Linkage analysis and association mapping for plant height in maize
Texto
Imagem
Documentos relacionados
Best estimates of each term in the model, time, T (linear and quadratic), enzyme/substrate (E/S) ratio, R (linear and quadratic) and interaction thereof (linear), and corre-
Com o objetivo de desenvolver as habilidades comunicativas em LI através da negociação de sentidos para atingir a compreensão recíproca, o professor utilizou,
Coexistente a esta compreensão, Galiazzi (2003) articula o educar pela pesquisa com um viés habermasiano que coloca a pesquisa como estruturante da aula; com a abordagem
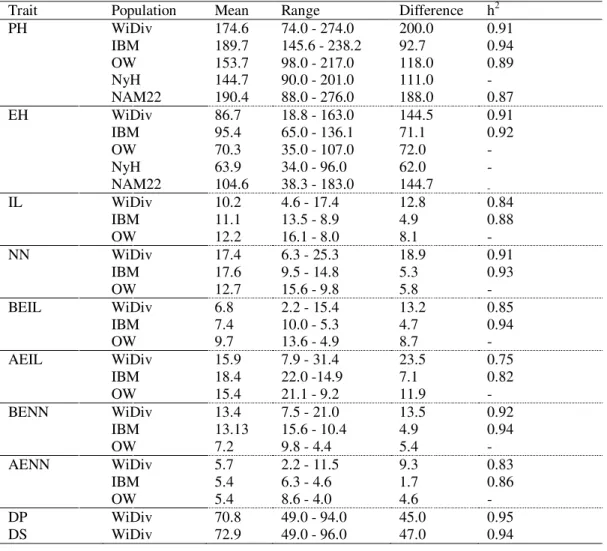
Table 1 - Analysis of variance (MS - mean square) for the traits plant height (PH), ear insertion height (EIH), stem diameter (SD), number of leaves per plant (NL), time to
Based on the experiment on planting densities it can be concluded that gliricidia showed greater plant height than sabiá, but the two species did not differ in canopy diameter,
(BP), plant height (PH, m), ear height (EH, m), and number of days until female flowering (FF) in white grain maize populations and hybrids with high quality protein, according to
stages at PGR application – early tillering phase and panicle primordium differentiation phase – on gas exchange rates, plant height, yield components, and
The development of earlier hybrids, with shorter plant height, lower leaf number, upright leaves, smaller tassels and more synchronized floral development improved maize ability
