular localization that will not only impact function but also turnover rate of miRNAs [52].
Interestingly, in our study, most of the mature miRNAs shown to be down-regulated in the Abtime-course assay were also down-regulated invivoin hippocampi of APP23 mice. Ablevels are not as high in mouse brain as those used to acutely treat hippocampal cultures, but never the less a strong decrease in miRNA expression was also seen in seven month-old APP23 mice. At this age, mice reach the critical period of Abplaque formation where insoluble Ab42 peptides increase five-fold compared to younger animals and the first scarce small plaques can be seen in hippocampus and neocortex. In addition, the mice display major cognitive deficits affecting visuo-spatial learning abilities [31]. Not many miRNAs were altered in two month-old APP23 mice, representing a juvenile age prior to deposition of Ab plaques and in which no changes in behavioral performance is evident. Similarly, most miRNAs were comparable between APP23 and WT at thirteen months of age where AD pathology is evident with plaque deposits having increased in size and number and most histopathological, biochemical, cognitive and behavioral alterations characteristic for AD being present. Interestingly, miR-9 maintained its down-regulation in older mice and miR-409-3p was the only miRNA to be consistently down-regulated in APP23 from a very young age right through to older animals. Normalization of miRNA levels in thirteen month-old mice may be the result of several mechanisms
Figure 3. Invivoanalysis of the Abplaque-forming APP23 mouse model. A. Insituhybridization showingAPPexpression pattern in brains
of three month-old APP23 versus wild-type mice. Note the high expression of the transgene in cortex and hippocampus.B. Immunohistochemistry with the 6E10 antibody showing high levels of Abin the hippocampus of three month-old APP23 mice compared to littermate controls. Scale bar = 500mm.C–E. miRNA expression in hippocampus of two- (C.), seven- (D.) and thirteen- (E.) month-old APP23 mice compared to wild-type littermate controls (n = 4) represented as fold change (FC) as determined using real-time PCR with TaqMan assays. Down-regulated miRNAs have been highlighted in green and up-regulated ones in red. Reactions were normalized to snoRNA135 and P-value significance was calculated using the Students T-test: P-value = *,0.05, **,0.01, ***,0.001.
including the induction of compensatory physiological responses induced by prolonged acute over-expression of proteins in transgenic mice or qualitative changes in Absuch as aggregation of Ab into oligomeric or fibrillar species. However, the exact mechanisms responsible for these patterns of miRNA deregulation occurring invivoare likely complex and remain to be determined. Of great interest is the possible overlap in miRNA deregulation between the models used in our study with the existing profiling studies performed on human AD brain (Table 1). In general, miRNA expression studies on AD patients revealed either no or only very little overlap in miRNA changes (reviewed in [10]). However, similar to our finding, Hebert et al showed that in AD temporal cortex the deregulated miRNAs were also mostly down-regulated compared to controls [13]. Importantly, this human study showed that miR-9, 181c and Let-7i were down-regulated in AD brain. miR-9 has also been reported to be down-regulated in an independent human profiling study of various brain regions including hippocampus [25]. In addition, this study showed that miR-30c was down-regulated in hippocampus at an early stage of disease (Braak stages 3 and 4). A recent study by Nunez-Iglesias et al found forty-eight significantly deregulated miRNAs in human AD parietal lobe cortex, of which miR-148b, 20b and 181c were down-regulated [33]. Our invivoanalysis of APP23 hippocampus showed down-regulation of miR-9, 181c, 30c, 20b, 148b and Let-7i, all of which were altered in human AD brain. The overlap between human AD and our invitroand invivoAD models indicates that amongst the complex pathology in human AD brain, down-regulation of miR-9, miR-181c, miR-30c, miR-20b, miR-148b and Let-7i could be attributed at least in part to the presence of Ab.
miR-9, the most abundant human brain miRNA [53], is a recurring candidate from several AD profiling studies. In contrast to the above studies including ours, miR-9 was found to be up-regulated in human AD CA1 [34] and temporal cortex [35]. Studies performed in zebrafish and mice revealed that miR-9 is essential in patterning, neurogenesis and differentiation and thus ideally placed to impact various aspects of brain function. Over-expression of miR-9 accelerates neuronal differentiation, while its inhibition in the medial pallium of E11.5 mouse embryos results in defective differentiation of Cajal-Retzius cells, the first neurons to populate the embryonic cortex. Similarly, loss of miR-9 in zebrafish embryos decreases the relative numbers of differentiated neurons in the anterior hindbrain [54,55,56]. Neurogenesis is not only important in the developing brain but is a process which continues in the adult hippocampus, a region heavily affected by Ab pathology in AD [57]. Interestingly, AD patients exhibit altered expression of early neuronal markers in the hippocampus which has been attributed to increased neurogenesis [58]. Decreased expression of miR-9 may therefore impact adult brain function.
It is encouraging to see that most of the pathways predicted to be affected by miR-9 target genes are related to brain function. In comparison, miR-21, miR-181 and Let-7 have well characterized roles in cancer and it is not surprising therefore that their target genes result in enrichment for cancer-related pathways as well. TheMAPK pathwaywas one of the top candidate pathways to be affected by Ab-mediated down-regulation of miRNAs. This signaling cascade is involved in various cellular functions including hippocampal synaptic plasticity and learning. Indeed, even very low concentrations of oligomeric Ab42 activate MAPK in human neuroblastoma cells [59]. In addition, MAPK activation was observed in hippocampal slice cultures of Ab-forming Tg2576 mice [60]. In rodent hippocampus, MAPK is essential for LTP formation, and several APP mutant mouse strains exhibit deficits in hippocampal LTP and hippocampus-dependent associative learning paradigms, including contextual fear conditioning and
escape training in the Morris water maze [30,61,62]. Thus, Ab 42-induced miRNA deregulation of the MAPK cascade may in part underlie the learning and memory deficits attributed to hippo-campal dysfunction in AD.
Axon guidancewas the other major pathway over-represented in our enrichment analysis. It represents a key stage in the formation of neuronal networks known to be disrupted in AD. The down-regulated miRNAs miR-9, miR-30 and miR-20 were all strongly predicted to affect target genes involved in axonal guidance. Interestingly, dihydropyrimidinase-related protein 2, DPYSL2, a highly abundant protein in brain, is targeted by miR-30, 20 and 181 and has been shown to be up-regulated in proteomic studies on APP23 mice already at a very early age [63]. Also called collapsin response mediator protein 2 (CRMP2), DPYSL2 is a signal mediator of Semaphorin 3A in the guidance of axonal growth. Dysregulation of DPYSL2 has also been reported in other AD proteomic studies [64,65,66,67], along with its aberrant phosphor-ylation [68] and association with NFTs [69]. However, whether or not miRNAs play a role in its regulation remains to be determined. Together, our work provides insight into previously unknown effects of Abon neuronal miRNA networks. We show that Abis a powerful regulator of miRNA expression, causing a rapid decrease of certain mature miRNA populations, which could have profound impacts on biological processes affecting the pathogen-esis of AD. Abis well positioned to target several mechanisms that affect the stability of mature miRNAs, including alterations in cis-acting modifications, protein complex formation or the exposure to nucleases, to name a few [70]. However, the exact mechanism of the rapid Ab-mediated down-regulation of mature brain miRNAs remains to be determined. The close overlap of our miRNA profiles in the cell culture model, APP23 hippocampus and human AD suggests the established APP23 mouse model is an ideal system to investigate further the role of Ab-induced miRNA deregulation in AD. Our study uncovers an unexplored mecha-nism of how Ab may impact the pathology of AD and the identification of key miRNAs affected by Ab will allow further analysis of target genes and biological pathways contributing to pathomechanisms in AD.
Supporting Information
Table S1 miRNA changes in mouse primary hippocampal cells evoked by Ab42 treatment. Expression profiling of microRNAs in mouse primary hippocampal cells with and without Ab42 treatment using Rodent TaqMan Low Denisty miRNA Arrays. Shown are the results for 230 miRNAs in ascending order out of the 381 miRNAs present on the TLDA whose amplification plots where above the cutoff threshold in the triplicate analysis. miRNA expression levels can be gauged using Average (Ave) Ct values. miRNAs highlighted in bold are those significantly deregulated. T-test P-value significance: **P,0.01, *P,0.05.
Found at: doi:10.1371/journal.pone.0011070.s001 (0.47 MB DOC)
Acknowledgments
We thank Denise Nergenau for technical assistance, Dr Christine Biben for help with in-situ hybridization and Dr Romaric Bouveret for fruitful discussions.
Author Contributions
References
1. Haass C (2004) Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J 23: 483–488. 2. Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, et al. (2006)
Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet 78: 936–946.
3. Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, et al. (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: 24–26.
4. Podlisny MB, Lee G, Selkoe DJ (1987) Gene dosage of the amyloid beta precursor protein in Alzheimer’s disease. Science 238: 669–671.
5. Gotz J, Schild A, Hoerndli F, Pennanen L (2004) Amyloid-induced neurofibrillary tangle formation in Alzheimer’s disease: insight from transgenic mouse and tissue-culture models. Int J Dev Neurosci 22: 453–465.
6. Gotz J, Ittner LM, Schonrock N, Cappai R (2008) An update on the toxicity of Abeta in Alzheimer’s disease. Neuropsychiatr Dis Treat 4: 1033–1042. 7. Gotz J, Deters N, Doldissen A, Bokhari L, Ke Y, et al. (2007) A decade of tau
transgenic animal models and beyond. Brain Pathol 17: 91–103.
8. Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, et al. (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 94: 13287–13292. 9. Walsh DM, Selkoe DJ (2007) A beta oligomers - a decade of discovery.
J Neurochem 101: 1172–1184.
10. Hebert SS, De Strooper B (2009) Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci 32: 199–206.
11. Lau P, de Strooper B (2010) Dysregulated microRNAs in neurodegenerative disorders. Semin Cell Dev Biol.
12. Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, et al. (2008) The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci 28: 1213–1223.
13. Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, et al. (2008) Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A 105: 6415–6420.
14. Hebert SS, Horre K, Nicolai L, Bergmans B, Papadopoulou AS, et al. (2009) MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis 33: 422–428.
15. Fath T, Ke YD, Gunning P, Gotz J, Ittner LM (2009) Primary support cultures of hippocampal and substantia nigra neurons. Nat Protoc 4: 78–85. 16. Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Gotz J (2003) beta-Amyloid
induces paired helical filament-like tau filaments in tissue culture. J Biol Chem 278: 40162–40168.
17. Habicht G, Haupt C, Friedrich RP, Hortschansky P, Sachse C, et al. (2007) Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Abeta protofibrils. Proc Natl Acad Sci U S A 104: 19232–19237.
18. Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, et al. (2008) Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J Mol Med 86: 1255–1267.
19. Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108.
20. Biben C, Harvey RP (1997) Homeodomain factor Nkx2-5 controls left/right asymmetric expression of bHLH gene eHand during murine heart development. Genes Dev 11: 1357–1369.
21. Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20.
22. Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3.
23. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57.
24. Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30.
25. Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, et al. (2008) Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 14: 27–41. 26. Gotz J, Ittner LM, Fandrich M, Schonrock N (2008) Is tau aggregation toxic or
protective: a sensible question in the absence of sensitive methods? J Alzheimers Dis 14: 423–429.
27. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, et al. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63.
28. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, et al. (2008) The impact of microRNAs on protein output. Nature 455: 64–71.
29. Gotz J, Schonrock N, Vissel B, Ittner LM (2009) Alzheimer’s disease selective vulnerability and modeling in transgenic mice. J Alzheimers Dis 18: 243–251. 30. Van Dam D, Vloeberghs E, Abramowski D, Staufenbiel M, De Deyn PP (2005)
APP23 mice as a model of Alzheimer’s disease: an example of a transgenic approach to modeling a CNS disorder. CNS Spectr 10: 207–222.
31. Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, et al. (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17: 388–396.
32. Fineberg SK, Kosik KS, Davidson BL (2009) MicroRNAs potentiate neural development. Neuron 64: 303–309.
33. Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ (2010) Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One 5: e8898.
34. Lukiw WJ (2007) Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 18: 297–300.
35. Sethi P, Lukiw WJ (2009) Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett 459: 100–104.
36. Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, et al. (2007) A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317: 1220–1224. 37. Schaefer A, O’Carroll D, Tan CL, Hillman D, Sugimori M, et al. (2007)
Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med 204: 1553–1558.
38. Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM (2006) MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell 24: 157–163.
39. Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL (2008) The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J Neurosci 28: 14341–14346.
40. Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, et al. (2008) A microRNA-based gene dysregulation pathway in Huntington’s disease. Neuro-biol Dis 29: 438–445.
41. Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, et al. (2007) microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol 8: R27.
42. Kuhn DE, Nuovo GJ, Martin MM, Malana GE, Pleister AP, et al. (2008) Human chromosome 21-derived miRNAs are overexpressed in down syndrome brains and hearts. Biochem Biophys Res Commun 370: 473–477.
43. Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, et al. (2006) Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20: 2202–2207.
44. Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, et al. (2009) Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res 37: 2461–2470.
45. Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, et al. (2009) Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol 11: 1157–1163.
46. Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S, et al. (2009) Selective stabilization of mammalian microRNAs by 39adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev 23: 433–438. 47. Winter J, Jung S, Keller S, Gregory RI, Diederichs S (2009) Many roads to
maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11: 228–234.
48. Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, et al. (2009) Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23: 1313–1326.
49. Chatterjee S, Grosshans H (2009) Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 461: 546–549.
50. Diederichs S, Haber DA (2007) Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131: 1097–1108.
51. Schratt G (2009) microRNAs at the synapse. Nat Rev Neurosci 10: 842–849. 52. Nelson PT, Wilfred BR (2009) In situ hybridization is a necessary experimental
complement to microRNA (miRNA) expression profiling in the human brain. Neurosci Lett 466: 69–72.
53. Mattick JS, Makunin IV (2005) Small regulatory RNAs in mammals. Hum Mol Genet 14 Spec No 1: R121–132.
54. Shibata M, Kurokawa D, Nakao H, Ohmura T, Aizawa S (2008) MicroRNA-9 modulates Cajal-Retzius cell differentiation by suppressing Foxg1 expression in mouse medial pallium. J Neurosci 28: 10415–10421.
55. Zhao C, Sun G, Li S, Shi Y (2009) A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol 16: 365–371.
56. Leucht C, Stigloher C, Wizenmann A, Klafke R, Folchert A, et al. (2008) MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat Neurosci 11: 641–648.
57. Hallbergson AF, Gnatenco C, Peterson DA (2003) Neurogenesis and brain injury: managing a renewable resource for repair. J Clin Invest 112: 1128–1133. 58. Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, et al. (2004) Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A 101: 343–347.
59. Young KF, Pasternak SH, Rylett RJ (2009) Oligomeric aggregates of amyloid beta peptide 1-42 activate ERK/MAPK in SH-SY5Y cells via the alpha7 nicotinic receptor. Neurochem Int 55: 796–801.
nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 21: 4125–4133.
61. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, et al. (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102.
62. Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, et al. (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271–276.
63. Guerreiro N, Staufenbiel M, Gomez-Mancilla B (2008) Proteomic 2-D DIGE profiling of APP23 transgenic mice brain from pre-plaque and plaque phenotypes. J Alzheimers Dis 13: 17–30.
64. David DC, Ittner LM, Gehrig P, Nergenau D, Shepherd C, et al. (2006) Beta-amyloid treatment of two complementary P301L tau-expressing Alzheimer’s disease models reveals similar deregulated cellular processes. Proteomics 6: 6566–6577.
65. Lubec G, Nonaka M, Krapfenbauer K, Gratzer M, Cairns N, et al. (1999) Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down
syndrome and Alzheimer’s disease brain is downregulated at the mRNA and dysregulated at the protein level. J Neural Transm Suppl 57: 161–177. 66. Sizova D, Charbaut E, Delalande F, Poirier F, High AA, et al. (2007) Proteomic
analysis of brain tissue from an Alzheimer’s disease mouse model by two-dimensional difference gel electrophoresis. Neurobiol Aging 28: 357–370. 67. Vercauteren FG, Clerens S, Roy L, Hamel N, Arckens L, et al. (2004) Early
dysregulation of hippocampal proteins in transgenic rats with Alzheimer’s disease-linked mutations in amyloid precursor protein and presenilin 1. Brain Res Mol Brain Res 132: 241–259.
68. Gu Y, Hamajima N, Ihara Y (2000) Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry 39: 4267–4275.
69. Yoshida H, Watanabe A, Ihara Y (1998) Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. J Biol Chem 273: 9761–9768.