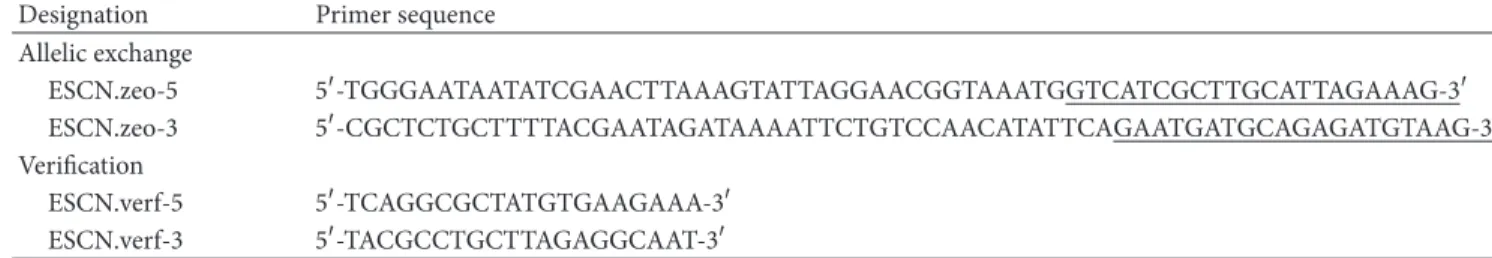Research Article
Analysis of the Virulence of an Atypical Enteropathogenic
Escherichia coli
Strain
In Vitro
and
In Vivo
and the Influence
of Type Three Secretion System
Suely C. F. Sampaio,
1Fabiana C. Moreira,
1Ana M. A. Liberatore,
2Mônica A. M. Vieira,
1Terezinha Knobl,
3Fabiano T. Romão,
1Rodrigo T. Hernandes,
1,4Claudete S. A. Ferreira,
3Antônio P. Ferreira,
3Aloísio Felipe-Silva,
5Rita Sinigaglia-Coimbra,
6Ivan H. J. Koh,
2and Tania A. T. Gomes
11Departamento de Microbiologia, Imunologia e Parasitologia, Universidade Federal de S˜ao Paulo, Escola Paulista de Medicina,
Rua Botucatu 862, 3rd floor, Vila Clementino, 01023-062 S˜ao Paulo, SP, Brazil
2Departamento de Cirurgia, Universidade Federal de S˜ao Paulo, Escola Paulista de Medicina, Rua Pedro de Toledo 781, 9th floor,
Vila Clementino, 01039-032 S˜ao Paulo, SP, Brazil
3Departamento de Ornitopatologia, Faculdade de Medicina Veterin´aria, Universidade de S˜ao Paulo,
Avenida Prof. Dr. Orlando Marques de Paiva 87, Cidade Universit´aria, 05508-900 S˜ao Paulo, SP, Brazil
1Departamento de Microbiologia e Imunologia, Instituto de Biociˆencias, UNESP, Distrito de Rubi˜ao Junior s/n, Botucatu,
18618-970 S˜ao Paulo, SP, Brazil
5Fleury Medicina e Sa´ude, Avenida General Valdomiro de Lima 508, 01311-903 S˜ao Paulo, SP, Brazil
6Centro de Microscopia Eletrˆonica, Universidade Federal de S˜ao Paulo, Escola Paulista de Medicina, Rua Botucatu 862,
1 st floor, 01023-062 S˜ao Paulo, SP, Brazil
Correspondence should be addressed to Tania A. T. Gomes; [email protected]
Received 7 February 2014; Accepted 5 April 2014; Published 28 April 2014
Academic Editor: Chensong Wan
Copyright © 2014 Suely C. F. Sampaio et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Atypical enteropathogenicEscherichia coli(aEPEC) inject various effectors into intestinal cells through a type three secretion system (T3SS), causing attaching and effacing (A/E) lesions. We investigated the role of T3SS in the ability of the aEPEC 1711-4 strain to interact with enterocytesin vitro(Caco-2 cells) andin vivo(rabbit ileal loops) and to translocate the rat intestinal mucosain vivo. A T3SS isogenic mutant strain was constructed, which showed marked reduction in the ability to associate and invade but not to persist inside Caco-2 cells. After rabbit infection, only aEPEC 1711-4 was detected inside enterocytes at 8 and 24 hours pointing to a T3SS-dependent invasive potentialin vivo. In contrast to aEPEC 1711-4, the T3SS-deficient strain no longer produced A/E lesions or induced macrophage infiltration. We also demonstrated that the ability of aEPEC 1711-4 to translocate through mesenteric lymph nodes to spleen and liver in a rat model depends on a functional T3SS, since a decreased number of T3SS mutant bacteria were recovered from extraintestinal sites. These findings indicate that the full virulence potential of aEPEC 1711-4 depends on a functional T3SS, which contributes to efficient adhesion/invasionin vitroandin vivoand to bacterial translocation to extraintestinal sites.
1. Introduction
Atypical enteropathogenic Escherichia coli (aEPEC) are emerging agents of diarrhea. They differ from typical EPEC (tEPEC) strains mainly by the absence of the EAF (EPEC adherence factor) plasmid [1,2]. Like tEPEC, aEPEC strains
inject various effector proteins into enterocytes through a type three secretion system (T3SS) leading to the formation of attaching-effacing (A/E) lesions [3–5]. The assembly of T3SS is dependent on an ATPase encoded by escN, and conse-quently,escNmutants are incapable of assembling or inject-ing effector proteins via T3SS into the host cell cytoplasm
[6]. Tir (translocated intimin receptor) is a T3SS-dependent effector protein, which is inserted in the eukaryotic cell membrane and interacts with an EPEC outer membrane adhesive protein (intimin) [7]. Tir-intimin interaction leads to the establishment of A/E lesions [8]. Many other T3SS-dependent effector proteins, such as Map (mitochondrial-associated protein) and EspF, have important roles in aEPEC pathogenesis. These proteins have redundant functions and can cause epithelial barrier disruption by interacting with tight junctions, leading to cell death by apoptosis [9,10].
Bacterial translocation (BT) is defined as the phe-nomenon by which live bacteria and/or their products cross the intestinal barrier reaching normally sterile extraintestinal sites, such as the liver, spleen, and mesenteric lymph nodes (MLN). The translocation of certain indigenous bacteria from the gastrointestinal tract to the MLN and various organs had been previously demonstrated in a gnotobiotic mouse model [11]. There is much circumstantial proof that translocation is associated with an increased occurrence of postoperative septic complications, and E. coli has been reported to be one of the most common BT-associated organisms isolated from surgical patients with postoperative sepsis [12, 13]. In humans, one of the most well-studied translocation events is that observed in cirrhotic patients with spontaneous bacterial peritonitis (SBP) [14].
We recently demonstrated that an aEPEC strain (1711-4) is able to invade and induce inflammatory responses in intestinal Caco-2 cell lines [15]. This strain is also able to invade these cellsin vitroand to escape from the intracellular compartment on the basolateral side [16]. In addition, we have demonstrated that in an experimental BT-rat model, aEPEC 1711-4 can reach the MLN, liver, and kidneys [17]. We also showed that aEPEC 1711-4 infected-animals had intestinal mesenteric microcirculation injury and systemic hypoperfusion similar to those observed with the virulent murineE. coli strain R6 [17, 18]. In the BT-rat model, the latter strain was recovered from the MLN, liver, and spleen and impaired mesenteric microcirculation [19].
The role of T3SS-dependent effector proteins in the ability of aEPEC to invade and persist in the intracellular compartment in vitroand to cross the intestinal barrierin vitroandin vivois not yet established. The objective of this study was to determine the role of T3SS in the ability of aEPEC 1711-4 to invade and persist inside polarized intestinal cells in vitro (Caco-2 cells), to promote A/E lesions and invadein vivo(rabbit ligated ileal loop model), and to pass through the intestinal barrier in an in vivo experimental model (bacterial translocation model).
2. Materials and Methods
2.1. Ethics Statement. This study was carried out in strict accordance with the recommendations of the Ethical princi-ples of the Sociedade Brasileira de Ciˆencia em Animais de Laborat´orio (COBEA). The protocol was approved by the Committee on Research Ethics of the Universidade Federal de S˜ao Paulo (Permit number: 0235/12). All surgery was performed under Telazol anesthesia (rabbits) or xylazine
hydrochloride plus ketamine hydrochloride (rats), and all efforts were made to minimize suffering.
2.2. Bacterial Strains and Growth Conditions. aEPEC 1711-4 (serotype O51:H1711-40), which was isolated from a child with diarrhea in the city of S˜ao Paulo [20], an isogenic mutant deleted in the escN gene (1711-4 ΔescN), and a complemented mutant 1711-4ΔescN(pEscN) were used. The nonpathogenicE. colistrain HS was used as a negative control (Table 1). The strains were cultivated overnight at 37∘C in 5 mL of Luria-Bertani (LB) broth. The 1711-4ΔescNand the 1711-4 ΔescN (pEscN) strains were cultivated in LB broth containing zeocin (60𝜇g mL−1) and zeocin-chloramphenicol (30𝜇g mL−1), respectively.
2.3. Construction of an Isogenic escN Deficient Mutant of aEPEC 1711-1 and Mutant Complementation. The escN-deficient mutant was constructed by homologous recombi-nation using the Lambda Red system as previously described [15, 22]. Primers ESCN.zeo5 and ESCN.zeo3 were used to amplify the zeocin resistance gene (Table 2). The amplified product was electroporated into the 1711-4 strain containing the pKOBEG-Apra plasmid. Transformants were selected on LB agar containing zeocin (60𝜇g mL−1). Deletion of the escN gene was confirmed using primers ESCN.verf5 and ESCN.verf3, targeting regions flanking this gene (Table 2). For complementation, the plasmid pEscN (pACYC184 vector carrying theescNgene) was electroporated into 1711-4ΔescN and transformants were selected on LB agar containing chloramphenicol (30𝜇g mL−1) [6].
2.1. Fluorescent-Actin Staining (FAS) Test in HeLa Cells. This test allows an indirect evaluation of the pathogen’s ability to induce A/E lesions evidenced by actin nucleation underneath the site of intimate bacterium-enterocyte interaction [23]. Bacteria were grown in 5 mL of LB broth for approximately 18 h, in ambient air, at 37∘C. Caco-2 cells were grown in 24-well plates (Corning) containing glass coverslips. They were cultivated in Dulbecco’s modified Eagle’s minimal essen-tial medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a 5% CO2atmosphere at36 ± 1∘C. Cells were grown up to 80% confluence. Cells were then washed three times with phosphate-buffered saline (PBS) before DMEM supplemented with 10% FBS containing 40𝜇L of bacterial suspension (∼108CFU mL−1) was added. Three hours after infection, cells were washed with PBS before they were fixed with 3% formaldehyde and permeabilized with 1% Triton X-100 for 4 min. Cells were washed with PBS and then incubated with PBS containing 5𝜇g/mL fluorescein isoth-iocyanate (FITC)-conjugated phalloidin (Sigma-Aldrich) for 20 min in a dark chamber. Cells were then washed three times with PBS every 10 min. Coverslips were removed, dried, and placed inverted onto glass slides containing 10𝜇L of 80% glycerol in PBS. Preparations were examined under fluorescence microscopy.
Table 1: Bacterial strains and plasmids used in this study.
Genotype and characteristics Genotype and characteristics Source/reference
1711-4 aEPEC O51:H40; wild-type Gomes et al., 2004 [21]
1711-4ΔescN escN::zeo (Zeor) This study
1711-4ΔescN (pEscN) 1711-4ΔescNcarrying pEscN (Zeor, Clor) This study pEscN pACYC184 carrying theescNgene from tEPEC E2348/69 strain Gauthier et al., 2003 [6] pKOBEG-Aprar Derivative (Aprar) of pKOBEG plasmid encoding the𝜆phage red operon Chaveroche et al., 2000 [22]
MC4160-malTΔ224::zeo-(F+) Source of zeocin cassette Gift from J. M. Ghigo
r
Means that the strain is resistant to that antibiotic.
Table 2: Primers used for construction and verification of mutation.
Designation Primer sequence Allelic exchange
ESCN.zeo-5 5-TGGGAATAATATCGAACTTAAAGTATTAGGAACGGTAAATGGTCATCGCTTGCATTAGAAAG-3 ESCN.zeo-3 5-CGCTCTGCTTTTACGAATAGATAAAATTCTGTCCAACATATTCAGAATGATGCAGAGATGTAAG-3 Verification
ESCN.verf-5 5-TCAGGCGCTATGTGAAGAAA-3 ESCN.verf-3 5-TACGCCTGCTTAGAGGCAAT-3
aUnderlined bases correspond to 5and 3regions of theSh blegene, which encodes zeocin resistance.
with∼1×107colony forming units (CFU) mL−1in each well of a 6-well cell culture plate. The number of cell-associated bacteria was determined three hours after infection. Cells were washed with phosphate-buffered saline pH 7.2 (PBS) before they were lysed with 1% (v/v) Triton X-100. Bacterial suspensions were plated on LB agar to determine the number of CFU. Bacterial invasion and persistence were assessed using gentamicin (100 and 10𝜇g mL−1, resp.) to kill extracel-lular bacteria before eukaryotic cell lysis for determination of the number of viable bacteria. All tests were performed twice in triplicate. The percentage of bacteria recovered after 48 h (persistence index) was calculated taking the number of CFU at three hours as 100% [15].
2.6. Rabbit Ligated Ileal Loop Model. Prior to the assays, New Zealand White rabbits (weighing 1.8 to 2.5 kg and 4 to 8 weeks of age) were examined for the presence of A/E lesion-producing E. coli by PCR using primers that identify theeaegene [24]. All bacterial strains were tested in three animals. Rabbits were fed only 10% (w/v) glucose solution for 48 h prior to the test. Rabbits were anesthetized with an intramuscular injection of Telazol (Fort Dodge Animal Health, Iowa, USA) (0.2 mL kg−1) and sedated with Nilperidol (0.3 mL kg−1, Crist´alia, S˜ao Paulo, Brazil). Anti-sepsis with 70% (v/v) ethanol was performed after shaving the abdomen. The mid-ileum was exposed by a midline laparotomy, and through a small incision made on the ileum wall, the distal portion was gently washed using a syringe with sterile saline to minimize the presence of luminal feces and resident microbiota. Immediately afterwards, five separated ileum segments, measuring 5 cm long and 3 cm apart, were constructed by ligatures, and 0.3 mL of a bacterial suspension (1 × 108CFU mL−1) in sterile LB broth was injected into each ligated loop using a 25-gauge needle. The ileum was then returned to the abdominal cavity, and the peritoneal
membrane and the abdominal wall were sutured. Animals were kept fasting for eight or 24 h and then were sacrificed with 3% (w/v) pentobarbital and zolazepam hydrochloride (0.4 mL kg−1). Ileal fragments including the whole intestinal wall were excised and fixed in 3% (w/v) glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2, for electron microscopy procedures.
2.7. Transmission Electron Microscopy (TEM). After fixing with 2.5% (v/v) glutaraldehyde for 24 h at 4∘C, the ileal fragments were rinsed with 0.1 M cacodylate buffer, pH 7.4, and postfixed in 1% (w/v) osmium tetroxide. Specimens were then exposed to a graded ethanol series and to propylene oxide. After embedding in Epon resin and polymerization at 60∘C for 48 h, ultrathin sections were stained with 2.0% (w/v) aqueous uranyl acetate and 2.5% (w/v) lead citrate. The specimens were then examined under a transmission electron microscope (JEOL 1200 EX II) at 80 kV.
2.8. Histopathological Analyses. Transverse segments of rab-bit ileum were fixed in buffered formalin before they were processed and embedded in paraffin. Sections were stained with hematoxylin-eosin before they were examined by a pathologist without previous knowledge of the details of the rabbit ileal loop experiments. Microscopy was carried out with a Zeiss microscope model Axio Lab.A1.
2.9. Bacterial Translocation Assays. Prior to the assays, adult female Wistar-EPM rats weighing 200–250 g (𝑛 =
17
11
-4
WT
17
11
-4
Δ
es
cN
(pEs
cN)
17
11
-4
Δ
es
cN
(a)
C
el
l ass
o
cia
te
d
b
ac
ter
ia (CFU)
∗
1711-4 ΔescN 1711-4 ΔescN (pEscN)
1711-4(WT)
1.0 × 100 1.0 × 103 3.4 × 107
1.0 × 106
3.3 × 104
3.2 × 101
T
T∗
(b)
Figure 1: Lack of T3SS renders aEPEC 1711-4 unable to aggregate actin in HeLa cells and association with Caco-2 cells is decreased in the absence of T3SS. The ability of the wild-type strain 1711-4, its isogenic mutant deficient inescN, and the complemented mutant 1711-4ΔescN
(pEscN) as well to promote actin aggregationin vitro(evidence of A/E lesion formation) was examined by FAS. For actin accumulation, cells were stained with fluorescein isothiocyanate (FITC)-conjugated phalloidin (green), and bacterial (white arrow) and HeLa cell DNA was stained with DAPI (blue). No actin nucleation was observed with 1711-4ΔescN mutant, whereas the ability of the complemented mutant to induce actin nucleation was restored (white arrowhead) (Figure 1(a)). Bacterial association was evaluated six hours after infection of differentiated Caco-2 cells. The number of viable bacteria recovered from cells infected with 1711-4ΔescNmutant (∼4.0×104CFU/well) was significantly lower compared with the wild-type strain (1.2 × 107CFU/well) (𝑃 < 0.05).∗The association capacity of theescNmutant was restored in the 1711-4ΔescN (pEscN) complemented strain and no statistically significant difference was observed when compared to the wild-type strain aEPEC 1711-4 (𝑃 > 0.05) (Figure 1(b)).
0.1 mL per 100 g body weight, intramuscular). After antisepsis with 70% (v/v) ethanol and midline laparotomy, the terminal ileum was ligated, the second portion of the duodenum was repaired, and an oroduodenal catheter was inserted. Subsequently, an inoculum of 1010CFU mL−1 (5 mL per 100 g body weight) was injected through the catheter and confined to the entire small bowel segment by the duodenum ligature. In six animals, saline was used instead of bacterial suspension (sham). The abdominal wall was closed with stitches after catheter removal. Sodium dipyrone (25 mg per kg body weight) was used for analgesia. After a period of two hours, animals were again subjected to laparotomy under anesthesia and samples were collected for analysis: one milliliter of blood from the inferior cava vein, MLN, spleen, and liver. Upon completion of the procedures, the animals were sacrificed by sectioning the aorta, still under anesthesia. Organs were weighed separately, crushed, macerated, and suspended in sterile saline, and the filtrate was plated on MacConkey agar to determine the number of translocated bacteria. Twenty-four hours after incubation in ambient air at 37∘C, the translocated bacteria in the plate were counted and CFU/g/compartment were determined [25].
2.10. Statistical Analyses. Data were analyzed using Prism program version 5.03 from GraphPad Software. Analysis of variance (ANOVA) with Bonferroni post hoc test was applied to evaluate all results. ANOVA and Fisher’s exact test were used for analysis of the BT results.
3. Results
3.1. T3SS Mutant of aEPEC 1711-1 (1711-1ΔescN) Is Unable to Cause A/E Lesion and Is Required for Efficient Association of aEPEC 1711-1 with Differentiated Caco-2 Cells In Vitro While escN Complementation Restores These Features. The ability of the wild-type strain 1711-4, its T3SS isogenic mutant (deficient inescN), and complemented mutant 1711-4ΔescN(pEscN) as well to cause A/E lesionsin vitrowas evaluated using the FAS test. As expected and in contrast to the wild-type strain, no actin nucleation was observed with the 1711-4ΔescNmutant, whereas the ability of the complemented mutant to induce actin nucleation was restored as inFigure 1(a). Although we have obtained the same results in Caco-2 cells, the presence of microvilli in polarized cells hampered the generation of a sharp image.
Bacterial association was evaluated three hours after infection of Caco-2 cells. The number of viable bacteria recov-ered from cells infected with the 1711-4ΔescN mutant was on average4 × 104 CFU/well. This number was significantly lower (𝑃 < 0.05) than that obtained with the wild-type strain (1.2 × 107CFU/well). The association capacity of the escN mutant was restored in the 1711-4 ΔescN (pEscN) comple-mented strain, with no statistically significant difference (𝑃 >
0.05) when compared to the wild-type strainFigure 1(b).
(a) (b) (c)
(d) (e) (f)
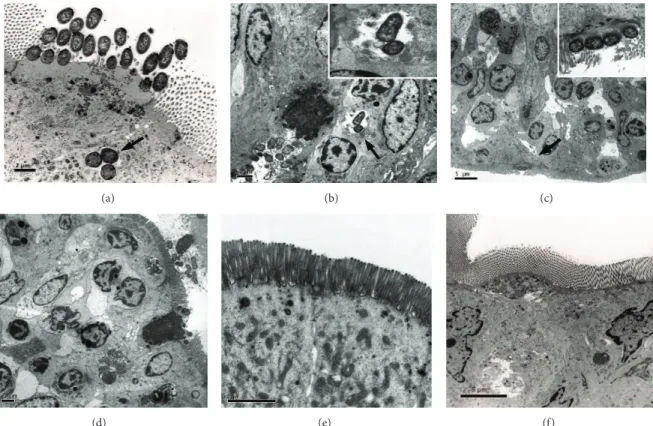
Figure 2: TEM images of rabbit ileal loops infected with aEPEC 1711-4, an isogenic T3SS-mutant or non-pathogenicE. coliHS. (a) wild-type strain at 8 h after infection; (b), (c), and (d)—1711-4 wild-type strain at 24 h after infection. Note an epithelial disorganization at 24 h after infection (c) and (d) in tissue infected with aEPEC 1711-4 strain but not 1711-4ΔescN(e) or nonpathogenicE. colistrain HS (f). Of note, aEPEC 1711-4 was detected inside an enterocyte at 8 h (a) and 24 h (b) after infection (black arrowheads). Note actin accumulation leading to pedestal formation (black arrow) (c).
Table 3: Intracellular bacteria at three and 48 hours after infection of Caco-2 cells.
Strain Number of intracellular bacteria at 3 hours (mean±SD)
Number of intracellular bacteria at 48 hours (mean±SD)
Mean bacterial persistence index
1711-4 WT 41,666±16,093.5 3,333±1,310.9 7.9%
1711-4ΔescN 1350±129.1 433±110.1 32.1%
1711-4ΔescN(pEscN) 37,500±3,535.5 3,266±503.3 8.7%
The mean number of CFU/well recovered from Caco-2 cells infected with the wild-type strain was approximately 30-fold higher than that observed with the 1711-4ΔescNmutant. In addition, the mean CFU number obtained with the comple-mented mutant 1711-4ΔescN(pEscN) was approximately 27-fold higher than that observed with the 1711-4ΔescNmutant (Table 3). These differences were statistically significant (𝑃 <
0.05).
For bacterial persistence evaluation, monolayers were washed with PBS three hours after infection and incubated with DMEM containing gentamicin (10𝜇g mL−1) to elimi-nate extracellular bacteria. The number of CFU recovered 48 h after infection with the 1711-1ΔescNmutant was approx-imately 7-fold lower (𝑃 < 0.05) than that observed with the wild-type strain, while the complemented mutant 1711ΔescN (pEscN) showed restored ability to persist inside enterocytes, which did not significantly differ compared to the wild-type strain (𝑃 > 0.05). The persistence rate was 7.9% for wild-type
strain 1711-4, 32.1% for the 1711-4ΔescNmutant, and 8.7% for the complemented 1711ΔescNmutant (Table 3).
100 𝜇m 100 𝜇m 100 𝜇m
PMN
PMN PMN
(a) (b) (c)
(a1) (b1) (c1)
Figure 3: Histopathological analyses of the H&E-stained infected rabbit ileum sections. Squares indicate areas magnified in (Figure 3(a1), (b1), and (c1)). (a) and (a1)—Ileal loop infected with aEPEC 1711-4; (b) and (b1) ileal loop infected with 1711-4ΔescN; (c) and (c1) ileal loop infected withE. coliHS. Note intense intraluminal polymorphonuclear leukocytes (PMN) infiltrate, red arrow, in (Figure 3(a)). Note moderate PMN tissue infiltration in (a1) and (b1).
3.1. aEPEC 1711-1 Stimulates an Acute PMN Infiltrate in the Rabbit Ileal Loop Model. Sections of the ileum infected with the wild-type aEPEC strain 1711-4 showed a moderate intraepithelial polymorphonuclear leukocytes (PMN) infil-trate, a large number of intraluminal PMN, and intraluminal bleeding (Figure 3(a)-(a1)). The ileum infected with the 1711-4
ΔescNhad an intraepithelial and intraluminal PMN infiltrate but to a lesser extent than that observed with the wild-type strain (Figure 3(b)-(b1)). In contrast, the ileum infected with the nonpathogenic strain HS showed a discrete polymor-phonuclear infiltration (Figure 3(c)-(c1)).
3.5. T3SS Is Necessary for Efficient In Vivo Translocation of aEPEC 1711-1 in the Rat Model. The Most striking difference—3 log10—was observed in the number of CFU recovered from MLN of animals infected with wild-type strain 1711-4 when compared to that recovered from animals infected with the T3SS-deficient mutant (1711-4ΔescN), but marked reduction in the CFU number was also observed in spleen and liver, since this mutant was not recovered even from these organs (𝑃 < 0.05) (Figure 4).
4. Discussion
In this study, we analyzed the ability of aEPEC 1711-4 as well as its isogenic mutant deficient in T3SS to adhere to, invade, and persist inside intestinal Caco-2 cells in vitro. We also evaluated the ability of these strains to invade and elicit an
inflammatory infiltrate in a rabbit ligated ileal loop modelin vivoand to translocate through the intestinal mucosa in a rat model.
During in vitro or in vivo interactions, aEPEC strains translocate effector proteins into enterocytes through a T3SS, resulting in the formation of A/E lesions [26]. According to Gauthier et al., 2003, the EscN protein functions as an ATPase, whose absence prevents T3SS assembly, blocking translocation of some structural and effector proteins into the eukaryotic target cell [6]. Our results demonstrated that the 1711-4escNmutant was unable to cause A/E lesion in HeLa cells and to adhere effectively to Caco-2 cells thus indicating that T3SS contributes to aEPEC 1711-4 adhesion. This was expected since Tir uses T3SS to reach the eukaryotic cell cyto-plasm before inserting into the host cell membrane to serve as an intimin receptor [8]. However, some intimin subtypes have alternative receptors in the eukaryotic membrane [27–
29] and flagella also play a role in bacterial adhesion [15,16,
30]. These previous findings may explain whyescNdeletion decreased but did not abolish the ability of aEPEC 1711-4 to adhere to Caco-2 cellsin vitro.Additionally, previous studies conducted in our laboratory have demonstrated that aEPEC strains can produce several fimbrial adhesion structures, which could contribute to the adherence process at least in epithelial cellsin vitro[31].
Sham 5
4
3
2
1
0
B
ac
ter
ial load (log
10
CFU
g
−1
)
MLN Spleen Liver
HS
1711-4
1711-ΔescN
∗
∗ ∗
Figure 4: Bacterial translocation assays. Bacterial recovery after BT in mesenteric lymph node (MLN), spleen, and liver 2 h after infection. Statistically significant differences (∗) were observed between aEPEC 1711-4 and 1711-4ΔescNin all compartments (𝑃 <
0.05). Bacteria were not detected in sham animals. HS strain was only detected in MLN.
1711-4 but not with the nonpathogenicE. coli HS. On the other hand, some authors reported that an escN deficient mutant derived from the tEPEC prototype strain E2348/69 was able to induce very high levels of IL-8 in a flagella-dependent pathway [32,33]. Consequently, we would expect a more exuberant PMN infiltrate in ileal loops infected with the 1711-4 ΔescN strain, but surprisingly the more pronounced intraepithelial and intraluminal PMN infiltrates were observed with the wild-type strain. These data taken together indicate that IL-8 production by enterocytes may not be the main factor determining epithelial infiltration by PMN during aEPEC infection in vivo. Another CXC-type chemokine, such as CXCL1 or CXCL5 [34, 35] known to be produced by enterocytes and to have their transcription driven by NF𝜅B signaling pathways triggered via toll-like receptors, may be more important in neutrophil recruitment in the rabbit ileal loop model.
Although the total CFU of the escN mutant detected in the intracellular compartment at 48 h was reduced when compared to the wild-type 1711-4 strain, the persistence index (the percentage of intracellular bacteria detected at 48 h) indicated that the 1711-4 ΔescN mutant had an increased capacity to persist inside Caco-2 cells (32.1%). Possibly a nonfunctional T3SS reduces the induction of nitric oxide synthase (iNOS) allowing more efficient bacterial persistence, suggesting that iNOS induction may be the result of one or more T3SS-dependent effectors [36]. Persistence may be essential to establish the carrier state, allowing colonization of other parts of the intestinal epithelium by bacteria that escape from infected enterocytes or allowing bacteria to go unde-tected by phagocytes and antibodies. aEPEC location inside vacuoles with pedestal formation, as demonstrated in HeLa
and Caco-2 cells may be another protective factor allowing intracellular persistence [37]. Several studies have shown that E. coliis the most commonly isolated pathogen in bacterial translocation events [38–41]. Moreover, some authors have demonstrated the occurrence of bacterial translocation in patients who developed sepsis after surgery [39]. It has been shown that a specific E. coli strain isolated from a fatal case of human hemorrhagic pancreatitis was more efficiently translocated to MLNs, blood, and peritoneal fluid [42].
To date, no cases of bacteremia due to EPEC have been described in humans, but strains harboring the eae gene have been detected in E. coli cultivated from bacteremic neonatal calves [43]. In previous studies by our group, we have shown that some aEPEC strains have the potential to invade and persist in enterocytes (Caco-2 and T84 cells) in vitro [16] and to translocate in the rat model [17]. In this study, we demonstrated by electron microscopy that the 1711-4 strain was able to colonize and to form A/E lesions in rabbit intestinal cells in the ileal loop modelin vivo. This event was not observed with theescN-deficient mutant. These results suggest the involvement of T3SS in the translocation eventin vivo. Liberatore et al. demonstrated that EPEC are able to translocate through the small bowel epithelium and reach not only the MLN but the spleen and the liver as well in a rat model. In this model, translocation of wild-type aEPEC 1711-4 has also been associated with damage to mesenteric microcirculation and hypoperfusion in the liver, small intestine, and kidneys [17]. We demonstrated in this study that a functional T3SS is necessary for efficient bacterial translocation, since a decreased number of bacteria were recovered from the liver and spleen of rats infected with the 1711-4 escN mutant. Martinez-Argudo et al. reported that T3SS is required for inducing loss of intestinal barrier function and allowing translocation ofSalmonella enterica strains through M cells [44]. The effector proteins injected via the T3SS that could contribute to driving the translocation events need to be characterized. Although findings in animal models should be extrapolated to humans with caution, our results indicate that aEPEC, an infectious agent theoretically restricted to the intestinal mucosa, has the potential to cross the intestinal barrier under overgrowth conditions which can occur in many clinical situations, such as immunosup-pression, antibiotic therapy, biliary obstruction, and other processes that cause changes in the intestinal microbiota.
5. Conclusion
Research carried out within vitroandin vivomodels have added to our understanding of how bacteria interact with and modify host cells functions leading to the establishment of disease. Our findings indicate that the full virulence potential of aEPEC 1711-4 depends on a functional T3SS, which contributes to efficient adhesion/invasionin vitroand in vivoand to bacterial translocation to extraintestinal sites.
Conflict of Interests
Acknowledgments
This work was supported by Grant nos. 2008/53812-4 and 2011/12664-5, S˜ao Paulo Research Foundation (FAPESP) and Conselho Nacional de Desenvolvimento Cient´ıfico e Tec-nol´ogico (Grant 304453/2011-0 and fellowship 150833/2012-1 to Suely C. F. Sampaio). The authors thank Jean-Marc Ghigo (Unit´e de G´en´etique des Biofilms, Department de Microbiologie, Institut Pasteur) for providing KOBEG-Apra plasmid.
References
[1] J. B. Kaper, J. P. Nataro, and H. L. T. Mobley, “Pathogenic
Escherichia coli,”Nature Reviews Microbiology, vol. 2, no. 2, pp.
123–140, 2004.
[2] L. R. Trabulsi, R. Keller, and T. A. Tardelli Gomes, “Typical and atypical enteropathogenicEscherichia coli,”Emerging Infectious
Diseases, vol. 8, no. 5, pp. 508–513, 2002.
[3] H. Deborah Chen and G. Frankel, “Enteropathogenic
Escherichia coli: unravelling pathogenesis,”FEMS Microbiology
Reviews, vol. 29, no. 1, pp. 83–98, 2005.
[4] S. C. Clarke, R. D. Haigh, P. P. E. Freestone, and P. H. Williams, “Virulence of enteropathogenic Escherichia coli, a global pathogen,”Clinical Microbiology Reviews, vol. 16, no. 3, pp. 365–378, 2003.
[5] P. Dean and B. Kenny, “The effector repertoire of enteropathogenicE. coli: ganging up on the host cell,”Current
Opinion in Microbiology, vol. 12, no. 1, pp. 101–109, 2009.
[6] A. Gauthier, J. L. Puente, and B. B. Finlay, “Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization,”Infection and Immunity, vol. 71, no. 6, pp. 3310– 3319, 2003.
[7] A. E. Jerse, J. Yu, B. D. Tall, and J. B. Kaper, “A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells,”Proceedings of the National Academy of Sciences of the
United States of America, vol. 87, no. 20, pp. 7839–7843, 1990.
[8] B. Kenny and B. Brett Finlay, “Intimin-dependent binding of enteropathogenicEscherichia colito host cells triggers novel signaling events, including tyrosine phosphorylation of phos-pholipase C-𝛾1,”Infection and Immunity, vol. 65, no. 7, pp. 2528– 2536, 1997.
[9] J. K. Crane, B. P. McNamara, and M. S. Donnenberg, “Role of EspF in host cell death induced by enteropathogenicEscherichia
coli,”Cellular Microbiology, vol. 3, no. 4, pp. 197–211, 2001.
[10] P. Dean and B. Kenny, “Intestinal barrier dysfunction by enteropathogenicEscherichia coliis mediated by two effector molecules and a bacterial surface protein,”Molecular
Microbi-ology, vol. 54, no. 3, pp. 665–675, 2004.
[11] R. D. Berg and A. W. Garlington, “Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model,”Infection and Immunity, vol. 23, no. 2, pp. 403– 411, 1979.
[12] C. J. O’Boyle, J. MacFie, C. J. Mitchell, D. Johnstone, P. M. Sagar, and P. C. Sedman, “Microbiology of bacterial translocation in humans,”Gut, vol. 42, no. 1, pp. 29–35, 1998.
[13] J. MacFie, C. O’Boyle, C. J. Mitchell, P. M. Buckley, D. Johnstone, and P. Sudworth, “Gut origin of sepsis: a prospective study
investigating associations between bacterial translocation, gas-tric microflora, and septic morbidity,”Gut, vol. 45, no. 2, pp. 223–228, 1999.
[14] F. Bert, J. R. Johnson, B. Ouattara et al., “Genetic diversity and virulence profiles ofEscherichia coliisolates causing spon-taneous bacterial peritonitis and bacteremia in patients with cirrhosis,”Journal of Clinical Microbiology, vol. 48, no. 8, pp. 2709–2714, 2010.
[15] S. C. F. Sampaio, T. A. T. Gomes, C. Pichon et al., “The flagella of an atypical enteropathogenicEscherichia colistrain are required for efficient interaction with and stimulation of interleukin-8 production by enterocytes in vitro,”Infection and Immunity, vol. 77, no. 10, pp. 4406–4413, 2009.
[16] S. C. F. Sampaio, J. R. C. Andrade, J. L. M. Sampaio, C. R. W. Carneiro, E. Freym¨uller, and T. A. T. Gomes, “Distinct interaction of two atypical enteropathogenic Escherichia coli
strains with enterocytes in vitro,”Open Microbiology Journal, vol. 5, no. 1, pp. 65–71, 2011.
[17] A. M. A. Liberatore, F. C. Moreira, T. A. T. Gomes, J. L. Menchaca-Diaz, and I. H. J. Koh, “Typical and atypical enteropathogenicEscherichia colibacterial translocation asso-ciated with tissue hypoperfusion in rats,”Brazilian Journal of
Medical and Biological Research, vol. 44, no. 10, pp. 1018–1024,
2011.
[18] I. H. J. Koh, R. Guatelli, E. F. S. Montero et al., “Where is the site of bacterial translocation—small or large bowel?”
Transplantation Proceedings, vol. 28, no. 5, p. 2661, 1996.
[19] I. H. J. Koh, J. L. Menchaca-Diaz, S. H. P. Farsky et al., “Injuries to the mesenteric microcirculation due to bacterial translocation,”Transplantation Proceedings, vol. 34, no. 3, pp. 1003–1004, 2002.
[20] M. A. M. Vieira, J. R. C. Andrade, L. R. Trabulsi et al., “Phenotypic and genotypic characteristics ofEscherichia coli
strains of non-EnteropathogenicE. coli(EPEC) serogroups that carry eae and lack the EPEC adherence factor and shiga toxin DNA probe sequences,”Journal of Infectious Diseases, vol. 183, no. 5, pp. 762–772, 2001.
[21] T. A. T. Gomes, K. Irino, D. M. Girao et al., “Emerging enteropathogenicEscherichia colistrains?”Emerging Infectious
Diseases, vol. 10, no. 10, pp. 1851–1855, 2004.
[22] M. K. Chaveroche, J. M. Ghigo, and C. d’Enfert, “A rapid method for efficient gene replacement in the filamentous fungus
Aspergillus nidulans,”Nucleic Acids Research, vol. 28, no. 22, p.
E97, 2000.
[23] S. Knutton, T. Baldwin, P. H. Williams, and A. S. McNeish, “Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagicEscherichia coli,”Infection and Immu-nity, vol. 57, no. 4, pp. 1290–1298, 1989.
[24] M. A. M. Vieira, T. A. T. Gomes, A. J. P. Ferreira, T. Kn¨obl, A. L. Servin, and V. L.-L. Moal, “Two atypical enteropathogenic
Escherichia colistrains induce the production of secreted and
membrane-bound mucins to benefit their own growth at the apical surface of human mucin-secreting intestinal HT29-MTX cells,”Infection and Immunity, vol. 78, no. 3, pp. 927–938, 2010. [25] I. H. J. Koh and R. M. Silva, “Novelin vitro small intestinal
graft model for study of bacterial translocation in the rat,”
Transplantation Proceedings, vol. 28, no. 5, pp. 2667–2668, 1996.
[26] R. T. Hernandes, W. P. Elias, M. A. M. Vieira, and T. A. T. Gomes, “An overview of atypical enteropathogenicEscherichia
coli,”FEMS Microbiology Letters, vol. 297, no. 2, pp. 137–149,
[27] G. Frankel, O. Lider, R. Hershkoviz et al., “The cell-binding domain of intimin from enteropathogenicEscherichia colibinds to𝛽1 integrins,”Journal of Biological Chemistry, vol. 271, no. 34, pp. 20359–20364, 1996.
[28] G. Frankel, A. D. Phillips, L. R. Trabulsi, S. Knutton, G. Dougan, and S. Matthews, “Intimin and the host cell—is it bound to end in Tir(s)?”Trends in Microbiology, vol. 9, no. 5, pp. 214–218, 2001. [29] J. F. Sinclair, E. A. Dean-Nystrom, and A. D. O’Brien, “The established intimin receptor Tir and the putative eucaryotic intimin receptors nucleolin and 𝛽1 integrin localize at or near the site of enterohemorrhagicEscherichia coli O157:H7 adherence to enterocytesin vivo,”Infection and Immunity, vol. 74, no. 2, pp. 1255–1265, 2006.
[30] J. A. Gir´on, A. G. Torres, E. Freer, and J. B. Kaper, “The flagella of enteropathogenicEscherichia colimediate adherence to epithelial cells,”Molecular Microbiology, vol. 44, no. 2, pp. 361–379, 2002.
[31] R. T. Hernandes, I. Velsko, S. C. F. Sampaio et al., “Fimbrial adhesins produced by atypical enteropathogenicEscherichia coli
strains,”Applied and Environmental Microbiology, vol. 77, no. 23, pp. 8391–8399, 2011.
[32] R. Sharma, S. Tesfay, F. L. Tomson, R. P. Kanteti, V. K. Viswanathan, and G. Hecht, “Balance of bacterial pro- and anti-inflammatory mediators dictates net effect of enteropathogenic
Escherichia colion intestinal epithelial cells,”American Journal
of Physiology—Gastrointestinal and Liver Physiology, vol. 290,
no. 4, pp. G685–G694, 2006.
[33] M. A. Khan, S. Bouzari, C. Ma et al., “Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenicEscherichia coliand Citrobacter rodentium,”
Infection and Immunity, vol. 76, no. 4, pp. 1410–1422, 2008.
[34] S. Keates, A. C. Keates, E. Mizoguchi, A. Bhan, and C. P. Kelly, “Enterocytes are the primary source of the chemokine ENA-78 in normal colon and ulcerative colitis,”American Journal of
Physiology—Gastrointestinal and Liver Physiology, vol. 273, no.
1, pp. G75–G82, 1997.
[35] S. J. H. van Deventer, “Review article: chemokine production by intestinal epithelial cells: a therapeutic target in inflammatory bowel disease?” Alimentary Pharmacology and Therapeutics,
Supplement, vol. 11, no. 3, pp. 116–121, 1997.
[36] R. B. Canani, P. Cirillo, V. Buccigrossi et al., “Nitric oxide produced by the enterocyte is involved in the cellular regulation of ion transport,”Pediatric Research, vol. 54, no. 1, pp. 64–68, 2003.
[37] R. T. Hernandes, R. M. Silva, S. M. Carneiro et al., “The localized adherence pattern of an atypical enteropathogenicEscherichia coliis mediated by intimin omicron and unexpectedly promotes HeLa cell invasion,”Cellular Microbiology, vol. 10, no. 2, pp. 415– 425, 2008.
[38] C.-G. Nettelbladt, M. Katouli, T. Bark, T. Svenberg, R. M¨ollby, and O. Ljungqvist, “Orally inoculatedEscherichia colistrains colonize the gut and increase bacterial translocation after stress in rats,”Shock, vol. 20, no. 3, pp. 251–256, 2003.
[39] J. MacFie, B. S. Reddy, M. Gatt, P. K. Jain, R. Sowdi, and C. J. Mitchell, “Bacterial translocation studied in 927 patients over 13 years,”British Journal of Surgery, vol. 93, no. 1, pp. 87–93, 2006. [40] C. Macutkiewicz, G. Carlson, E. Clark, U. Dobrindt, I. Roberts, and G. Warhurst, “Characterisation ofEscherichia colistrains involved in transcytosis across gut epithelial cells exposed to metabolic and inflammatory stress,”Microbes and Infection, vol. 10, no. 4, pp. 424–431, 2008.
[41] M. Katouli, N. L. Ramos, C. G. Nettelbladt et al., “Host species-specific translocation ofEscherichia coli,”European Journal of
Clinical Microbiology and Infectious Diseases, vol. 28, no. 9, pp.
1095–1103, 2009.
[42] C.-G. Nettelbladt, M. Katouli, T. Bark, T. Svenberg, R. M¨ollby, and O. Ljungqvist, “Evidence of bacterial translocation in fatal hemorrhagic pancreatitis,”Journal of Trauma—Injury, Infection
and Critical Care, vol. 48, no. 2, pp. 314–315, 2000.
[43] G. Fecteau, J. M. Fairbrother, R. Higgins et al., “Virulence factors inEscherichia coliisolated from the blood of bacteremic neonatal calves,”Veterinary Microbiology, vol. 78, no. 3, pp. 241– 249, 2001.
[44] I. Martinez-Argudo, C. Sands, and M. A. Jepson, “Translocation of enteropathogenicEscherichia coliacross anin vitroM cell model is regulated by its type III secretion system,”Cellular
Submit your manuscripts at
http://www.hindawi.com
Stem Cells
International
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
INFLAMMATION
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Behavioural
Neurology
Endocrinology
International Journal of Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Disease Markers
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014 BioMed
Research International
Oncology
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014 Oxidative Medicine and Cellular Longevity Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
PPAR Research
The Scientific
World Journal
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Immunology Research Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Journal of
Obesity
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Computational and Mathematical Methods in Medicine
Ophthalmology
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Diabetes Research
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Research and Treatment
AIDS
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Parkinson’s
Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014