Artigo
*e-mail: [email protected]
ANÁLISE CONFORMACIONAL POR CÁLCULOS TEÓRICOS DA DISTINCTINA, PEPTÍDEO ANTIMICROBIANO ISOLADO DE ANUROS DA ESPÉCIE Phyllomedusa distincta
Victor H. de Oliveira Munhoz, Antônio F. de Carvalho Alcântara e Dorila Piló-Veloso*
Departamento de Química, Universidade Federal de Minas Gerais, 31270-901 Belo Horizonte – MG, Brasil
Recebido em 7/2/08, aceito em 8/4/08, publicado na web em 29/4/08
CONFORMATIONAL ANALYSIS BY THEORETICAL CALCULATIONS OF DISTINCTIN, AN ANTIMICROBIAL PEPTIDE ISOLATED FROM Phyllomedusa distincta. Various studies demonstrate that different frog species produce distinct classes of biologically active peptides. These peptides can act as alternative agents against pathogenic bacteria and fungi by membrane permeability. Although studies have recently demonstrated that this process is utterly related to the secondary structure adopted by the peptide (in this case, the α-helical structure) when in contact with the bacterial membrane, the detailed mechanism is still unknown. In this work we describe a conformational analysis of distinctin, a heterodimeric peptide isolated from the skin of
Phyllomedusa distincta, an anuran found in the Brazilian Atlantic Forest. The study yielded a stable geometry with a high content of the α-helical structure both in chains 1 and 2 of distinctin, showing strong interaction between them.
Keywords: Phyllomedusa distincta; antimicrobial peptide; conformational analysis.
INTRODUÇÃO
Peptídeos biologicamente ativos são encontrados em diferen-tes espécies de anuros,1 atuando como agentes alternativos contra bactérias patogênicas e fungos, por permeabilização da membrana bacteriana.2,3 O processo inicia-se basicamente com o peptídeo desestruturado em solução que adota uma estrutura α-helicoidal, com seus resíduos catiônicos atraídos eletrostaticamente à mem-brana.4 O mecanismo detalhado dessas permeabilizações por peptídeos antimicrobianos (AMP) é ainda pouco conhecido.5,6 No entanto, tem sido proposto que o peptídeo na forma α-helicoidal mantém suas regiões hidrofílicas em contato com os grupos hidrofílicos da membrana lipídica para formar poros. Uma outra proposta sugere interações de sítios hidrofóbicos do peptídeo com regiões hidrofóbicas localizadas na parte central da membrana. Neste último caso, as formas α-helicoidais não são requeridas para as interações do peptídeo com a membrana.7-10 Assim sendo, o ba-lanço hidrofóbico-hidrofílico de um peptídeo torna-se um fator determinante para compreender as interações lipídeo – peptídeo e, por extensão, interações peptídeo – membrana bacteriana.11-14
Diferentes metodologias têm sido empregadas para a análise conformacional de peptídeos,15,16 a qual tem sido uma estratégia muito importante na busca de informações sobre suas atividades biológicas, uma vez que muitas destas estão relacionadas a pro-cessos de permeabilização de membranas. Os métodos experi-mentais, principalmente Dicroísmo Circular (DC), difração de raios-X e RMN, têm sido eficientes para este fim. No entanto, o uso desses experimentos pode ser limitado por requerer materi-ais na forma de monocristmateri-ais (como no caso da difração de raios-X), em solução com solvente apropriado ou em meio sólido como no caso da RMN. Para todas essas análises, é necessário um alto grau de pureza e, as vezes, quantidades relativamente grandes de peptídeo. Isso pode restringir o trabalho, uma vez que AMP ocor-rem normalmente na natureza em micro-quantidades e, por outro lado, o custo para sintetizá-los é significativamente alto. Como alternativa e, em muitos casos, como técnica complementar, uma
ferramenta muito utilizada tem sido a busca em bancos de dados de seqüências peptídicas homólogas que apresentam proprieda-des químicas conhecidas. Essa ferramenta tem sido muito utili-zada, pois peptídeos com seqüências semelhantes possuem, em alguma extensão, estruturas secundárias também semelhantes entre si. As estruturas secundárias podem ser previstas ainda por metodologias estatísticas.17-19 Essas ferramentas podem ser acessadas pelos bancos de dados Expert Protein Analysis System (ExPASy),20,21 Basic Local Alignment Search Tool (Blast),22,23 Network Protein Sequence Analysis (NPSA)24,25 e MetaServer.26
A literatura descreve também o emprego de metodologias teóri-cas na análise conformacional de peptídeos.27 Cálculos teóricos de otimização de geometria em nível de Mecânica Molecular (MM) são os mais empregados normalmente devido à simplicidade de ope-ração e recursos computacionais.28-30 Os cálculos semi-empíricos (AM1, PM3 e MNDO) e ab initio (HF e DFT) têm sido relativamen-te pouco explorados por requererem maiores recursos computacionais, apesar de fornecerem normalmente resultados mais próximos aos experimentais.16
A distinctina (1, Figura 1) é um AMP isolado das glândulas da pele de Phyllomedusa distincta, anuro natural da Mata Atlântica brasileira. Esse peptídeo apresenta atividade contra bactérias Gram -positivas e negativas e sua estrutura heterodimérica é constituída por cadeias de 22 e 25 resíduos de aminoácidos, com ligação dissulfídica entre seus resíduos de cisteína.31 Análises por DC e IV de1 indicaram um alto conteúdo de folhas β-pregueadas em solu-ção aquosa.31 Por sua vez, as análises por RMN em 2D (TOCSY e NOESY) de 1 em meio aquoso indicaram a ocorrência de várias regiões com formas α-helicoidais em ambas as cadeias.32
Neste trabalho, nós descrevemos a análise conformacional de 1 por três métodos: a partir de informações de homólogos em bancos
E N R E V P P G F T A L I K T L R K C K I I (Cadeia 1) N L V S G L I E A R K Y L E Q L H R K L K N C K V (Cadeia 2)
Figura 1. Estrutura primária da distinctina (1), com indicação da ligação
de dados, por metodologias estatísticas também em bancos de da-dos e metodologias teóricas em nível de mecânica molecular (MMFF94) e semi-empírico (PM3). O procedimento foi baseado inicialmente na análise estrutural das cadeias tratadas isoladamen-te (Cadeia 1 e Cadeia 2) para, depois, realizar a análise de 1.
PARTE EXPERIMENTAL
As consultas a seqüências homólogas foram realizadas para as duas cadeias isoladas, empregando o banco de dados MetaServer. As informações são obtidas pelo emprego de diferentes técnicas de alinhamento dos resíduos, permitindo a comparação e procura por estruturas recorrentes entre os resultados gerados. As homologias de 100% são as próprias seqüências peptídicas estudadas e, por sua vez, as homologias de 0% são aquelas com nenhuma identidade com a seqüência estudada.
As previsões de estruturas secundárias por metodologias esta-tísticas foram realizadas pela consulta ao banco de dados NPSA para as duas cadeias isoladas.24 As metodologias de previsão em-pregadas foram DPM,33 GOR1,34 GOR2,35 GOR4,36 HNNC,37 PHD,38 SIMPA96,39 SOPM,40 SOPMA41 e Secondary Consensus.42 Outra previsão de estruturas secundárias foi realizada pelo banco de da-dos ExPASy, utilizando-se os métoda-dos de Chou e Fasman,43 Deleage e Roux,33 Levitt44 e de previsão da natureza das β-folhas (paralelas ou antiparalelas).45 Estas metodologias são mais confiáveis, pois se baseiam no alinhamento de várias seqüências peptídicas.
Para cadeias pequenas, com menos de 90 resíduos de aminoácidos, a metodologia de previsão mais indicada é a PHD.38,46 Essa metodologia baseia-se em um tratamento estatístico não-li-near, por meio de um sistema de redes neurais, prevendo qual a conformação mais provável por comparações entre seqüências homólogas. Essas previsões são resultantes apenas da estrutura primária e consideram implicitamente o efeito do solvente como meio, uma vez que advêm de dados experimentais. O método PHD é o mais indicado para servir de comparação na modelagem molecular das cadeias por apresentar maior exatidão em previsões de estruturas secundárias.
As regiões de hidrofilia de 1 foram investigadas para cada ca-deia tratada isoladamente, empregando as metolodologias segun-do OMH,47 Kyte e Doolittle,48 Abraham e Leo,49 Bull e Breese,50 Guy,51 Miyazawa,52 Roseman,53 Welling,54 Parker (HPLC),55 Cowan (HPLC pH 7,5 e 3,4),56 Wilson (HPLC),57 Chothia,58 Eisenberg,59 Hopps e Woods,60 Manavalan,61 Black,62 Fauchere,63 Janin,64 Rao e Argos,65 Wolfenden,66 Mobilidade de Rf67 e Rose.68 As regiões de mínimo de hidrofilia foram consideradas como aquelas regiões hidrofóbicas. Esses métodos baseiam-se normalmente na partição de aminoácidos ou seus análogos entre água e um solvente menos polar, bem como em análises estatísticas da distribuição de resídu-os de aminoácidresídu-os em proteínas solúveis de estrutura conhecida. Alguns métodos consideram também a influência da estrutura se-cundária sobre a hidrofobia de certas regiões, uma vez que formas helicoidais em especial têm polarizações devidas às interações intramoleculares do tipo ligação hidrogênio.
Os estudos teóricos foram realizados empregando o pacote computacional SPARTAN69para cálculos em nível de mecânica molecular MMFF94.70-76 O pacote computacional GAUSSIAN77 foi empregado para cálculos em nível semi-empírico PM3.78 Geome-trias otimizadas em nível MMFF94 foram utilizadas como modelo inicial para os cálculos de otimização da geometria da Cadeia 1 e Cadeia 2 em nível PM3. Em todos os casos, os cálculos foram realizados para estruturas não ionizadas em suas extremidades e no estado gasoso, sem considerar interações intermoleculares.
A análise conformacional da Cadeia 1 foi realizada por
cálcu-los de otimização de geometria (MMFF94 e PM3) de fragmentos dessa cadeia, em etapas sucessivas de adições de aminoácidos na extremidade N-terminal de sua seqüência peptídica e re-otimizações de geometria. Na análise conformacional da Cadeia 1, os resíduos de aminoácido foram sendo adicionados no sentido da extremida-deN-terminal (resíduo de ácido glutâmico) para a extremidade C -terminal (resíduo de isoleucina). A busca conformacional de cada fragmento da seqüência peptídica foi realizada por variação do ângulo diedro \ (Ni – Cα – C’ – N
i+1) envolvendo o último resíduo adicionado. As rotações do ângulo \ foram realizadas de 90 em 90 graus a partir da geometria do fragmento com \ igual a 180 graus, resultando em quatro geometrias otimizadas para cada fragmento. Assim, após otimização de geometria do primeiro resíduo, foi adicionado o segundo, conforme seqüência peptídica dada na Fi-gura 1, e feitas re-otimizações das quatro geometrias desse frag-mento contendo apenas dois resíduos. A geometria re-otimizada de menor energia foi utilizada para a adição do terceiro resíduo de aminoácido da seqüência da Cadeia 1 e posterior otimização das quatro geometrias desse fragmento. Esse procedimento foi repeti-do até otimização de geometria completa da Cadeia 1. Procedi-mento idêntico foi empregado para a análise conformacional da Cadeia 2.
Para otimização de geometria de 1, as geometrias otimizadas de ambas as cadeias em níveis MMFF94 e PM3 foram ligadas pe-los resíduos de cisteína. Quatro diferentes geometrias foram gera-das variando-se de 90 em 90 graus o ângulo diedro C–S–S–C, que foram otimizadas em níveis de cálculos correspondentes. As análi-ses da estabilidade de formas D-helicoidal e folhas-E na estrutura de1 foram realizadas considerando ambas as cadeias totalmente nessas duas formas de estrutura secundária.
RESULTADOS E DISCUSSÃO
Resultados das consultas em bancos de dados
A busca por homólogos foi realizada empregando o banco de dadosMetaServer. Pelas limitações dos programas de busca, a es-trutura dimérica de 1 não foi considerada, realizando-se as buscas das cadeias tratadas isoladamente. Ambas as cadeias apresentam 100% de homologia com seqüências depositadas em bancos de dados e que se apresentam predominantemente em formas D -he-licoidais, conforme análises por RMN em solução.32
A consulta ao banco de dados NPSA mostrou que todos os méto-dos indicam uma região D-helicoidal entre os resíduos A11 e L16 da Cadeia 1. Exceto os métodos DPM e GOR4, os demais métodos indi-cam ainda uma expansão da forma D-helicoidal que inclui os resíduos F9 e T10. Nas regiões E1 – P6 e K18 – I22 da Cadeia 1, os métodos indicam estruturas secundárias randômicas e formas indefinidas, res-pectivamente. Na consulta sobre a Cadeia 2, todos os métodos indi-cam região D-helicoidal entre os resíduos E8 e R18. No entanto, as previsões da estrutura secundária em ambas as extremidades diferem consideravelmente entre as metodologias empregadas.
A previsão de sítios hidrofílicos nas duas cadeias foi realizada por consultas ao banco de dados ExPASy, empregando 23 metodologias diferentes. O sítio de maior hidrofilia da Cadeia 1 lo-caliza-se nos resíduos R3 e L16 (52,2 e 30,4% dos resultados, res-pectivamente). O mínimo de hidrofilia (máximo de hidrofobia) lo-caliza-se no resíduo A11 e P7 (78,3 e 13,0% dos resultados, respec-tivamente). Na Cadeia 2, o sítio de maior hidrofilia localiza-se nos resíduos L20, L16 e K19, com 43,5; 13,0 e 13,0% dos resultados, respectivamente. A hidrofobia localiza-se nos resíduos G5 e S4 (50,0 e 39,3% dos resultados, respectivamente). Como pode ser observa-do, as previsões de hidrofilia e hidrofobia por diferentes metodologias convergem para determinadas regiões de cada cadeia.
Resultados dos cálculos teóricos
A Figura 2 mostra a geometria otimizada em nível PM3 da Ca-deia 1. A maior parte de sua estrutura secundária apresenta-se na formaD-helicoidal, principalmente as regiões que contêm os resídu-os de lisina (K14, K18 e K20), leucina (L12 e L16) e isoleucina (I13 e I21).79 A distorção torna-se mais acentuada próxima à extremidade N-terminal contendo resíduos de prolina e glicina, localizadas em P6, P7 e G8, que desfavorecem normalmente a formação de D -héli-ces. A geometria otimizada em nível MMFF94 da Cadeia 1 apresen-ta-se semelhante à geometria mostrada na Figura 2. Entretanto, a região próxima ao resíduo C19 apresenta uma forma D-helicoidal mais distorcida em relação geometria otimizada em nível PM3.
Cálculos de otimização de geometria em nível PM3 da Cadeia 1 foram também feitos, partindo de estruturas totalmente na forma D-helicoidal e folhas-E, não realizando variações de ângulos diedros ou de ligação para busca conformacional. A geometria otimizada partindo da estrutura totalmente na forma D-helicoidal mostra-se menos distorcida. As formas D-helicoidais predominam na geome-tria, com exceção da região P6 – G8. No entanto, os cálculos MMFF94 mostram que essa geometria apresenta uma energia de 10,1 kcal/mol maior que a geometria dada na Figura 2.
Os cálculos de otimização de geometria em nível PM3 da
Ca-deia 1 partindo da estrutura na forma totalmente folhas-E não con-vergiram, indicando tratar-se de uma conformação pouco favorecida termodinamicamente ou estado de transição. Por sua vez, a geome-tria otimizada em nível MMFF94 partindo também da estrutura to-talmente folhas-E mostrou-se em forma de fita altamente estirada, com acentuada torção da cadeia na região contendo os dois resíduos de prolina (P6 e P7), conseqüência do impedimento estérico da ca-deia em adquirir uma estrutura com formas E-pregueadas. Os cálcu-los MMFF94 mostraram que essa geometria apresenta uma energia de 53,02 kcal/mol maior que a geometria dada na Figura 2.
A geometria otimizada em nível PM3 da Cadeia 2 (Figura 3) mostra predominância da forma D-helicoidal, com uma acentuada distorção próxima aos resíduos L16 – K19. A geometria otimizada em nível MMFF94 mostra-se semelhante à geometria otimizada em nível PM3, porém apresentando a região próxima à extremida-deN-terminal com forma D-helicoidal menos explícita. Nesta re-gião estão presentes resíduos de leucina (L2, L6), valina (V3) e serina (S4), que favorecem formas D-helicoidais.
Os cálculos de otimização de geometria da Cadeia 2 partindo da geometria na forma totalmente D-helicoidal foram realizados em ambos os níveis de cálculos. A geometria otimizada em nível PM3 apresentou desestruturações nas extremidades (N1 – L6 e R18 – N22), sugerindo que as formas D-helicoidais nessas regiões apre-sentam-se menos favorecidas. A geometria otimizada em nível MMFF94 manteve-se totalmente na forma D-helicoidal. Os cálcu-los MMFF94 mostraram que essa geometria apresenta uma ener-gia de 22,11 kcal/mol maior que a geometria dada na Figura 3.
Os cálculos de otimização de geometria em nível PM3 da Cadeia 2 partindo da estrutura na forma totalmente folhas-E não convergiram. Os cálculos MMFF94 resultaram em uma geometria linear com sua-ves dobramentos ao longo da seqüência peptídica. Os cálculos MMFF94 mostraram que essa geometria apresenta uma energia de 101,36 kcal/mol maior que a geometria dada na Figura 3.
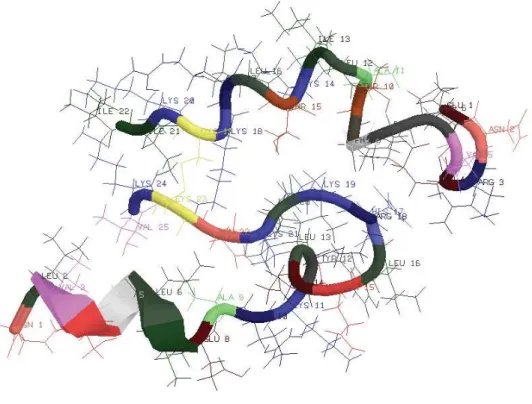
A Figura 4 mostra a geometria otimizada em nível PM3 da distinctina1. Essa geometria apresenta-se essencialmente randômica, com algumas regiões que podem ser classificadas como D-hélice
Figura 2. Geometria otimizada em nível PM3 da cadeia 1 da distinctina, cálculos considerando estrutura no estado gasoso, sem interações intermoleculares
distorcida. A geometria otimizada em nível MMFF94 mostra a Ca-deia 2 com regiões D-helicoidais menos evidentes. Em ambas as cadeias, formas desestruturadas são observadas nas regiões próxi-mas à ligação dissulfídica. A Cadeia 1 apresenta uma dobra na re-gião T10 – T15, favorecendo interações entre E4 da Cadeia 1 e a ramificação de H17 da Cadeia 2, bem como interações do anel aro-mático de F9 da Cadeia 1 com a ramificação de K19 da Cadeia 2.
Os cálculos de otimização de geometria em nível PM3 de 1 par-tindo da estrutura na forma totalmente D-helicoidal não convergiram.
A geometria otimizada em nível MMFF94 apresenta em sua maior parte formas D-helicoidais distorcidas, exceto nas regiões próximas às extremidades e aos resíduos de prolina (P6 e P7) e glicina (G8) da Cadeia 1, em que predominaram estruturas randômicas. Os cálculos MMFF94 mostraram que essa geometria apresenta uma energia de 56,76 kcal/mol maior que a geometria dada na Figura 4.
Analisando a estrutura da distinctina 1 obtida por RMN em água, depositada no PDB sob o código 1XKM,80 percebe-se que suas Cadeias 1 e 2 possuem um alto conteúdo de formas D-hélice.
Figura 3. Geometria otimizada em nível PM3 da Cadeia 2 da distinctina, cálculos considerando estrutura no estado gasoso, sem interações intermoleculares
e extremidades não-ionizadas
Figura 4. Geometria otimizada em nível PM3 da distinctina, cálculos considerando estrutura no estado gasoso, sem interações intermoleculares e extremidades
Na Cadeia 1, essa estrutura é quebrada devido à presença de uma região flexível compreendida entre os resíduos de P6, P7 e G8. A Cadeia 2 apresenta formas D-hélice por toda a sua extensão. Com-parando-se primeiramente esses resultados com os obtidos pela metodologia teórica para as estruturas das Cadeias 1 e 2, calcula-das separadamente, verifica-se que neste caso ambas apresentam apenas certa tendência em adotar a forma D-helicoidal (Figuras 2 e 3, respectivamente). No entanto, o conteúdo dessa forma nas estru-turas obtidas por RMN é mais significativo.
No caso da comparação da estrutura de 1 depositada no PDB com a calculada teoricamente, podem ser percebidas semelhanças um pouco maiores entre ambas. Assim, a Cadeia 1 no dímero apre-senta maiores torções causadas pela presença dos resíduos de prolina, enquanto a Cadeia 2 apresenta um maior conteúdo de for-masD-hélice na região próxima à extremidade N-terminal (Figura 4). A partir das geometrias otimizadas pôde-se estabelecer a por-centagem de resíduos que participam de formas D-hélice (conteú-do de forma D-hélice) das Cadeias 1 e 2, bem como da distictina 1. Na Cadeia 1 as formas D-hélice apresentaram-se distorcidas tanto quando tratada isoladamente, quanto no dímero. Entretanto, a Ca-deia 2 apresenta um conteúdo de forma D-hélice de 12% quando tratada isoladamente e de 24% quando no dímero.
CONCLUSÕES
Os resultados obtidos a partir do banco de dados ExPASy fo-ram condizentes com a previsão fornecida pelo banco de dados NPSA para a análise estrutural de ambas as cadeias da distinctina. Na Cadeia 1, observa-se um conteúdo significativo de D-hélices. Na região contendo os resíduos de prolina e glicina, observa-se uma quebra dessa estrutura secundária, conforme o esperado, dado o impedimento desses resíduos à adoção da forma helicoidal. Na Cadeia 2, um conteúdo ainda maior de formas helicoidais pode ser previsto, em conformidade com as metodologias de ambos os ban-cos de dados. As previsões específicas de folhas-E de 1indicaram uma preferência em se adotar folhas E-pregueadas antiparalelas. Esses resultados estão de acordo com as análises por CD e IV de 1, que indicaram um alto conteúdo dessa estrutura secundária em meio aquoso. No entanto, as formas em folhas E-pregueadas não foram observadas nas estruturas otimizadas, de acordo com as previsões realizadas para as cadeias separadas.
A presença de resíduos apolares como centros hidrofílicos (re-síduo L20 da Cadeia 2 e L16 de ambas as cadeias) pode ser devida às suas estruturas secundárias. A polarização inerente às formas D -helicoidais e as ligações hidrogênio específicas envolvendo os áto-mos dos grupos amidas da cadeia peptídica favorecem interações com as moléculas de água. Essas previsões fornecem informações importantes no estudo da interação da distinctina com membranas celulares e no mecanismo de ação sobre bactérias patogênicas.
As geometrias otimizadas de 1partindo de estruturas na forma totalmenteD-helicoidal mostram-se menos distorcidas. As formas D-helicoidais predominam na geometria, com exceção da região P6 – G8 da Cadeia 1 e L16 – L19 da Cadeia 2, estando de acordo com os resultados obtidos a partir dos bancos de dados. Esses re-sultados estão também de acordo com as análises por RMN da distinctina (1) em meio aquoso, que indicaram a ocorrência de vá-rias regiões com formas D-helicoidais em ambas as cadeias. Por outro lado, as geometrias otimizadas de 1partindo de estruturas na forma totalmente folhas-E mostraram-se menos favorecidas termodinamicamente. Esses resultados indicam ligações de hidro-gênio envolvendo tanto formas helicoidais, com interações intra-cadeias mais estáveis, quanto formas folha-E, com interações inter-cadeias relativamente menos estáveis.
Como a estrutura da distinctina (1) depositada no PDB se apre-senta com alto conteúdo de formas D-hélice, os resultados dos cál-culos MMFF94 e PM3 realizados no estado gasoso podem ser con-siderados uma boa aproximação, pois indicam sua tendência em adotar estruturação D-helicoidal. Assim, a metodologia teórica uti-lizada mostra-se apropriada à análise de 1, podendo, portanto, ser empregada convenientemente no estudo de suas propriedades.
AGRADECIMENTOS
Ao CNPq, FAPEMIG e CAPES pelas bolsas e apoios financei-ros concedidos.
REFERÊNCIAS
1. Zasloff, M.; Nature2002,415, 389.
2. Rinaldi, A. C.; Curr. Opin. Chem. Biol.2002,6, 799. 3. Csordas, A.; Michl, H.; Montsh. Chem.1970,101, 182. 4. Bechinger, B.; Biochim. Biophys. Acta1999,1462, 157. 5. Shai, Y.; Biopolymers2002,6, 236.
6. Bechinger, B.; Crit. Rev. Plant Sci.2004,23, 1. 7. Brogden, K. A.; Nature Rev. Microbiol.2005,3, 238. 8. Oren, Z.; Shai, Y.; Biopolymers1998,47, 451.
9. Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K.; Biochemistry1996,
35, 11361.
10. Shai, Y.; Biochim. Biophy. Acta1999,1462, 55. 11. Rao, A. G.; Arch. Biochem. Biophys.1999,361, 127. 12. Toke, O.; Peptide Sci.2005,80, 717.
13. Kitamura, A.; Kiyota, T.; Tomohiro, M.; Umeda, A.; Lee, S.; Inoue, T.; Sugihara, G.; Biophys. J. 1999,76, 1457.
14. Verly, R. M.; Rodrigues, M. A.; Daghastanli, K. R. P.; Denadai, A. M. L.; Cuccovia, I. M.; Bloch, Jr., C.; Frezard, F. J. G.; Santoro, M. M.; Piló-Veloso, D.; Bemquerer, M. P.; Peptides2008,29, 15.
15. Spartan 2004 Windows: Tutorial and User’s Guide Wavefunction, Inc., Portland, 2003.
16. Tsai, C. S.; An Introduction to Computational Biochemistry, Wiley-Liss Inc.: New York, 2002.
17. Ostberg, N.; Kaznessis, Y.; Peptides2005,26, 197. 18. Apatoff, A.; Kim, E.; Kliger, Y.; PLoS ONE2006,1, 113.
19. Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.; Nucleic Acids Res.2003,
31, 3381.
20. http://us.expasy.org, acessada em Janeiro 2008.
21. Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R. D.; Bairoch, A.;Nucleic Acids Res.2003,31, 3784.
22. http://www.ncbi.nlm.nih.gov/blast, acessada em Janeiro 2008. 23. McGinnis, S.; Madden, T. L.; Nucleic Acids Res.2004, 32, W20. 24. http://npsa-pbil.ibcp.fr, acessada em Janeiro 2008.
25. Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G.; Trends Biochem. Sci. 2000,25, 147.
26. https://genesilico.pl/meta2/, acessada em Janeiro 2008.
27. Schueler-Furman, O.; Wang, C.; Bradley, P.; Misura, K.; Baker, D.; Science 2005,310, 638.
28. Ponder, J. W.; Case, D. A.; Adv. Prot. Chem.2003,65, 27. 29. Weber, I. T.; Harrison, W. R.; Protein Eng.1996,9, 679.
30. Lomize, A. L.; Flippen-Anderson, J. L.; George, C.; Mosberg, H. L.; J. Am. Chem. Soc. 1994,116, 1.
31. Batista, C. V. F.; Scaloni, A.; Riegden, D. J.; Lindomar, R. S.; Romero, A. R.; Dukor, R.; Ceben, A.; Talamo, F.; Bloch, Jr.; C.; FEBS Lett. 2001,494,
85.
32. Raimondo, D.; Andreotti, G.; Saint, N.; Amodeo, P.; Renzone, G.; Sanseverino, M.; Zocchi, I.; Molle, G.; Motta, A.; Scaloni, A.; PNAS 2005,
102, 6309.
33. Deleage, G.; Roux, B.; Protein Eng. 1987,1, 289.
34. Garnier, J.; Osguthorpe, D. J.; Robson, B.; J. Mol. Biol. 1978, 120, 97. 35. Garnier, J.; Robson, B. Em Prediction of Protein Structure and the
Principles of Protein Conformation; Fasman, G. D., ed., Plenum Press: New York, 2003.
36. Garnier, J.; Gibrat, J-F.; Robson, B.; Methods Enzym.1996, 266, 540. 37. Guermeur, Y.; Tese de Doutorado, Université de Paris 6, 1997. 38. Rost, B.; Sander, C.; J. Mol. Biol. 1993,232, 584.
39. Levin, J. M.; Robson, B.; Garnier, J.; FEBS Lett.1986, 205, 303. 40. Geourjon, C.; Deleage, G.; Protein Eng. 1994, 7, 157.
42. Deleage, G.; Blanchet, C.; Geourjon, C.; Biochimie1997, 79, 681. 43. Chou, P. Y.; Fasman, G. D.; Adv. Enzym.1978, 47, 45.
44. Levitt, M.; Biochem.1978,17, 4277. 45. Lifson, S.; Sander, C.; Nature1979,282, 109. 46. King, R. D.; Sternberg, M. J.; Protein Sci.1996,5, 2298. 47. Sweet, R. M.; Eisenberg D.; J. Mol. Biol.1983,171, 479. 48. Kyte, J.; Doolittle, R. F.; J. Mol. Biol.1982,157, 105.
49. Abraham, D. J.; Leo, A. J.; Proteins: Struct. Funct. Gen.1987, 2, 1302. 50. Bull, H. B.; Breese, K.; Arch. Biochem. Biophys.1974, 161, 665. 51. Guy, H. R.; Biophys. J.1985, 47, 61.
52. Miyazawa, S.; Jernigen, R. L.; Macromolecules1985,18, 534. 53. Roseman, M. A.; J. Mol. Biol.1988,200, 513.
54. Welling, G. W.; Weijer, W. J.; Van der Zee, R.; Welling-Wester, S.; FEBS Lett.1985,188, 215.
55. Parker, J. M. R.; Guo, D.; Hodges, R. S.; Biochemistry1986,25, 5425. 56. Cowan, R.; Whittaker, R. G.; Peptide Res.1990,3, 75.
57. Wilson, K. J.; Honegger, A.; Stotzel, R. P.; Hughes, G. J.; Biochem. J. 1981,
199, 31.
58. Chothia, C.; J. Mol. Biol. 1976, 105, 1.
59. Eisenberg, D.; Schwarz, E.; Komarony, M.; Wall, R.; J. Mol. Biol.1984,
179, 125.
60. Hopp, T. P.; Woods, K. R.; Proc. Natl. Acad. Sci. U. S. A.1981,78, 3824. 61. Manavalan, P.; Ponnuswamy, P. K.; Nature1978,275, 673.
62. Black, S. D.; Mould, D. R.; Anal. Biochem.1991,193, 72. 63. Fauchere, J. -L.; Pliska, V. E.; Eur. J. Med. Chem.1983, 18, 369. 64. Janin, J.; Nature1979, 277, 491.
65. Rao, M. J. K.; Argos, P.; Biochim. Biophys. Acta1986,869, 197. 66. Wolfenden, R. V.; Andersson L.; Cullis P. M.; Southgate, C. C. F.;
Biochemistry1981,20, 849.
67. Aboderin, A. A.; Int. J. Biochem. 1971, 2, 537.
68. Rose, G. D.; Geselowitz, A. R.; Lesser, G. J.; Lee, R. H.; Zehfus, M. H.;
Science1985,229, 834.
69. Spartan 2002: Wavefunction, Inc., Irvine CA & Schrödinger, Inc., Portland, 1999.
70. Halgren, T. A.; J. Comput. Chem.1996,17, 490. 71. Halgren, T. A.; J. Comput. Chem.1996,17, 520. 72. Halgren, T. A.; J. Comput. Chem.1996,17, 553. 73. Halgren, T. A.; J. Comput. Chem.1996,17, 616.
74. Halgren, T. A.; Nachbar, R. B.; J. Comput. Chem.1996,17, 587. 75. Halgren, T. A.; J. Comput. Chem.1999,20, 720.
76. Halgren, T. A.; J. Comput. Chem.1999,20, 730.
77. Gaussian 2003, Revision B.04; Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen ,W.; Wong, M. W.; Gonzalez, C.; Pople, J. A; Gaussian, Inc., Pittsburgh PA, 2003.
78. Stewart, J. J. P.; J. Comp. Chem.1989,10, 209.
79. Csordes, F. S.; Bright, J. N.; Samson, S. P.; J. Mol. Biol. 2002,323, 951. 80. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig,