Arilson José do Nascimento Silva
Estudo da regeneração de aditivos para catalisadores de craqueamento
aluminofosfatos (ALPO’s) e silicoaluminofosfatos (SAPO’s)
_______________________________________
Dissertação de Mestrado
Natal/RN, novembro de 2007
CENTRO DE CIÊNCIAS EXATAS E DA TERRA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
Centro de Ciências Exatas e da Terra
Programa de Pós-Graduação em Química
DISSERTAÇÃO DE MESTRADO
Arilson José do Nascimento Silva
Natal / RN Novembro – 2007
Estudo da Regeneração de aditivos para
Catalisadores de Craqueamento
ESTUDO DA REGENERAÇÃO DE ADITIVOS PARA
CATALISADORES DE CRAQUEAMENTO
ALUMINOFOSFATOS (ALPO’S) E
SILICOALUMINOFOSFATOS (SAPO’S)
Dissertação de Mestrado apresentada ao Programa de Pós-Graduação em Química da Universidade Federal do Rio Grande do Norte como parte dos requisitos para obtenção do título de Mestre em Química
Orientador: Prof. Dr. Valter José Fernandes Junior
Catalogação da Publicação na Fonte. UFRN / SISBI / Biblioteca Setorial Especializada do Centro de Ciências Exatas e da Terra – CCET.
Silva, Arilson José do Nascimento.
Estudo da regeneração de aditivos para catalisadores de craqueamento aluminofosfatos (ALPO’s)e silicoaluminofosfatos (SAPO’s) / Arilson José do Nascimento Silva. – Natal, 2007.
125 f.: il.
Orientador: Prof. Dr. Valter José Fernandes Junior.
Dissertação (Mestrado) – Universidade Federal do Rio Grande do Norte. Centro de Ciências Exatas e da Terra. Programa de Pós-Graduação em Química.
1. Síntese hidrotérmica – Dissertação. 2. Materiais microporosos – Dissertação. I. Fernandes Junior, Valter José. II. Título.
Ao meu Senhor Jesus Cristo,
Sua e Nossa Mãe, Nossa Senhora,
A minha amada filha Clarisse e minha
amada esposa Janaina,
A meus pais Hermano e Marta e aos meus
Agradecimentos
À Universidade Federal do Rio Grande do Norte e ao Programa de Pós-Graduação em Química do Departamento de Química por proporcionarem o apoio técnico e os recursos necessários para a realização desta pesquisa.
À ANP pela concessão de uma bolsa de estudos.
Obviamente seria uma “mqi” registrar o nome de todos aqueles que de uma forma ou outra contribuíram para a realização deste trabalho. A todas essas pessoas, os meus mais sinceros agradecimentos e desde já as minhas desculpas por não as ter citado. Contudo, algumas dessas devem ser destacadas com todo o meu carinho pela inestimável contribuição que deram a essa tese. Desejaria agradecer de modo especial:
Ao Prof. Dr. Valter José Fernandes Júnior, pela valiosa e inesquecível orientação, amizade e apoio demonstrado ao longo deste trabalho;
Ao Programa de Pós-graduação em Química da UFRN (PPGQ), especialmente a saudosa coordenadora Profª. Rosangela e a secretária Gisele pelo apoio durante este trabalho;
Aos Professores do Laboratório de Catálise e Petroquímica Antonio Souza de Araújo e José Melo de Carvalho pelo companheirismo e troca de idéias científicas;
Aos colegas do Laboratório de Catálise da UFRN, Viviane, Luzia Patrícia, Stevie, Hellyda, Larissa, Maria, Ricardo e especialmente as minhas grandes amigas Joana, Solange e Marcelo pela amizade e apoio constante;
Ao Laboratório de Combustíveis e Lubrificantes da UFRN (LCL) pela utilização de gases essenciais às análises químicas e aos seus integrantes em especial, Regina, Amanda, as secretárias e Nadir pelo companheirismo ao longo da realização da pesquisa, e Iranir, por todo carinho, preocupação e dedicação;
Ao Centro de Ciências Exatas e da Terra (CCET) pela infra-estrutura para realização das análises de Difração de Raios-X, em especial a Artejose e Ériko;
À minha amada filha Clarisse Silva Stuckert, e a razão da minha vida;
A minha esposa Janaina Silva Stuckert que me apóia e incentiva em todos os momentos, mostrando sempre uma luz no fim do túnel;
A meu Pai Hermano, que me ajudou em mais essa realização, permanecendo sempre ao meu lado com muito carinho, dedicação;
À minha mãe Marta e meus irmãos Andréa, Hermano que sempre torcem por mim.
Estou esquecendo de muitos, mas percebo agora que é “realmente impossível lembrar de todos, portanto”...
Resumo
Foram sintetizados catalisadores heterogêneos do tipo aluminofosfatos e silicoaluminofosfatos pelo método hidrotérmico a partir de alumina hidratada (pseudobohemita), ácido fosfórico 85%, sílica gel, água e diisopropilamina (DIPA) usada como direcionador estrutural orgânico. Estes reagentes foram misturados a fim de obter géis com as seguintes composições: 2.9 Al +3.2 P + 3.5 DIPA +32.5 H20; para ALPO e 2.9 Al +3.2 P + 0.5 Si + 3.5 DIPA +32.5 H20 para SAPO. O processo de cristalização ocorreu à temperatura de 170 0C durante 48 h, quando foi possível obter as fases puras para ALPO – 11 e SAPO – 11. Os materiais obtidos foram lavados com água deionizada, secos e calcinados para remover as moléculas do direcionador. Os materiais foram caracterizados por difração de raios-X (DRX), microscopia eletrônica de varredura (MEV), espectroscopia de absorção na região do infravermelho (FTIR), análise térmica via TG/DTG e adsorção de nitrogênio (BET). As propriedades ácidas foram determinadas usando adsorção de n-butilamina seguida de termodessorção programada. Este método revelou que o ALPO – 11 possuem sítios ácidos fracos devido a defeitos estruturais, já a amostra SAPO – 11 apresenta uma acidez tipicamente fraca a moderada. Entretanto, uma pequena quantidade de sítios ácidos fortes foi detectada. A desativação dos catalisadores foi conduzida pela reação de craqueamento do n-hexano em um microrreator catalítico de leito fixo com fluxo contínuo acoplado em linha com um cromatógrafo a gás. Como principais produtos foram obtidos: etano, propano, isobutano, n-butano,e n-pentano, isopentano. Para determinar a regeneração e a remoção do direcionador orgânico foi aplicado o método cinético Vyazovkin (Model Free).
Palavras Chaves: Síntese hidrotérmica, Materiais microporosos,.
BANCA EXAMINADORA
Presidente:-Prof. Dr. Valter José Fernandes Júnior
Membros:-Prof. Dr.Jodé Marcos Melo de Carvalho
Abstract
Heterogeneous catalysts such as aluminophosphate and silicoaluminophosphate, molecular sieves with AEL of ALPO-11 and SAPO-11, were synthesized by the hydrothermal method with the following molar composition: 2.9 Al +3.2 P + 3.5 DIPA +32.5 H20 (ALPO-11); 2.9 Al +3.2 P + 0.5 Si + 3.5 DIPA +32.5 H20 (SAPO-11)
starting from silica (only in the SAPO-11), pseudoboehmite, orthophosphoric acid (85%) and water, in the presence of a di-isopropylamine organic template. The crystallization process occurred when the reactive hydrogel was charged into a vessel and autoclaved at 170ºC for a period of 48 hours under autogeneous pressure. The obtained materials were washed, dried and calcined to remove the molecular sieves of DIPA. The samples were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), infrared spectroscopy (FT-IR), thermo gravimetric differential thermal analysis (TG/DTA) and nitrogen adsorption (BET). The acidic properties were determined using adsorption of n-butylamine followed by programmed thermodessorption. This method revealed that ALPO-11 has weaker acid sites due to structural defects, while SAPO-11 shows an acidity that ranges from weak to moderate. However, a small quantity of strong acid sites could be detected there. The deactivation of the catalysts was conducted by the cracking of the n-hexane in a fixed bed continuous flow microrreator coupled on line to a gas chromatograph. The main products obtained were: ethane, propane, isobutene, n-butane, n-pentane and isopentane. The Vyazovkin (model-free) kinetics method was used to determine the regeneration and removal of the organic template.
Keywords: Hydrothermal synthesis, Micro porous materials.
MEMBERS OF THE BOARD OF EXAMINATION:
President:- Prof. Dr. Valter José Fernandes Júnior
Members:- Prof. Dr.José Marcos Melo de Carvalho
Principais Trabalhos do Autor
1. SILVA, A. J. N.; LIMA, S. H.; SILVA, A. O. S. da.; SOUZA, M. J. B.; ARAUJO, A. S., FERNANDES,V. J. Jr.; CARVALHO, J. M. de., Caracterização e aplicação catalítica das zeólitas HZSM-5 e HZSM-12 na desidratação de etanol. Terceiro Congresso Brasileiro de Petróleo, Salvador. 2005.
2. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; FERNANDES,V. J. Jr.;
SOBRINHO,E.V.,Cinética Isotérmica da Degradação Térmica de óleos Lubrificante
Automotivos. In: ABRATEQ, 2004, Poços de Caldas. CD. 2004.
3. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; FERNANDES,V. J. Jr.;
SOBRINHO,E.V.,Thermoanalytical and rheological characterization of
automotive mineral lubrificants after thermal degradation., Fuel, IN PRESS, 2004.
4. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; FERNANDES,V. J. Jr.;
SOBRINHO,E.V., Estudo Reológicos e Térmicos de óleos sem aditivos. In: XI
ENCONTRO DE INICIAÇÃO CIENTIFICA DA UFPB, João Pessoa. 2003.
5. SILVA, A. J. N.; SANTOS, J. C. O.; SANTOS, I. M. G. dos.; SOUZA, A. G de.; SOBRINHO, E. V.; Kinetic Study On Thermal decomposition reactions of mineral lubrificant oils by.; IICONGRESSO BRASILEIRO DE P&D EM PETROLEO & GAS, 2003, Rio de Janeiro. CD Rom.2003.
6. SILVA, A. J. N.; SANTOS, J. C. O.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.;
FERNANDES,V. J. Jr.; Estudo Reológico e Térmico de Óleos Lubrificantes Minerais e Sintéticos. In: X ENCONTRO DE INICIAÇÃO CIENTIFICA DA UFPB, 2002, João Pessoa. 2002.
7. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; Influência da Adição de Álcoois Lineares Sobre as Propriedades físico-químicas da Gasolina Base, ABQ, 2002, Rio de Janeiro. 2002.
9. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; FERNANDES,V. J. Jr.;
Característica reologica da degradação térmica de óleos lubrificantes automotivos. In: Primeiro Congresso Brasileiro de Petróleo, 2001, Natal. 2001.
10. SILVA, A. J. N.; SANTOS, J. C. O.; SINFRONIO, F. S.; CONCEIÇAO, M. M.; SANTOS, I. M. G. dos.; SOUZA, A. G. de.; FERNANDES,V. J. Jr.;
Lista de Tabelas
Capítulo 2. Revisão da Literatura
Tabela 2.1 Tamanho de poros para a família de aluminofosfatos...14
Tabela 2.2 Tipos de ligações em ALPO’s modificados por Si e Metais...16
Tabela 2.3 propriedades das peneiras moleculares baseadas em aluminofosfatos...17
Capítulo 3. Materiais e Métodos Tabela 3.1 Composição dos géis de síntese das amostras...32
Capítulo 4. Resultados e Discussão Tabela 4.1 isoconverção para aluminofosfatos...54
Tabela 4.2 isoconverção para silicoaluminofosfatos...54
Tabela 4.3 Grau de cristalinidade das amostras sintetizadas...57
Tabela 4.4 Atribuições para as principais bandas dos espectros de FT-IR das amostras de ALPO-11 e SAPO-11...59
Tabela 4.5 Acidez das amostras de ALPO-11 e SAPO-11...67
Tabela 4.6 Dados relativos à distribuição de produtos para ALPO-11...74
Tabela 4.7 Dados relativos à distribuição de produtos para SAPO-11...74
Tabela 4.8 Temperatura (0C) para remoção do coque em função do tempo em diferentes conversões para aluminofosfatos...77
Lista de Figuras
Capítulo 2. Revisão da Literatura
Figura 1Produção de derivados de Petróleo no Brasil. ...7
Figura 2Unidade de craqueamento de hidrocarbonetos...7
Figura 3 Estrutura de peneira molecular do tipo AEL...15
Capítulo 3. Materiais e Métodos Figura 4 Modelo de autoclave usado na síntese hidrotérmica do ALPO-11 e SAPO-11... 29
Figura 5 Fluxograma geral da síntese dos materiais ALPO e SAPO...31
Figura 6 Sistema de calcinação para remoção de DIPA das materiais...33
Figura 7 Perfil de aquecimento das amostras calcinadas...33
Figura 8Equipamento utilizado para analise de raios-x...34
Figura 9 Microscópico eletrônico de varredura...35
Figura 10Termobalança de forno horizontal da Mettler...36
Figura 11Equipamento para analise de BET...37
Figura 12 Sistema de adsorção de base para medir propriedades ácidas...39
Figura 13Microrreator catalítico...41
Figura 14 Esquema representativo para formulação da lei de Bragg...44
Capítulo 4. Resultados e Discussão
Figura 16 Esquema representativo da remoção do direcionador dos poros de
materiais com arranjo microporoso...52
Figura 17Curvas TG/DTG da amostra de ALPO-11 não calcinada obtida em diferentes razões de aquecimento (β)....52
Figura 18Curvas TG/DTG da amostra de SAPO-11 não calcinada obtida em diferentes razões de aquecimento (β)....53
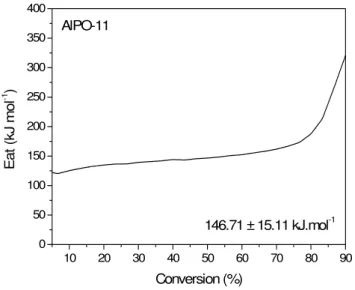
Figura 19Energia de ativação aparente para remoção da DIPA em ALPO-11...55
Figura 20Energia de ativação aparente para remoção da DIPA em SAPO-11...55
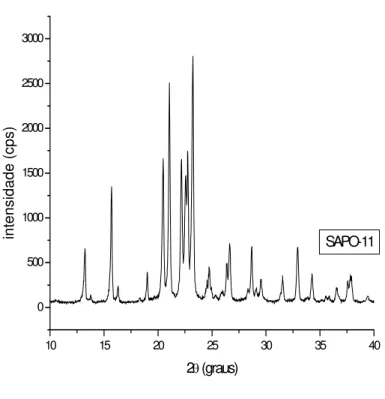
Figura 21Análise de DRX dos sólidos obtidos durante cristalização do ALPO-11...56
Figura 22Análise de DRX dos sólidos obtidos durante cristalização do SAPO-11...57
Figura 23 Espectros de FT-IR para ALPO-11 e SAPO-11 não calcinados obtidos durante a cristalização...58
Figura 24 Espectros de FT-IR para ALPO-11 e SAPO-11 calcinados obtidos durante a cristalização...58
Figura 25 Micrografia eletrônica de Varredura para amostra de ALPO-11 não calcinadas...60
Figura 26 Micrografia eletrônica de Varredura para amostra de ALPO-11 não calcinada...60
Figura 27 Micrografia eletrônica de Varredura para amostra de ALPO-11 calcinada...61
Figura 29 Micrografia eletrônica de Varredura para amostra de
SAPO-11 não calcinada...62
Figura 30 Micrografia eletrônica de Varredura para amostra de
SAPO-11 não calcinada...62
Figura 31 Micrografia eletrônica de Varredura para amostra
de SAPO-11 calcinada...63
Figura 32 Micrografia eletrônica de Varredura para amostra de
SAPO-11 calcinada...63
Figura 33 Curva TG da amostra de ALPO-11, saturada
com n-butilamina...65
Figura 34 Curva TG da amostra de SAPO-11, saturada
com n-butilamina...65
Figura 35 Curvas da TG/DTG com a razão de β= 10 0 C/min para
dessorção da n-butilamina da amostra de ALPO-11...66
Figura 36 Curvas da TG/DTG com a razão de β= 10 0 C/min para
dessorção da n-butilamina da amostra de SAPO-11...66
Figura 37 Isotermas de adsorção de N2 e distribuição do diâmetro de
poros do suporte microporoso ALPO-11...67
Figura 38 Isotermas de adsorção de N2 e distribuição do diâmetro de
poros do suporte microporoso SAPO-11...68
Figura 39 Conversão em função do tempo de reação para a amostra de
ALPO-11...69
Figura 40Seletividade dos produtos em função do tempo de reação
para amostra de ALPO – 11....70
Figura 41 Conversão em função do tempo de reação para a amostra de
Figura 42Seletividade dos produtos em função do tempo de reação para
amostra de SAPO – 11....71
Figura 43. Cromatograma típico da análise dos produtos da reação de craqueamento de n-hexano sobre ALPO-11...72
Figura 44. Cromatograma típico da análise dos produtos da reação de craqueamento de n-hexano sobre SAPO-11...72
Figura 45 Curvas de TG/DTG para o ALPO-11 coqueada na razão de aquecimento β=10 0C min-1 com o fluxo de ar continuo de 120 Cm3 min-1...75
Figura 46Curvas de TG/DTG para o SAPO-11 coqueada na razão de aquecimento β=10 0C min-1 com o fluxo de ar contínuo de 120 Cm3 min-1...75
Figura 47Curvas de TG da amostra de ALPO-11 coqueada nas taxas de aquecimento: a - 5, b - 10 e c – 20 0C min-1...78
Figura 48Curvas de TG da amostra de SAPO-11 coqueada nas taxas de aquecimento: a - 5, b - 10 e c – 20 0C min-1...78
Figura49 Curvas de conversão vs. temperatura para termoxidação do coque em ALPO-11...79
Figura50 Curvas de conversão vs. temperatura para termoxidação do coque em SAPO-11...79
Figura51 Curva de energia de ativação do ALPO-11...80
Figura52 Curva de energia de ativação do SAPO-11...80
Figura53 Curvas de conversão para ALPO-11......81
Sumário
1. Introdução e objetivos
...21.1. Introdução...2
1.2. Objetivos...4
1.2.1. Objetivo geral...4
1.2.2. Objetivos específicos...4
2. Revisão da literatura
.
...62.1. A Indústria do Petróleo...6
2. 1.1. Craqueamento de frações de petróleo...7
2.1.2. Craqueamento Catalítico...8
2.2. Desativação de catalisadores...10
2.2.1. Regeneração dos catalisadores...11
2.3. Peneiras Moleculares...12
2.3.1. Aluminofosfatos - ALPO’s...14
2.3.2. Silicoaluminofosfatos - SAPO’s...15
2.3.3. Síntese de aluminofosfatos e silicoaluminofosfatos...18
2.3.4. Composição da Mistura Reacional...19
2.3.5. Preparação da Mistura Reacional...19
2.4. Caracterização das peneiras moleculares...19
2.4.1. Análise Termogravimétrica...19
3. Materiais e métodos
...283.1. Materiais precursores...28
3.1.1. Síntese hidrotérmica dos Suportes ALPO-11 e SAPO-11...28
3.1.2. Síntese das peneiras moleculares ALPO-11 e SAPO-11...29
3.1.3. Calcinações dos Materiais...32
3.2. Caracterização dos catalisadores...34
3.2.1. Difração de raios – X...34
3.2.2. Microscopia eletrônica de varredura...35
3.2.3. Análise Térmica via TG/DTG...36
3.2.4. Adsorção de Nitrogênio...37
3.2.5. Espectroscopia de absorção na região do infravermelho...38
3.2.6. Estudo das propriedades ácida das amostras...39
3.2.7 Teste catalítico de craqueamento do n-hexano...40
3.2.8. Estudo das Propriedades Catalíticas para a Reação Modelo de Craqueamento do n-hexano...42
3.2.9. Difração de raios-X...42
3.3 Adsorção de nitrogênio...44
3.4. Microscopia eletrônica de varredura...47
3.5. Espectroscopia na região do infravermelho...49
4. Resultados e discussão
...514.1. Caracterização dos catalisadores ...51
4.1.1. Análise Térmica...51
4.1.2. Difração de raios-X...56
4.1.3. Microscopia eletrônica de varredura...59
4.1.4. Determinação da acidez superficial...64
4.2. Reação de Craqueamento do n-Hexano...68
4.2.1. Teste catalítico do craqueamento do n-hexano...68
4.3. Cinética de Regeneração...75
5. Conclusões
...
...836. Sugestões para futuros trabalhos
...
...87Referências
..
...89Capítulo I
1. Introdução e objetivos
1.1. Introdução
Este trabalho teve como finalidade sintetizar os aditivos para catalisadores de FCC (Fluid Catalytic Cracking) à base de peneiras moleculares microporosas do tipo aluminofosfatos e silicoaluminofosfatos, usando o método hidrotermal. Os materiais microporosos citados apresentam uma área superficial e diâmetro de poros de acordo com a literatura melhorando a eficiência do craqueamento industrial e petroquímico.
Os materiais estudados (ALPO´S e SAPO´S), têm sido utilizados em diversas áreas científicas: física, bioquímica, catálise, e em várias outras áreas tecnológicas. Desta forma eles têm despertado grandes interesses por muitos grupos industriais como a PETROBRÁS, no craqueamento catalítico e outras indústrias nas diversas áreas de atuação.
O método hidrotérmico é amplamente utilizado para síntese de materiais cristalinos, especificamente zeólitas e outros contendo silicatos. Este processo de cristalização ocorre a temperaturas baixas no meio aquoso. A maioria das fases cristalinas que se obtêm em condições hidrotérmicas, sob pressão autógena, são metaestáveis. Se o tempo de cristalização não for suficiente para a formação dos cristais, muitas fases cristalinas desaparecem e se formam outras de estabilidade relativa.
1.2. Objetivos
1.2.1. Objetivo geral
O objetivo deste trabalho foi sintetizar e caracterizar peneiras moleculares do tipo ALPO-11 e SAPO-11 e determinar os parâmetros cinéticos na degradação do coque depositado nos catalisadores, utilizando o método de Vyazovkin (“model free
kinetics”).
1.1.2. Objetivos específicos
• Síntese de peneiras moleculares do tipo aluminofosfatos e
silicoaluminofosfatos pelo método hidrotérmico;
• Otimização dos parâmetros de calcinação para remoção do direcionador orgânico dos materiais microporosos através da termogravimetria;
• Caracterização físico-química dos materiais obtidos através dos seguintes métodos: análise térmica (TG/DTG), espectroscopia de absorção no infravermelho, difração de raios-X (DRX) e adsorção de nitrogênio pelo método de BET, microscopia eletrônica de varredura (MEV);
• Avaliação catalítica dos materiais obtidos na reação de craqueamento do n-hexano em micro reator catalítico de leito fixo com fluxo contínuo;
Capítulo II
2. Revisão da Literatura
2.1 A Indústria do Petróleo
A importância do petróleo e do gás natural para a sociedade moderna, como fonte não só de combustíveis, mas também de matérias-primas industriais que são bastante conhecidas, é evidente. O mundo produz e consome hoje, por dia, cerca de 72 milhões de barris de petróleo e cerca de 6,3 bilhões de m3 de gás Natural. No Brasil, a produção chega a 1,1 milhões de barris/dia de petróleo e 32,5 milhões de m3/dia de gás natural. O percurso dessa matéria-prima até o consumidor inclui a localização das jazidas de petróleo ou gás natural no subsolo, à extração, o transporte, o armazenamento, o processamento (refino) e a distribuição por empresas autorizadas.
O conjunto desses processos complexos constitui a indústria do petróleo. Os derivados do petróleo podem ser utilizados em aplicações energéticas ou não-energéticas, ressaltando-se que os derivados energéticos são também chamados combustíveis. Eles geram energia térmica (calor ou luz) por meio do processo de combustão.
Além dos derivados dos combustíveis, existem outros derivados, com aplicações não-energéticas, tais como, nafta e gasóleos petroquímicos; solventes domésticos e industriais; parafinas; lubrificantes básicos; asfalto e coque. Embora conhecido desde tempos antigos, o petróleo era pouco aproveitado até meados do século 19, quando descobertas em maior escala propiciaram seu uso como combustível para iluminação, criando o interesse pela busca de novas jazidas. Porém, a sua grande ascensão se deu com a descoberta do motor de combustão interna, daí a substituição de combustíveis como o carvão, a lenha, a turfa e o alcatrão, bastante empregados na época.
A Figura 1 apresenta a distribuição percentual da produção de derivados do petróleo nas refinarias nacionais.
Figura 1. Produção de derivados de Petróleo no Brasil.
2. 1.1. Craqueamento de frações de petróleo
Atualmente o craqueamento é um dos processos mais utilizados na indústria de refino do petróleo. O objetivo do craqueamento é transformar hidrocarbonetos de alto peso molecular em hidrocarbonetos de baixo peso molecular, de maior valor agregado experimental. Os fragmentos pequenos apresentam alta volatilidade, são usados em grandes escala como combustíveis.
Atualmente, na indústria do petróleo, existem várias configurações para estes processos, dentre eles podemos citar como os mais empregados:
• Craqueamento térmico: é um dos processos mais antigos que existe. Este
processo tem como objetivo a redução da massa molecular da mistura de hidrocarbonetos pela simples aplicação de calor sem sofisticações adicionais como, por exemplo, a adição de hidrogênio. O craqueamento térmico pode assumir três configurações básicas: quebra da viscosidade, craqueamento de correntes de vapor. Esse processo é usado em refinarias para produzir hidrocarbonetos olefínicos de várias cargas de alimentação para a manufatura de petroquímicos. (Heinshaw, 1989).
• Craqueamento catalítico: a quebra das cadeias longas de hidrocarbonetos
ocorre tanto pelo efeito térmico, quanto pela presença de um catalisador, que vai atuar diminuindo a energia de ativação, melhorando a seletividade a determinados produtos durante o processo de craqueamento catalítico. Por sua maior importância, esse processo será abordado a seguir.
2.1.2. Craqueamento Catalítico
As reações de craqueamento de hidrocarbonetos referem-se à ruptura da ligação carbono-carbono, que é termodinamicamente favorecida em altas temperaturas por ser um processo endotérmico, e é definido como reverso da reação de alquilação e de polimerização. As equações apresentadas abaixo, são as reações envolvidas no craqueamento de parafinas, olefinas e hidrocarbonetos aromáticos.
CnH2n + 2 → CmH2m + 2 + CpH2p+ 2
n = m + p
CnH2n →CmH2m + CpH2p
n = m + p
O Craqueamento catalítico, segundo Bond, G. C; (1987) ocorre na presença de catalisadores ácidos e, portanto, é uma reação que apresenta baixa seletividade, o que significa que existe uma série de reações paralelas, tais como:
• Desidrogenação para formação de alcenos
R1CH2 - CH2R2 → R1CH=R2 + H2
• Formação do íon carbônio
R1CH= CHR2 + H+ → R1CH2 - +CHR2
• Migração do grupo metila
CH3 – CH2 - +CHR6 → +CH2 – CH (CH3)R6 → CH3 - C(CH3)R6
• Transferência de prótons
R1CH2 – CHR2 + R3CH(CH3) – CH2R4 → R1CH2 - CH2R2 + R3+C(CH3) - CH2R4
• Cisão – β
R3+C(CH3) - CH2 - CH2R5 → R3C(CH3) = CH2 + CH2R5
2.2. Desativação de catalisadores
O principal problema para a operação de catalisadores heterogêneos na indústria petroquímica é a perda da atividade catalítica com o tempo de operação do catalisador causando desta forma a formação de compostos indesejáveis que leva ao bloqueio dos sítios ativos, levando à desativação. Estas combinações são denominadas de coque (Christoph Kern, Andreas Jess; 2005).
Existem muitos exemplos de reações nas quais os catalisadores perdem sua atividade com o decorrer do tempo de reação por deposição de coque sobre sua superfície, (J. Haber et al., 1995; C. N. Satterfield). Geralmente estes produtos carbonáceos são eliminados por combustão controlada que chamamos de regeneração ou termoxidação do coque.
A desativação ocorre por diversos fatores de natureza química e física, por exemplo: envenenamento, sinterização e mecanismo de bloqueio para a formação do coque (Pio Forzatti, Luaca Lietti 1999):
• O envenenamento é causado por substâncias que reagem com os sítios
catalíticos desativando-os;
• Mecanismo de bloqueio é dado pelo entupimento dos poros;
• Sinterização ocorre em uma menor extensão, pela formação de pontos específicos de alta temperatura, que promove a degradação térmica do catalisador que pode causar modificação na natureza química através distribuição de coque na estrutura porosa do catalisador.
2.2.1. Regeneração dos catalisadores
A deposição de coque sobre o catalisador provoca a sua desativação, ou seja, torna-o incapaz de exercer sua função. Neste caso é necessário aplicar um tratamento para que possa ser reativado. Este processo é denominado de regeneração, sendo necessário fazê-lo freqüentemente.
Um dos fatores mais comuns para que aconteça a desativação é a formação de coque, que desfavorece o funcionamento das unidades de FCC (Fluid Catalytic Cracking). Segundo (R.Von.Ballmoss, et al., 1997), observa-se no craqueamento catalítico, quatro tipos de coque:
• Carbono Conradson, onde ocorre a deposição diretamente dos
constituintes pesados da carga sobre o catalisador (tipicamente menos de 5% do coque total);
• Coque de contaminação, produzido pelos metais (em particular, o Ni) o
qual é depositado sobre o catalisador durante o craqueamento;
• Coque ocluso (occluded coke ou soft coke), constituído pelos
hidrocarbonetos gasosos não retirados no stripper. As melhorias do processo e no catalisador (porosidade) permitiram reduzir consideravelmente este tipo de coque;
• Coque catalítico (majoritário), que se forma no decorrer do
craqueamento catalítico e que deve ser minimizado.
2.3. Peneiras Moleculares
O termo peneira molecular foi introduzido por MacBain em 1932, para definir sólidos microporosos que exibiam propriedades para utilização em separação de substâncias de acordo com sua forma geométrica e seu tamanho. Esses materiais possuem diâmetro de poros bem definido que variam entre 3 e 20Å.
A síntese das peneiras moleculares que teve início na década de 40, com a descoberta dos aluminossilicatos sintéticos (zeólitas) com formas estruturais, gerada pela descoberta de uma nova família de materiais cristalinos e microporosos a base de aluminofosfato. Estes novos materiais foram expostos à comunidade científica em 1982, pelos cientistas da Union Carbide.
As peneiras moleculares possuem uma estrutura cristalina ordenada em três dimensões, a qual proporciona uniformidade nos seus poros. Por esta razão são capazes de selecionar moléculas que possuem forma e tamanho específicos tendo acesso ao interior de seus sistemas de canais microporosos.
Estes materiais além de ter a capacidade de fazer adsorção seletiva, apresentam excelente estabilidade térmica, proporcionando sua utilização em reações que requerem elevadas temperaturas, sem ocorrer nenhuma modificação na sua estrutura cristalina. Essas propriedades associadas mostram a potencialidade dos mesmos para servir como catalisadores ou como suporte para fases ativas.
As classes conhecidas de peneiras moleculares compreendem as zeólitas (Breck, D. W; 1974), aluminossilicatos substituídos por fósforo (Flanigen, E. M et al; 1971; Artioli 1982), sílica polimorfa (Grose, R. W. et al; 1981) os aluminofosfatos (ALPO’s) (Wilson, S.T et al; 1982) e silicoaluminofosfatos (SAPO's) (Hasha, D. et al; 1988; Wang, R. 1991).
Os ALPO’s e SAPO´s podem ser utilizados como suportes ou catalisadores para utilização em várias reações de interesse da indústria petroquímica, por exemplo: isomerização, alquilação e craqueamento.
A atividade catalítica dos SAPO´s para estes processos depende da concentração de silício substituído isomorficamente na estrutura do ALPO. Assim, o estudo sistemático da incorporação de Si no SAPO, com relação aos mecanismos de substituição isomórfica, merece especial atenção em catálise, pois permite uma melhor compreensão das suas propriedades estruturais, cristalográficas e ácidas.
Os silicoaluminofosfatos exibem uma grande diversidade estrutural, sendo que até o momento são conhecidas cerca de dezesseis estruturas tridimensionais para estes materiais. Estas estruturas incluem: SAPO-44, SAPO-41, SAPO-40; estruturas análogas a zeólita chabazita (SAPO-34), levinita (SAPO-35), faujasita (SAPO-37), zeólita do tipo A (SAPO-42) e erionita (SAPO-17). Há estruturas de silicoaluminofosfatos que não são relacionadas a nenhum tipo de zeólitas existentes, tais como: SAPO-31, SAPO-16, SAPO-11 e SAPO-5.
2.3.1. Aluminofosfatos - ALPO’s
A família destas peneiras moleculares apresenta uma grande variedade em sua estrutura, possuindo um volume de poros variando entre 0,04 e 0,35 cm3g-1 , podendo-se destacar quatro grupos de materiais conforme seu diâmetro de poro. Classificam-se como 3 ou 4, estando distribuídos em um intervalo de 0,3 a 1,25 nm. Entre as estruturas que apresentam um diâmetro muito grande, destaca-se a VPI-5 que possui anel de 18 membros e canais unidimensionais com diâmetro de 12 Å (Young, D e Davis, M. E 1991).
O ALPO-11 se enquadra nas estruturas que possuem diâmetros de poros médios com canais unidimensionais e com abertura elíptica de 0,64 x 0,44 nm, formado por anéis de 10 membros. Estas propriedades estão resumidas na Tabela 2.1.
Tabela 2.1 - Tamanho de poros para a família de aluminofosfatos.
Tamanho do poro Átomos “T” Presentes no anel TTIIPPOODDEEEESSTTRRUUTTUURRAA
Muito grande 18 VPI-5
Grande 12 5, 36, 37, 40, 46, 50
Médio 10 11, 31, 41
Pequeno 8 14, 17, 18, 22, 26. 34, 35
Na Figura 3 podem ser vistas peneiras moleculares com estrutura AEL (ALPO-11).
Figura 3. Estrutura de peneira molecular com estrutura AEL.
2.3.2. Silicoaluminofosfatos - SAPO’s
Os silicoaluminofosfatos SAPO’s são obtidos pela incorporação de silício na estrutura dos ALPO’s, tendo ligações do tipo Al-O-P, Al-O-Si e em alguns casos Si-O-Si dando origem à zonas ricas de silício na estrutura (zona chamada de zeolíticas).
Na síntese do SAPO-11, utilizam-se as mesmas condições de preparação de aluminofosfatos gerando novas estruturas, sendo algumas similares às apresentadas por zeólitas naturais e outras que não são encontradas em nenhum dos tipos de sistemas zeólitas e ALPO’s (Yu Fan. et al; 2006)
• Incorporação do silício na posição hipoteticamente ocupada por átomos de
alumínio (Si+4 → Al+3), dando origem uma estrutura com capacidade de troca aniônica (MS1);
• Deposição do átomo de silício em uma posição ocupada hipoteticamente por
um átomo de fósforo (Si+4→ P+5), gerando uma estrutura com capacidade de fazer troca catiônica (MS2);
• Introdução de dois átomos de silício, um na posição do alumínio e outro na
posição hipoteticamente por um átomo de fósforo (2Si+4 → Al+3 + P+5) permanecendo a eletroneutralidade no composto (MS3).
Na Tabela 2.2, estão relacionadas os tipos de ligações observadas em estruturas
baseadas em ALPO’s.
Tabela 2.2 Tipos de ligações em ALPO’s modificados por Si e Metais
Lig. Observada Lig. Não-observada
Al−O−P P−O−P
Si−O−Si P−O−Si
Si−O−Al Al−O−Al
Me−O−P Me−O−Me
Na Tabela 2.3 estão expostas propriedades estruturais de alguns aluminofosfatos.
Tabela 2.3 – Propriedades das peneiras moleculares baseadas em aluminofosfatos.
SAPO’s Estrutura Típica
Diâmetro do poro (nm)
No Tetraedros no anel (nm)
Volume de poro cm3g-1
O2 H2O
5 AlPO-5 0,8 12 0,23 0,31
11 AlPO-11 0,6 10 0,13 0,18
16 AlPO-16 0,3 6 - -
17 Erionita 0,43 8 0,25 0,35
20 Sodalita 0,3 6 0 0,40
31 AlPO-31 0,7 10 0,13 0,21
34 Chabazita 0,43 8 0,32 0,42
35 Levinita 0,43 8 0,26 0,48
37 Faujazita 0,8 12 0,37 0,35
40 Nova 0,7 10 0,31 0,33
41 Nova 0,6 10 0,10 0,22
42 Zeólita-A 0,43 8 - -
2.3.3. Síntese de aluminofosfatos e silicoaluminofosfatos
As peneiras moleculares baseadas em aluminofosfatos e silicoaluminofosfatos são sintetizadas por cristalização hidrotérmica a pressão autógena, a partir de géis reativos de alumínio, fósforo e silício, utilizando um direcionador orgânico, geralmente uma amina ou sal de amônio quaternário, e metais ou elementos adicionais (Me e Si), utilizando uma temperatura entre 1700C com duração de 72 horas. Alguns fatores afetam e controlam a formação da estrutura dos aluminofosfatos:
• Os materiais de partida são de grande importância para o resultado da síntese;
• O uso de sais de alumínio e aluminatos como fonte de alumínio, e
fosfato como fonte de fósforo não são aconselháveis para a síntese desses materiais, pois a presença de cátions além desses, provavelmente acarreta problemas de incorporação de impureza na estrutura cristalina, mudando a característica do material;
• A presença de ânions no meio reacional pode causar a formação de fases densas de aluminofosfatos, como a tridimita ou a cristobalita;
• A fonte mais usada de fósforo é o acido ortofosfórico e para a fonte de
alumínio é a pseudoboemita por não apresentar na estrutura cátions estranhos.
2.3.4. Composição da Mistura Reacional
As composições típicas da mistura reacional para ALPO’s e SAPO’s são:
ALPO’s: 2.9Al + 3.2P + 3.5DIPA + 32.5H20
SAPO’s: 2.9Al + 3.2P + 0.5Si + 3.5DIPA + 32.5 H20
2.3.5. Preparação da Mistura Reacional
Existe uma grande variedade de métodos de preparação da mistura reacional, descritos na literatura (A. K. Sinha et al. 2004). Alguns efeitos observados na ordem de adição dos componentes não estão bem esclarecidos como, por exemplo, a mistura reacional do direcionador com o ácido ortofosfórico antes da adição do alumínio. Como conseqüência ocorrerá uma variação no pH, afetando a velocidade da formação do fosfato de alumínio e formação de um gel menos viscoso.
2.4. Caracterização das peneiras moleculares
2.4.1. Análise Termogravimétrica
Na catálise, as técnicas mais empregadas são: Termogravimetria (TG), derivada da (DTG), e a calorimetria exploratória diferencial (DSC). Na análise termogravimétrica (TG), a variação na massa da amostra em termos de ganho ou perda é registrada continuamente em função da temperatura ou do tempo, onde a temperatura da amostra aumenta normalmente de forma linear com o tempo (Keattch et al., 1975).
O gráfico da massa ou percentual de massa em função da temperatura é denominado curva termogravimétrica. O equipamento utilizado para este experimento é denominado de termobalança e os resultados são mostrados na forma das curvas de TG e DTG.
A partir das curvas de TG pode-se obter um gráfico de variação de massa no eixo da ordenada versus temperatura ou tempo na abscissa. A derivada da curva termogravimétrica (DTG) nos mostra a taxa de variação de massa em função da temperatura ou do tempo. São conhecidos três modelos de termogravimetria:
• Termogravimetria isotérmica: onde a variação da massa é registrada em função do tempo à temperatura constante.
• Termogravimetria quase isotérmica: a variação da massa ocorre em função do
tempo a várias temperaturas constantes.
• Termogravimetria dinâmica: a variação da massa é registrada em função da temperatura. A amostra é aquecida em um meio cujo aumento de temperatura se dá a uma razão de aquecimento pré-determinada.
Para obtenção de uma análise termogravimétrica satisfatória, alguns fatores importantes devem ser considerados no equipamento, dentre os quais estão os seguintes:
• O equipamento deverá registrar a perda ou ganho de massa em função do tempo
e da temperatura;
• O forno da termobalança deverá possuir uma ampla faixa de operação, indo da temperatura ambiente até 1000, 1600 ou 2400 0C;
• A variação na massa da amostra e a temperatura deverão ser registradas com
uma precisão de 0,01%;
• Os defeitos físicos devido ao funcionamento normal do instrumento, como radiação e correntes de convecção e os efeitos magnéticos devido ao aquecimento do forno, não deverão afetar a precisão da balança;
• O posicionamento do cadinho sempre deverá ser o mesmo no interior da
balança para que a temperatura registrada corresponda à temperatura da amostra;
• O forno deverá permitir uma operação em várias atmosferas;
• A termobalança deverá ser o mais versátil quanto possível, para facilitar a mudança na razão de aquecimento simultaneamente com o controle automático da programação de temperatura.
Outra aplicação da termogravimetria nesta pesquisa foi a termoxidação do coque depositado nos materiais oriundos das reações de craqueamento do n-hexano. Foi aplicado o método cinético integral desenvolvido por Vyazovkin e Goryachko (1992) aplicando múltiplas razões de aquecimento que são usadas para se avaliar por TG reações de decomposição simples ou complexas. A taxa de reação neste caso depende da conversão (α), temperatura (T) e tempo (t).
A análise é baseada no princípio da isoconversão, em que é necessário para uma conversão constante, e a taxa de reação é unicamente função da temperatura (Vyazovkin e Lesnikovich, 1988; Vyazovkin e Wright, 1999). Em um experimento típico é necessário se obter no mínimo três razões de aquecimento diferentes (β) e suas respectivas curvas de conversão são avaliadas das curvas de TG.
2.4.2. Cinética de oxidação do coque método de Vyazovkin (“model free kinetics”)
Vyazovkin e Goryachko (1992) desenvolveram um método cinético integral no que múltiplas razões de aquecimento foram utilizadas para avaliar por termogravimetria quais as melhores condições de reações de decomposição do Coque.
Este modelo cinético livre é baseado no método de isoconversão (Vyazovkin e Lesnikovich, 1988; Vyazovkin e Wright, 1999), que calcula a energia de ativação efetiva como função da conversão (α) de uma reação química, ou seja, E = f(α). A conversão (α), temperatura (T) e tempo (t) são fatores que influenciam a velocidade da reação numa transformação química. A velocidade da reação, como função de α, é diferente para cada processo e precisa ser determinada experimentalmente.
somente um único valor de Ea para todo o processo de decomposição, não sendo possível detectar a complexidade das reações. Ao contrário, o método de isoconversão fornece um valor de Ea para cada α , permitindo visualizar o comportamento cinético da reação em todo intervalo de decomposição térmica, sendo sensível, desta forma, a processos que apresentam múltiplas etapas. Na teoria proposta por Vyazovkin nenhum modelo cinético é aplicado.
As curvas TG são obtidas em diferentes razões de aquecimento e a partir delas são obtidos os dados de massa e temperatura em função da taxa de conversão. A idéia de Vyazovkin tem como base as duas suposições a seguir:
• A energia de ativação é constante somente para uma determinada conversão (α).
• A equação de Arrhenius que relaciona a constante cinética com a temperatura é válida, ou seja:
(Equação 01)
onde: d α/dt é a taxa de reação; k0 é constante a uma temperatura infinita; f( α) é uma equação matemática que descreve o mecanismo de reação, E α é energia de ativação como uma função da conversão (α), e R é a constante universal dos gases.
Dividindo a Equação (1) pela taxa razão de aquecimento β = dT/dt, tem-se:
(Equação 02)
(Equação 03)
Integrando a Equação (3) entre os limites α=0 e α= α e os correspondentes de temperatura T=T0 e T=T,
(Equação 04)
Aproximando a integral e dt
T
T RT
E
o
∫
− α obtém-se a seguinte equação:(Equação 05)
Reordenando a eq.(5) e aplicando logaritmo, tem-se:
(Equação 06)
Partindo da equação básica para a cinética não isotérmica, temos: ) ( . ) ( α β α α α f k T kf
t ∂ =
∂ → = ∂ ∂ (Equação 07)
Onde k é a constante de velocidade (s-1) e β é a taxa de aquecimento (oC.s-1). Substituindo k pela equação de Arhenius: k = ko.e-E/RT, ficamos com:
T e k f RT E ∂ = ∂ − . ) ( 1 0 β α
α (Equação 08)
Integrando a Eq. (08), ficamos com:
∫
∫
∂ = = T − ∂T RT E T e k g f 0 0 0 . ) ( ) ( 1 β α α α α (Equação 09)
Desde que E/2RT >>1, a integral da temperatura pode ser aproximada para:
∫
− ∂ = − T T RT E RT E e T E R T e 0 2 (Equação 10)Substituindo a integral da temperatura da Eq. 10 na Eq. 11, arrumando e aplicando o. logaritmo neperiano, ficamos com:
α α α α α β T R E g E K R T 1 ) ( . . ln
ln 2 0 ⎥−
⎦ ⎤ ⎢
⎣ ⎡
= (Equação 11)
Para a aplicação deste modelo, se faz necessária a obtenção das curvas termogravimétricas das amostras de ALPO-11 e SAPO-11 em três diferentes razões de aquecimento (β = 5, 10 e 20 ºC.min-1). A solução destes algorítimos consiste em se determinar para cada conversão valores da inclinação da reta obtida de ln(β/Tα2) versus
3. Materiais e métodos
Este capítulo descreve a metodologia utilizada para a síntese dos catalisadores, o processo de caracterização físico-química, as condições empregadas para a avaliação das propriedades catalíticas dos mesmos e a cinética de remoção do coque
.
3.1. Materiais precursores
Os materiais empregados para a síntese hidrotérmica das peneiras moleculares baseados em aluminofosfatos e silicoaluminofosfatos, estão listados abaixo:
• Como fonte de alumínio, utiliza-se pseudobohemita, AlOOH (Catapal –B,
fornecida pela fábrica carioca de catalisadores S/A);
• Ácido fosfórico (85%, Merck);
• Utiliza-se como direcionador estrutural: Di-isopropilamina – DIPA, (C3H7)2NH
(98% - Riedel);
• Sílica (95% SiO2 e 5% H2O, Merck), como fonte de Si;
• Água destilada, como solvente;
• Todos os materiais utilizados nesta síntese eram de pureza elevada.
3.1.1. Síntese hidrotérmica dos Suportes ALPO-11 e SAPO-11
Figura. 4. Modelo de autoclave utilizado na síntese hidrotérmica do ALPO–11 e SAPO-11
.
3.1.2. Síntese das peneiras moleculares ALPO-11 e SAPO-11
Os materiais foram sintetizados através do método hidrotérmico, usando como fonte de alumina a pseudobohemita (Catapal - B), ácido fosfórico, direcionador estrutural e sílica gel exclusivamente para a síntese de SAPO’s. Estes reagentes foram adicionados em proporções estequiométricas de modo a se obter géis com composição molar:
2.9 Al +3.2 P + 3.5 DIPA +32.5 H20 (ALPO-11);
Agit. 30 min
Agit. 60 min Agit. 60 min Agit. 120 min
Agit Agit. 60 mim
Agit. 60 min. Agit. 60 mim
48 h
48 h
Figura. 5. Fluxograma geral da síntese dos materiais ALPO e SAPO AlOOH + 2/3
H2O
Fosfato de alumino H3PO4 +1/3
H2O
Forma gel viscoso
Adição do
SiO2
Adição do DIPA
Estufa
170 oC
SAPO-11
Autoclavação
Estufa 170 oC
ALPO-11
Tabela .3.1 Composição dos géis de síntese das amostras.
Amostra Composição Molar do gel de síntese
ALPO-11 2.9 Al +3.2 P + 3.5 DIPA +32.5 H20
SAPO-11 2.9 Al +3.2 P + 0.5 Si + 3.5 DIPA +32.5 H20
3.1.3. Calcinações dos Materiais
O processo de síntese dos materiais ocorreu na presença de um direcionador estrutural que fica retido dentro dos poros do material durante todo o processo de cristalização. Após o período de síntese, o direcionador deve ser removido para liberar os espaços intracristalinos.
A calcinação desta série de catalisadores ocorreu em duas etapas onde inicialmente a amostra foi submetida a uma rampa de aquecimento de 10 0C min-1 da temperatura ambiente até 550oC em atmosfera inerte de nitrogênio a uma vazão de 100 mL min-1. Após ter atingido a temperatura de 550oC , o sistema permaneceu nesta condição por 1 hora. Em seguida, o fluxo de nitrogênio foi trocado por ar sintético na vazão de 100 mL min–1 por tempo adicional de 5 horas. Este processo de calcinação visa à remoção do direcionador estrutural dos poros dos catalisadores.
Figura.6. Sistema de calcinação para remoção de DIPA dos materiais ALPO-11 e SAPO-11
3.2. Caracterizações dos catalisadores
3.2.1. Difração de raios – X
A caracterização DRX para materiais sólidos utiliza o método de pó, a partir de radiações eletromagnéticas de comprimento de onda na faixa de 0,1 a 25 Å. Esta técnica é utilizada para determinar a estrutura cristalina, desde que o material em estudo seja sólido e suficientemente cristalino para difratar os raios X e esteja presente em quantidades maiores que 5%. Os difratogramas de raios-X das amostras de ALPO-11 e SAPO-11 foram obtidos em uma varredura de 5 a 400 em um equipamento Shimadzu modelo XRD 6000.
Figura. 8. Equipamento utilizado para difração de raios-x
3.2.2. Microscopia eletrônica de varredura
Análises de microscopia eletrônica de varredura dos suportes mesoporosos ALPO-11 e SAPO-11 foram realizadas com o objetivo de observar-se a morfologia após a síntese hidrotérmica dos materiais, utilizando-se um equipamento Microscópio Eletrônico de Varredura Ambiental (ESEM), Modelo XL 30, Phillips.
Figura. 9. mostra o microscópico eletrônico de varredura (MEV)
3.2.3. Análise Térmica via TG/DTG
As análises termogravimétricas de cada amostra foram obtidas em uma termobalança Mettler Toledo TGA/SDTA 851.
Figura. 10 Termobalança de forno horizontal da Mettler
3.2.4. Adsorção de Nitrogênio
O método de BET foi utilizado para a determinação da área superficial e o volume total de poros, distribuição e o diâmetro médio dos poros, por meio da adsorção de N2 à temperatura do N2 líquido (77K). As isotermas de adsorção das amostras de
ALPO-11 e SAPO-11 calcinadas foram obtidos em um equipamento Nova 1200e
Quantachrome. Para isso, cerca de 100 mg de cada amostra foram previamente
tratadas a 350 oC durante 3h, sob vácuo, para em seguida serem submetidas à adsorção de nitrogênio a 77K. As isotermas de adsorção foram obtidas em uma faixa de p/po de
0,01 a 0,95. Os dados a cerca do volume de gás adsorvido em função da pressão parcial foram correlacionados por modelos matemáticos para a determinação da área superficial (Brunauer et al., 1938), volume e distribuição de poros (Barret et al., 1953).
3.2.5. Espectroscopia de absorção na região do infravermelho
A técnica de espectroscopia na região do infravermelho foi utilizada com o objetivo de identificar qualitativamente as freqüências vibracionais e suas respectivas atribuições referentes aos grupos funcionais inorgânicos presentes nos materiais microporosos do tipo ALPO-11 e SAPO-11 e aos grupos funcionais orgânicos presentes na estrutura do direcionador (DIPA), contido nos poros das amostras na forma não calcinada, além de monitorar a eficiência do processo de calcinação na remoção do direcionador pelo desaparecimento dessas bandas. O princípio de funcionamento desta análise é descrito resumidamente a seguir.
A radiação no infravermelho quando apresenta uma freqüência menor do que aproximadamente 100 cm-1, ao ser absorvida por uma molécula converte-se em energia de rotação molecular. O processo de absorção é quantizado e, em conseqüência o espectro de rotação das moléculas também, consistindo em uma série de linhas. Quando a radiação infravermelha está compreendida na faixa de 1000 a 100 cm-1, converte-se em energia de vibração molecular. O processo também é quantizado e o espectro vibracional aparece como uma série de bandas ao invés de linhas, porque a cada mudança de nível de energia vibracional corresponde uma série de mudanças de níveis de energia rotacional. As linhas se sobrepõem dando lugar às bandas observadas. E são estas bandas de vibração-rotacão, particularmente, as que ocorrem entre 4000 e 400 cm
-1
3.2.6. Estudo das propriedades ácida das amostras
As medidas de acidez das amostras dos catalisadores foram realizadas por meio de adsorção de n-butilamina em fase gasosa, seguido de dessorção por aumento de temperatura, usando a técnica de termogravimetria. Os sólidos ácidos, através dos seus sítios, têm a facilidade de adsorver bases orgânicas quando expostos, em um sistema de adsorção. Com o aumento da temperatura, os sítios liberam a molécula da base, através de um processo de dessorção que inicia a partir dos sítios ácidos mais fracos (ARAUJO, A. S; 1992). As bases mais utilizadas para determinar a força ácida de um sólido que apresenta função catalítica são n-butilamina, amônia e piridina. Devido à facilidade de difusão através dos canais dos materiais sólidos cristalinos microporosos, que constituem o peneiramento molecular, estudos mostram que a amônia e n-butilamina são mais eficientes para atuar como molécula sonda.
Figura 12. Diagrama esquemático do sistema de adsorção de bases utilizado para as medidas de
acidez. Onde: 1 - válvula para ajuste da vazão de N2, 2 e 3 - válvula de três vias, 4 - saturador
contendo n-butilamina, 5 - forno, 6 - controlador de temperatura do forno, 7 - reator contendo a
O procedimento consistiu inicialmente em aquecer da temperatura ambiente até 400ºC as amostras já calcinadas, para a ativação dos sítios ácidos, com um fluxo de N2
de 30 mL/min por 40 minutos. Após este período, a temperatura foi reduzida para 95ºC e os vapores de n-butilamina foram continuamente direcionados para a amostra pelo fluxo de N2 por 1 hora, para uma completa saturação dos sítios ácidos presentes no
material. Em seguida, as amostras saturadas foram purgadas com N2 na mesma
temperatura de saturação, por 30 minutos, para a remoção da base fisicamente adsorvida.
Após este tratamento, foi realizada a termodessorção da n-butilamina em uma termobalança a uma razão de aquecimento de 10ºC/min, da temperatura 30 até 900ºC, sob fluxo de nitrogênio de 25 mL min-1. A acidez total foi calculada em função da quantidade de n-butilamina termodessorvida em cada amostra de acordo com a Equação 12 F
M
M
M
P
A
.
.
.
1000
0=
(Equação 12)Onde:
A = Acidez (mmol g-1)
P = Perda de massa no evento M0 = Massa inicial da amostra (g)
MF = Massa final da amostra (g)
M = Massa molecular da n-butilamina (73 g mol-1)
3.2.7 Teste catalítico de craqueamento do n-hexano
A unidade é constituída por um painel seletor de gases, um saturador, um micro-reator de quartzo em U, um forno ligado a um programador/controlador de temperatura e um sistema de análises cromatográficas. (Figura 13). A unidade é muito versátil, pois permite submeter o catalisador presente no micro-reator a diferentes condições de operação, sem que o mesmo entre em contato com a atmosfera.
Sua vazão é ajustada por válvula micrométrica e medida no Fluxímetro portátil. Válvulas de retenção foram instaladas em todas as linhas, com o objetivo de impedir o retorno dos gases através das linhas que não estejam em operação. O saturador de 100 mL de capacidade é empregado como tanque de armazenamento da solução de n-hexano e foi confeccionado em vidro pyrex. O saturador é encamisado, possibilitando, se necessário, a circulação de uma solução líquida a uma temperatura fixa, utilizando-se, para isso uma bomba peristáltica. A temperatura do saturador é monitorada por um termopar. Controlando a temperatura do líquido circulante, pode-se manter constante a pressão de vapor da solução de n-hexano durante os testes catalíticos.
Figura 13 Os testes de avaliação catalítica foram conduzidos utilizando-se a unidade
experimental mostrada.
O micro-reator utilizado, do tipo U, foi confeccionado em quartzo de 0,64 cm de diâmetro externo e é provido de um bulbo (1,91 cm de diâmetro) onde, por colocação de lã de quartzo, o sólido é suportado. O bulbo possui um poço para colocação de termopar através do qual a temperatura do sistema é monitorada.
Varian CP3800 com detector de condutividade térmica (TCD) acoplada a um computador.
3.2.8. Estudo das Propriedades Catalíticas para a Reação Modelo de
Craqueamento do n-hexano
Os testes catalíticos foram realizados à pressão de 1 atm em um micro-reator catalítico diferencial de leito fixo com fluxo contínuo tipo PFR (Plug Flow Reactor). Em cada reação foram utilizadas cerca de 200 mg de catalisador.
Inicialmente, a amostra foi ativada antes da reação com o fluxo de nitrogênio de 35 ml/min, a 400 0C durante 2 horas. Após a ativação do catalisador, os vapores do n-hexano são arrastados por nitrogênio pela linha de fluxo do saturador até alcançar o micro-reator onde ocorreu a reação. O reator operou a uma razão F/W (fluxo molar de reagente por grama de catalisador) de 0,85 mmol/h.g e temperatura no leito de 400 0C. As vazões volumétricas foram medidas na saída do reator por um medidor digital de fluxo modelo ADM 1000 (Micronal). Os produtos e fluentes do reator, uma mistura de C2 a C4, juntamente com os reagentes não consumido, foram injetados no cromatógrafo a gás modelo Varian CP3800 com detector de condutividade térmica (TCD) acoplado a um computador para identificação de cada substância
3.2.9. Difração de raios-X
O método aplicado para pó é utilizado para a identificação de sólidos e consiste em reduzir a amostra em um pó bem fino, que conterá conter uma boa porção de pequenos cristais, denominados cristalitos, e uma camada delgada deste pó é inserida na trajetória dos raios-X. Cada partícula do pó trata-se de um pequeno cristal orientado eletronicamente em todas as direções possíveis com relação ao feixe incidente. Dessa forma, quando um feixe de raios X atravessa o material analisado, todo o conjunto torna-se susceptível a condição de Bragg para a reflexão de cada possível distância interplanar, que é então obedecida (Skoog, D. A. & Leary, J. J., 1992).
Geralmente, a intensidade dos picos pode ser utilizada satisfatoriamente na medida do percentual de cristalinidade por difração de raios-X, desde que os cristais que estão sendo analisados sejam maiores que 0,3 micrômetros. Abaixo deste valor, ocorre o alargamento dos picos de difração devido ao efeito de dispersão dos pequenos cristais (Szostak, R., 1989). O cálculo do percentual de cristalinidade das amostras em estudo é pela divisão entre a soma das intensidades (altura) dos picos selecionados nas respectivas amostras pela soma das intensidades dos mesmospicos da amostra padrão, a qual é assumida ser 100% cristalina, de acordo (Van hoof, J. H. C. et al., 1991).
% Cristalinidade = Soma das intensidades dos picos (Amostra) *100 Soma das intensidades dos picos (padrão)
(Equação 13)
Limitações da técnica:
• Usada apenas em materiais estruturados, cristalinos ou não. Os materiais
amorfos não reproduzem uma difração;
• A sobreposição de picos pode interferir na análise quantitativa para identificação;
• Materiais fortemente difratados podem encobrir os fracamente difratados, o que chama-se de efeitos de matriz;
• As amostras fluorescentes podem elevar a linha de difração ou pode causar
A figura representa um plano cristalino para a formulação da Lei de Bragg. A equação básica da difração (Santos, 1989) é apresentada como:
nλ=2.d.Sen (θ) (Equação 14)
Onde n é a ordem de reflexão (n = {1, 2, 3,...}), λ é o comprimento de onda, d é a distancia interplanar e θ é o ângulo de incidência entre os planos reticulados.
Figura 14 Esquema representativo para formulação da lei de Bragg.
3.3 Adsorção de nitrogênio
As energias liberadas são relativamente baixas, devido ao calor de adsorção proveniente das interações de Wan der Waals. As propriedades como área superficial, volume e distribuição de poros são baseados neste fenômeno. Segundo a IUPAC, a maior parte dos sólidos segue uma seqüência de seis tipos de isotermas de adsorção. Apenas quatro tipos dessas isotermas (I, II, IV e VI) são freqüentemente encontradas na caracterização de catalisadores (Everett et al., 1988; Roqueirol et al., 1994). A Figura 15 apresenta a classificação das isotermas de adsorção segundo a IUPAC.
Figura 15. Classificação das isotermas de adsorção gás-sólido segundo a IUPAC.
As isotermas de adsorção de nitrogênio para cada material específico, segundo a classificação da IUPAC, podem ser descritas como:
• As isotermas de tipo I são características de sistemas compostos por materiais
microporosos, onde o tamanho do poro não é muito maior que o diâmetro molecular do adsorbato. Ocorre à formação de uma monocamada completa, devido ao limite de saturação definido pelo completo preenchimento dos microporos dos adsorventes que fazem parte destes sistemas;
• A isoterma de tipo II é tipicamente de adsorventes macroporosos ou não
porosos, onde o ponto de inflexão mostra a região em que a monocamada está completa. Desta região inicia-se a adsorção em multicamadas (tendo-se a elevação da pressão relativa);
• As isotermas de tipo III são caracterizadas, principalmente por, calores de
• As isotermas de tipo IV são comuns em materiais mesoporosos. Para este caso ,
ocorre no início a cobertura de uma monocamada. O segundo estágio de adsorção indica uma adsorção na faixa de mesoporosos. A isoterma para este caso apresenta um “loop” de histerese. A isoterma não segue a mesma trajetória para adsorção e dessorção;
• A isoterma de tipo V é quando ocorre pouca interação entre o adsorvente e o adsorbato, como no tipo III. Entretanto, o tipo V é comparado a estruturas porosas que formam o mesmo estágio que nas isotermas do tipo IV;
• A isoterma de tipo VI é comum em materiais ultramicroporosos. A pressão na
qual adsorção depende fundamentalmente da interação entre a superfície e o adsorbato. Quando a adsorção é uniforme, e a pressão é bem definida. Caso a superfície poucos grupos de sítios uniformes, é possível se obter uma isoterma com degraus. Cada etapa na isoterma corresponde a um grupo específico de sítios.
Para os materiais microporosos do tipo aluminofosfatos (ALPO-11) e silicoaluminofosfatos (SAPO-11) as isotermas de adsorção mostram comportamento igual à isoterma do tipo I, ao adsorverem nitrogênio a 77K. Para obtenção da área superficial total de sólidos porosos, emprega-se o método proposto por Brunauer, Emmet e Teller (BET) (Brunauer, 1945; Brunauer et al., 1938). Esse método nos permite determinar a massa de gás a cobrir uma monocamada (Wm) a 77K.
Pode-se alternativamente adsorver outros gases como Argônio, Kriptônio e Hélio para que possa substituir o nitrogênio. Quando se pretende determinar áreas superficiais inferiores a 1 m2.g1, o método BET assume que:
• O calor de adsorção da primeira camada é constante e igual ao calor de
condensação;
• A interação lateral entre as moléculas adsorvida é desprezada;
• As moléculas adsorvidas atuam como novos centros de adsorção para novas
O método proposto por Barret, Joiyner e Halenda (BJH) (Barret et al. 1953) foi empregado para obtenção do volume de poro de cada material. Esta metodologia bastante usada é encontrada com facilidade nos softwares comerciais de tratamentos de dados nos equipamento de isotermas de adsorção obedecendo a ASTM D 4481/87.
Outros métodos que podem ser utilizados para estimar as propriedades superficiais de materiais micro, meso e macroporosos são encontrados na literatura, como por exemplo, t,α-plot para área superficial externa e volume total de poros para sólidos microporosos (DeBoer et al., 1965; Sing et al., 1970), MP para volume e distribuição de diâmetro de microporos (Mikhail et al., 1968), DFT para volume e distribuição de diâmetro microporoso (Seaton et al., 1989), KJS para distribuição de diâmetro de mesoporos (Kruk et al., 1997 a,b).
3.4 Microscopia eletrônica de Varredura
• Microscopia óptica: possibilita a análise de grandes áreas, além de ser de
utilização simples, e pouco dispendiosa;
• Microscopia eletrônica de varredura: permite-nos um grande ajuste no foco, nos auxiliando na análise de superfícies irregulares, dando uma idéia de morfologia e do diâmetro médio de partículas;
• Microscopia eletrônica de transmissão: permite-nos análises dos defeitos e das
fases internas dos materiais, falhas de empilhamentos, discordâncias, metais dispersos em poros de peneiras moleculares mesoporosas;
• Microscopia de campo iônico: permite uma excelente resolução, possibilitam estudos difíceis de serem realizados com outras técnicas, tais como defeitos puntiformes, formas de contornos e de interfaces.
Na microscopia eletrônica de varredura, a superfície sólida de uma amostra é varrida por um feixe de elétrons energéticos. Diversos tipos de sinais são produzidos a partir da superfície neste processo, e cada um deles pode ser coletado e utilizado para modular a nitidez em uma tela de raios catódicos. Em um microscópico eletrônico de varredura, são elétrons secundários que são coletados e utilizados para a formação das imagens, todavia, raios X também podem ser coletados para este fim. O feixe de elétrons tem energia variável, na faixa de 5 a 50 keV, e o diâmetro projetado sobre a amostra, nos instrumentos convencionais, é menor que aproximadamente 10 nm.
3.5 Espectroscopia de absorção na região do infravermelho
A espectroscopia de adsorção na região do infravermelho por transformada de Fourier (FT-IR) é uma das técnicas de caracterização mais comuns existentes. O seu uso permite caracterizar uma larga faixa de compostos inorgânicos e orgânicos. Esta se baseia fundamentalmente na medida de absorção em freqüências de infravermelho por uma amostra posicionada no caminho do feixe de radiação infravermelha. As radiações infravermelhas apresentam comprimentos de onda típicos que variam 0,78 a 1000 µm e números de onda variando de 13000 a 10 cm-1. O número de onda pode ser definido como o recíproco do comprimento de onda (Settle, 1997).
Os espectros de infravermelho são gráficos apresentados sob a forma de número de onda ou comprimento de onda (eixo das abscissas) versus absorbância ou transmitância (eixo das ordenadas). A absorbância e a transmitância estão relacionadas entre si pela Eq. 2.04. Ab=log10(1/Tr)
As principais aplicações para esta técnica são:
• Identificação de todos os tipos de compostos orgânicos e muitos
tipos de compostos inorgânicos;
• Determinação de grupos funcionais em substâncias orgânicas;
• Determinação quantitativa de compostos em misturas;
• Identificação de componentes de reação e estudos cinéticos das
reações.