de nanoos e aglomerados
me-tálios enapsulados por
nanotu-bos de arbono.
EduardoMoraes Diniz
metálios enapsulados por nanotubos de arbono.
Eduardo Moraes Diniz
Orientador: Prof. Dr. MárioSérgiodeCarvalhoMazzoni
Tese de Doutoradoapresentada à
UNIVERSIDADE FEDERALDE MINAS GERAIS
omo requisito parial para aobtenção do títulode
DOUTOR EM FÍSICA.
porque já reebi muita ajuda, eo quefaço hoje éapenas retribuir om osmesmos gestos.
Para as pessoas a quem não posso ajudar, seja por estarem longe ou por não ter
o-nheimento suiente para resolver seus problemas, meu gesto de gratidão se traduz em
palavras: Muito obrigado.
Espeiamentepara esta tese, gostariade dirigirmeus sineros agradeimentosa:
Deus, por me oneder saúde e força para seguir em frente. Que Sua presença possa ser
sentida por todos.
Meu orientador,MárioMazzoni,portodooaprendizadoquetive,suas horasdedediação,
sua determinação, perseverança, extrema paiênia e, prinipalmente, por não desistir de
mim. Que sua reompensa venha notempo quevoê mais preisar.
Meus pais, em espeial minha mãe, Maria da Graça, que me apoiou de todas as formas
possíveis. Que um diaeu possa retribuí-laà alturade sua grandiosidade.
Minha esposa Selma, por me ensinar que resemos não quando nossos planos dão erto,
mas quando onsertamososque deramerrado. Que nosso amorse fortaleçatodos osdias.
Orestantede minha família,emespeial minha avó,Terezinha, que sem seu apoioeu não
estaria onde estou hoje. Que todoo suor derramado por elasirvade exemplo para todos.
Osprofessores Chaham, Riardoe Simone,espeialmenteaoChaham eRiardo porseu
tempo e dediação nas disussões sobre o artigo. Que este trabalho seja o primeiro de
muitos outros.
Todososprofessores dodepartamento omquem tive oprazerde ter aulas. Que os
onhe-imentos adquiridospor mimsejamlevados às próximasgerações.
O pessoal da seretaria, em espeial Marlue e Iêda, e ao pessoal da bibliotea de
pós-graduação, em espeial Clarie e Shirley, pela presteza e atenção dediadas a mim. Que
seu exemplo de ompetêniaseja seguido portodos.
Todoo pessoal dolaboratóriode estrutura eletrnia pelaamizade eompanheirismo
du-ranteessa jornada: Alexandre,Ananias,André,Angélia,Frederio,Ingrid, Juliana,Joie,
Jonathan, Kagimura, Lídia, Moisés, Daniel, Matheus, Maurisan, Maros, Regiane,
Ro-naldo, Sabrina e Viviane. Que seus nomes, rostos e sorrisos estejam sempre em minha
memória.
Dos itados aima, gostaria de agradeer em espeial a duas pessoas que me ajudaram
muito: Matheus e Moisés. Ao Matheus por me ajudarem vários pontos de meu trabalho,
Linux, fazendoom queeu gostassedesse sistema. Saiba queoonheimento quevoê me
passoufoiampliadoeestoupassandoadiante. Aindaparaessesdois,gostariadeagradeer
por sua amizade aolhedorae sinera, já que ariqueza de um homem se mede pela
quali-dade das amizades. Que nossas amizades sejam sempre maioresque asdistânias que nos
separamou otempo quenão nos vemos.
AoCNPqpelaonançaem meederabolsa. Queeu possa ontribuirparaoresimento
Frase dita pelo Gen. Maximusno lmeGladiator (Gladiador)
Neste trabalho estudamos os efeitos de onnamento de nanoestruturas metálias em
nanotubosdearbono. Ainvestigaçãodas propriedadesestruturais,eletrniase
magnéti-asforamfeitasatravésdeálulosdeprimeirosprinípiosbaseadosnaTeoriadoFunional
daDensidade. As nanoestruturas enapsuladaspuderamser divididasemduas ategorias:
os metálios (formados por níquel ou ferro) e aglomerados (formados por prata ou
o-bre). No estudo do enapsulamento de os também foram investigadas as propriedades
do sistema sob uma deformação que onsistia no ahatamento da seção transversal do
nanotubo. Nossos álulos revelaram a existênia de estruturas metaestáveis nas quais
o nanotubo enontra-se ahatado. As interações entre os átomos metálios e os átomos
de arbono foramutilizadas para a justiativa dessa observação. Alémdisso, foi
enon-trada uma transição para estados de momentomagnétio nulo om o ahatamento. Para
os aglomerados, foi onstatado o desenvolvimento de magnetismo nos átomos de obre e
prata sob o efeitodo onnamento. A expliaçãopara ofenmeno foi dada emtermos de
In this work westudiedthe eets of onnementof metallinanostrutures inarbon
nanotubes. The investigation of the strutural, eletroni and magneti properties were
performed by means of rst-priniplesalulations based on the Density Funtional
The-ory. The enapsulated nanostrutures ould be divided in two ategories: metalli wires
(made of nikel or iron) and lusters (made of silver or opper). In the study of the wire
enapsulation, the properties of the system under a deformation onsisting in the
atte-ning of the nanotube's ross setion were also investigated. Our alulations showed the
existene of metastable strutures in whih the nanotube was found attened. The
inte-rationsbetween metalliand arbonatomswere usedtojustify thisobservation. Besides,
it was found a transition to states with a null magneti moment upon attening. As for
the lusters, itwasobserved the developmentof magnetismintheopperandsilveratoms
under the eets of onnement. The explanation of this phenomenon wasgiven interms
1 Introdução 1
2 Metodologia 5
2.1 Osistema de unidades atmio . . . 5
2.2 Aproximação de Born-Oppenheimer . . . 7
2.3 A densidade eletrnia . . . 12
2.4 A Teoria doFunionalda Densidade . . . 14
2.4.1 Osteoremas daDFT . . . 15
2.4.2 As equações básias daDFT . . . 18
2.4.3 Sobre ofunional de troa-orrelação . . . 25
2.5 Oteorema de Hellmann-Feynman . . . 28
2.5.1 Oteorema de Hellmann-Feynmanna DFT . . . 29
2.6 Amostragem de pontos noespaço reíproo . . . 31
2.7 Pseudopoteniais . . . 34
2.7.1 Pseudopoteniaisde normaonservada . . . 38
2.7.2 Pseudopoteniaisnão loais . . . 40
2.8 OSIESTA . . . 41
2.8.1 As bases de orbitais atmios . . . 42
2.8.2 Ohamiltoniano eletrnio doSIESTA. . . 46
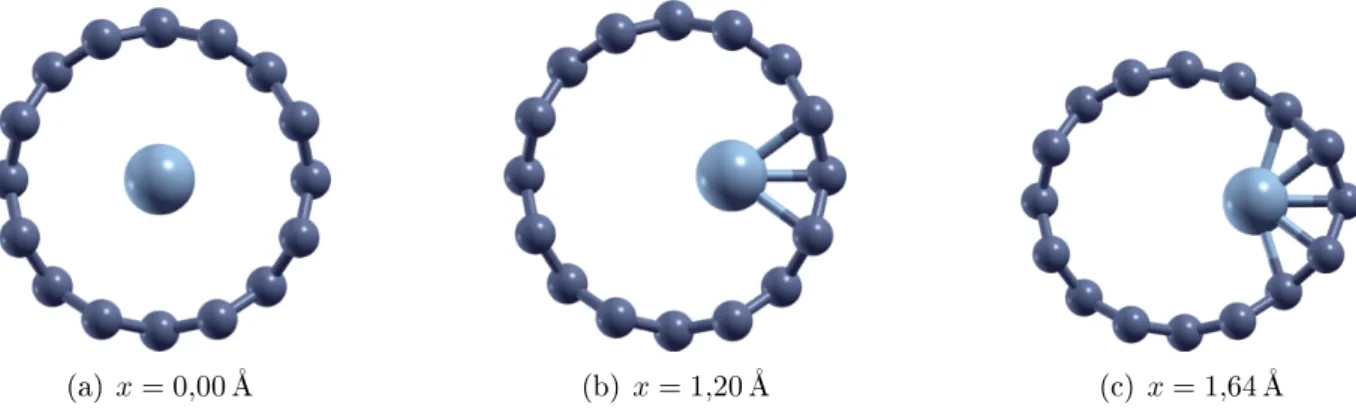
3 Nanoos enapsulados por nanotubos de arbono 48 3.1 Oenapsulamento de nanoos de níquel . . . 48
3.1.1 Introdução . . . 48
3.1.2 Metodologiaapliada . . . 50
3.1.3 Primeiromodelo: a adeiamonoatmia . . . 52
3.1.4 Osefeitos doahatamento . . . 57
3.2 Ummodelo mais realista para o enapsulamento . . . 66
3.2.1 Introdução . . . 66
3.2.2 Metodologiaapliada . . . 67
3.2.3 Resultados para o sistemaperfeito eahatado . . . 67
3.3 Oaso de nanotubosde diâmetros maiores . . . 73
3.3.1 Introdução . . . 73
3.3.4 Osefeitos doahatamento . . . 79
3.4 Oaso dos nanotubosde parede dupla . . . 86
3.4.1 Introdução . . . 86
3.4.2 Metodologiaapliada . . . 88
3.4.3 Resultados para o sistemaperfeito eahatado . . . 89
3.5 Oenapsulamento de nanoos de ferro . . . 93
3.5.1 Introdução . . . 93
3.5.2 Metodologiaapliada . . . 93
3.5.3 Resultados para o sistemaperfeito eahatado . . . 94
4 Aglomerados enapsulados por nanotubos de arbono 97 4.1 Introdução . . . 97
4.2 Modelos para os aglomeradosde obre e prata . . . 98
4.3 Metodologiaapliada . . . 100
4.4 Oaso do obre . . . 101
4.4.1 Propriedades estruturais . . . 101
4.4.2 Propriedades eletrniase magnétias . . . 101
4.5 Oaso da prata . . . 105
4.5.1 Propriedades estruturais . . . 105
4.5.2 Propriedades eletrniase magnétias . . . 106
5 Conlusões 111
2.1 Erros típios dos álulos de DFT para átomos, moléulas e sólidos
utili-zandoas aproximaçõesLDA eGGA. . . 28
3.1 Diferençasentreasenergiasdas geometriasparaoodeNienapsuladopor
umnanotubo(8,0)perfeito. Aposição
x
indiaoquantoooestádesloado doentrodo tubo. . . 523.2 Valores daarga transferida por élulaunitária e as populações eletrnias
dos orbitais
3d
e4s
dosistema Ni2
CNT(8,0). . . 54 3.3 Valores do momento magnétio total por élula unitária e as ontribuiçõesdevido aos orbitais
3d
e4s
.. . . 56 3.4 Valores daarga transferida por élulaunitária e as populações eletrniasdos orbitais
3d
e4s
dosistema Ni8
CNT(11,0). . . 76 3.5 Valores do momento magnétio total por élula unitária e as ontribuiçõesdevido aos orbitais
3d
e4s
para o sistemaNi8
CNT(11,0). . . 78 3.6 Valores daarga transferida por élulaunitária e as populações eletrniasdos orbitais
3d
e4s
dosistema Ni8
CNT(11,0)CNT(20,0). . . 91 3.7 Valores daarga transferida por élulaunitária e as populações eletrniasdos orbitais
3d
e4s
dosistema Fe8
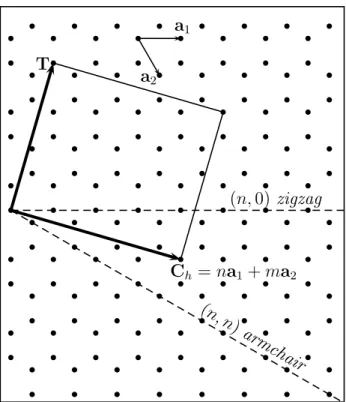
CNT(11,0). . . 95 3.8 Valores do momento magnétio total por élula unitária e as ontribuições1.1 Modelogeométrioparaaonstruçãodeumnanotuboapartirdeumafolha
de grafeno. Na gura, os pontos representam átomos de arbono. O vetor
C
h
é o vetor quiral,T
o vetor que dá a direção do eixo do nanotubo e osvetores de rede
a
1
ea
2
estão destaados. . . 2 1.2 Exemplos de nanotubos. . . 32.1 Cilo de autoonsistêniapara a aproximaçãode Born-Oppenheimer. . . . 11
2.2 Número de artigos publiados porano utilizando-se aDFT.. . . 15
2.3 Representação esquemátia doteoremade Hohenberg-Kohn. As setas
indi-amoaminhodasoluçãodaequaçãodeShrödinger. AindiadaporHK,
refere-se aoteoremade Hohenberg-Kohn. . . 17
2.4 Diagramaresumindo as prinipaisimpliaçõesdos teoremas de
Hohenberg-KohneométododeKohn-Sham. AssetasindexadasporHKeHK
s
referem-se aos teoremas de Hohenberg-Kohn apliados aos sistemas real e tíio,
respetivamente. A seta indexada por KS india o ansatz de Kohn-Sham
emonetar o sistemareal omotíioatravésdadensidade eletrniado
estadofundamental. . . 23
2.5 Cilo de autoonsistêniada DFT. . . 24
2.6 Distribuiçãode
4
×
4
pontosk
nas zonasde Brillouindeuma redequadrada (esquerda) ehexagonal (direita). Oontornodas zonas para ada redeestádestaado. Os írulos pretos representam os pontos da rede reíproa, as
ruzes representam ospontos geradospelo esquema de Monkhorst-Pak.. . 33
2.7 Comparaçãoentre o pseudo-orbital
ψ
ps
eos orbitaisAEψ
ae
e entre o pseu-dopotenialV
ps
e opotenial oulombiano. . . 40 3.1 Imagensde mirosopiade transmissão eletrniade uma adeiade átomosde platina e uma adeia de átomos de molibdênio enapsuladas por um
nanotubo. . . 49
3.2 Formas da seção reta transversal para o nanotubo perfeito (esquerda) e o
modelo adotadopara simular o ahatamento radial donanotubo (direita).. 51
3.3 Estruturasobtidasparaoolineardeníquel enapsuladoporumnanotubo
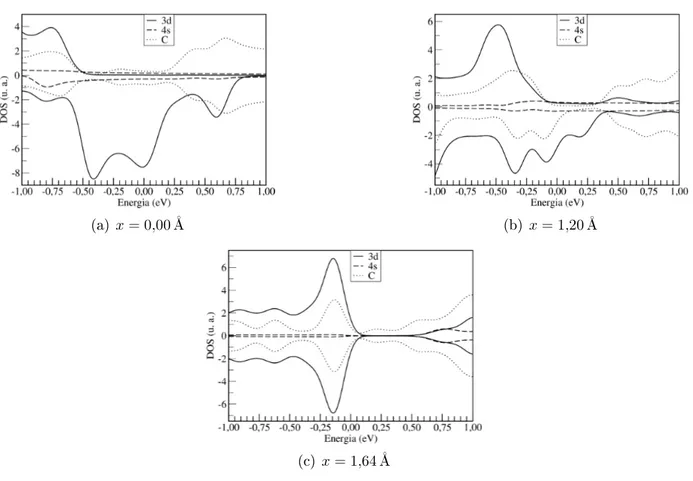
3.4 DOS projetadas nos orbitais
3d
e4s
e nos elétrons dos átomos de arbono para osistema Ni2
CNT(8,0). Osvalores da DOS positivos referem-seaoselétronsom spinup, enquanto queos valores daDOSnegativosreferem-se
aos elétrons om spindown. A energia de Fermi é denida omo o zero da
esala de energia. . . 56
3.5 Estruturasrelaxadasparaoolinearenapsuladoonentriamenteporum
nanotuboom vários níveis de ahatamento. . . 58
3.6 Energiaemfunçãode
η
paraoounidimensionalde níquelenapsuladopor um nanotubo. . . 593.7 Cargatransferida doo linearpara o nanotubo. . . 62
3.8 Populaçãoeletrnia dasestruturasmais estáveisenontradasparatodosos
níveis de ahatamentoinvestigados. . . 63
3.9 DOSprojetadanos orbitaisde valênia doníquel enos elétrons doarbono
para aestrutura autoahatadade menorenergia.. . . 64
3.10 Momentosmagnétiosemfunçãode
η
paraoolineardeníquelenapsulado porum nanotubo.. . . 653.11 Modelo do ode 8 átomosde níquel. . . 67
3.12 Estruturas relaxadas para Ni
8
CNT(9,0) (a) sem ahatamento e (b) omη
= 0,22
de ahatamento.. . . 68 3.13 Energia para Ni8
CNT(9,0) em função do ahatamento. O gráo menoromparaasinlinaçõesdasenergiasparaonanotubopreenhidoevazioom
oahatamento. . . 69
3.14 Cargatransferida doo de Ni para o nanotubo (9,0) emfunção do
ahata-mento. . . 70
3.15 População eletrnia dos orbitais
3d
e4s
para o sistemaNi8
CNT(9,0) em função doahatamento. . . 713.16 Dependênia dos momentos magnétios om o ahatamento para o sistema
Ni
8
CNT(9,0). . . 723.17 Estruturas relaxadas para osistema Ni
8
CNT(11,0). . . 753.18 DOSprojetadanosorbitais
3d
,4s
enoselétronsdosarbonosparaosistema Ni8
CNT(11,0). Os valores da DOS positivos referem-se aos elétrons omspin up, enquanto que os valores da DOS negativos referem-seaos elétrons
om spindown. . . 77
3.19 Estrutura ahatada relaxada para o o de 8 átomos de níquel enapsulado
um nanotubo. . . 79
3.20 Energiapara Ni
8
CNT(11,0) em função doahatamento. . . 803.21 Comparação da variação da energia em função do ahatamento para o
na-notubo (11,0)vazio epreenhido om oo de oitoátomos de níquel. . . 81
3.22 Carga transferida do o de Ni para o nanotubo (11,0) em função do
aha-tamento. . . 82
3.24 DOSprojetadanosorbitais
3d
,4s
enoselétronsdosarbonosparaosistema Ni8
CNT(11,0) autoahatado. . . 853.25 Dependênia dos momentos magnétios om o ahatamento para o sistema
Ni
8
CNT(11,0).. . . 863.26 Formas da seção reta transversal para o nanotubo de parede dupla
per-feito (esquerda) e o modelo adotado para simular o ahatamento radial do
nanotubo(direita). . . 88
3.27 Geometrias para o sistema Ni
8
CNT(11,0)CNT(20,0) om os nanotubosperfeitos e ambosahatados. . . 90
3.28 Estruturas relaxadas para Fe
8
CNT(11,0). . . 944.1 Alguns exemplos de aglomerados enapsulados por nanotubos de arbono
observados experimentalmente. . . 98
4.2 Geometrias de menor energia para aglomerados de oito átomos de obre e
prata. . . 99
4.3 Geometriasiniiais (não relaxadas)para ossistemas Cu
8
CNT(6,6)(supe-rior) eAg
8
CNT(6,6) (inferior).. . . 1004.4 Geometrias relaxadaspara o sistemaCu
8
CNT(6,6). . . 1024.5 Isosuperfíies de spin para osistema Cu
8
CNT(6,6). Ospin up estárepre-sentado pelaor vermelha eo spin down pela azul. . . 102
4.6 Estrutura de faixas para o sistema Cu
8
CNT(6,6). Na gura, os símbolosN
representam ospin up, enquanto que osH
, o down. . . 103 4.7 DOS projetada para o sistema Cu8
CNT(6,6). Na gura, os estados dosspinsup estãonapartepositivadaDOS,enquantoqueosestadosdos spins
down estão na regiãonegativa. Onívelde Fermi foi oloadoem 0eV. . . . 104
4.8 DOS projetada sobre os orbitais
s
ed
do átomos de obre para o sistema Cu8
CNT(6,6). Na gura, osestados dos spins up estão na parte positivadaDOS, enquanto queos estados dos spins down estão naregião negativa.
Onívelde Fermi foioloado em0eV. . . 105
4.9 Geometrias relaxadaspara o sistemaAg
8
CNT(6,6). . . 1064.10 Isosuperfíies de spin para o sistemaAg
8
CNT(6,6). Ospin up estárepre-sentado pelaor vermelha eo spin down pela azul. . . 107
4.11 Estrutura de faixas para o sistema Ag
8
CNT(6,6). Na gura, os símbolosN
representam ospin up, enquanto que osH
, o down. . . 108 4.12 DOS projetada para o sistema Ag8
CNT(6,6). Na gura, os estados dosspinsup estãonapartepositivadaDOS,enquantoqueosestadosdos spins
down estão na regiãonegativa.. . . 109
4.13 DOS projetada sobre os orbitais
s
ed
do átomos de prata para o sistema Ag8
CNT(6,6). Na gura, os estados dos spins up estão na parte positivaIntrodução
O mundo assim paree tão pequeno
Kid Abelha - Eu sópenso emvoê
Ummaronoestudodesistemasnaesalananométria(1nm=10
−
9
m)foiadesoberta
dofulerenoC
60
porKroto eolaboradores [1℄emmeadosdadéadade 1980. Desde então,graçasaavançosexperimentaissigniativos,foipossíveliradavezmaisfundonamatéria.
Issoabriuasportasparaumnovoramodapesquisaientía,onheidoomonanoiênia.
Dentretodososfrutos geradosporessananoiênia,osnanotubosdearbono(CNTs)
sãoomertezaum dosmaisimportantes. OsprimeirosCNTsforamobservadosporIijima
em 1991 [2℄, sendo os mesmos de paredes múltiplas (MWCNT). A síntese de CNTs de
parede simples (SWCNT) só foi possível em 1993 [3; 4℄. Desde então, muitas pesquisas
têm sido feitas nesta área não apenas emCNTs, mas em nanotubos formados por outros
elementos, omonitretode boro, arsenetode gálio, ouro,dióxidode titânioet [59℄. Para
araterizargeneriamenteumCNT, bastaidentiardoisíndies
n
em
,onheidos omo índies quirais [10℄, que são o número de vezes que os vetores de rede do grafeno sãorepetidosdendo omoafolhaseráenroladaparaformaroCNT.A onstruçãogeométria
de um CNT é mostrada na gura 1.1, onde temos os vetores
C
h
, uja denição está nab
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
C
h
=
n
a
1
+
m
a
2
T
(n,
0)
zigzag(
n, n
)
armhair
a
1
a
2
Figura 1.1: Modelo geométrio para a onstrução de um nanotubo a partir de uma folha
de grafeno. Na gura, os pontos representam átomos de arbono. O vetor
C
h
é o vetor quiral,T
o vetor que dá adireção do eixodo nanotubo eos vetores de redea
1
ea
2
estão destaados.Quando um CNT tem o segundo índie nulo, ou seja,
(n,
0)
, ele édito zigzag; quando os índies são iguais, istoé,(n, n)
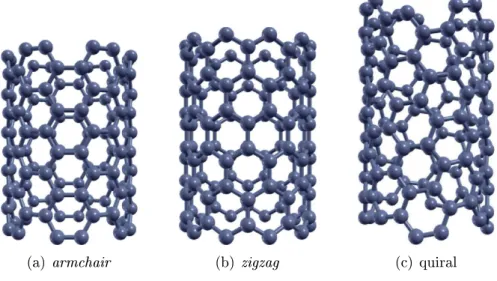
, eleé dito armhair;quando os índies são diferentes e não-nulos, eleé dito quiral. A razão dessa nomenlaturaé emvirtude da forma dabordadotubo. Exemplosdesses três tipospodem ser vistos nagura 1.2.
OsCNTspodemsermetáliosousemiondutores,dependendoapenasdesuageometria.
A regra para a determinação doaráter metáliode um CNT éa seguinte: se a diferença
entreosíndiesforigualazeroouummúltiplodetrês,otuboserámetálio,asoontrário,
será semiondutor. Isso implia em armar que todos os tubos do tipo armhair são
metálios,assim omo todos os dotipo zigzag formados poríndies múltiplos de 3.
Outra linha de pesquisa bastante ativa atualmente é aera de nanoestruturas
Figura1.2: Exemplos de nanotubos.
de átomos. Para o primeiro aso, o interesse prinipal do ponto de vista experimental
é a síntese estável desses os e sua araterização, enquanto que no âmbito teório, o
foo é sobre as novas propriedades apresentadas por eles, que são o mais próximo que se
pode hegar de um sistema rigorosamente unidimensional. O segundo tipo de estrutura
(aglomerados om pouos átomos) vema ser interessante devido ao fato de apresentarem
propriedadesintermediáriasentre moléulasesólidos. Emespeial,osaglomerados
forma-das pelos metais nobres (ouro, prata e obre) despertam uma grande uriosidade devido
ao fato desses elementos terem uma distribuição eletrnia singular, já que possuem uma
amada
d
ompletamente preenhida (3d
para o obre,4d
para a prata e5d
para oouro) e um elétrons
desemparelhado na última amada. Outro fato muito interessante sobre esses metaisé omagnetismo apresentado poreles, já quepara aglomeradosom pequenasquantidades de átomos, estudos teórios reentes revelaram que esses metais apresentam
um momento magnétio não-nulo[1113℄, difereniando-se de suas araterístias quanto
noestado sólido.
Para estudar essas linhas de pesquisa bastante rias, preisamos de uma ferramenta
quântio, dependendo do tamanho do sistema e/ou a quantidade de átomos envolvidos,
uma ou outra teoria pode ser utilizada. A primeira abordagem é feita utilizando-se um
hamiltoniano-modelo para se desrever o sistema. Neste hamiltoniano, o potenial real é
substituído por um onjunto de poteniais parametrizados que visam reproduzir
resulta-dos experimentais através do ajuste desses parâmetros. O segundo método parte de um
hamiltonianoreal quântio, onde não háaneessidade de se fazerajustes de nenhum tipo
a m de se reproduzir resultados experimentais. Por não ser neessário o onheimento
prévio (experimental) do sistema, esse proedimento também é hamado de ab-initio ou
primeiros prinípios. Aqui será tratado apenas o método quântio, sendo que os
álu-los de primeiros prinípios foram realizados por meio do ódigo SIESTA [14℄, que é um
programa de aesso livreamplamenteutilizado por vários gruposde pesquisa ao redor do
mundo. Maisdetalhesaeradométododeprimeirosprinípiosabordadonestatesee
tam-bémsobre oSIESTA estão presentes nosegundo apítulo,onde onstarãoasmetodologias
empregadas nos estudos propostos.
No apítulo de resultados serão mostradas as prinipais observações aera das
mu-dançasnas propriedadesestruturais,eletrnias emagnétias de nanoestruturas formadas
por metais de transição enapsuladas por CNTs. As nanoestruturas envoltas pelos CNTs
foram: os lineares de níquel ouferro (om apliação ounão de uma pressãoradial sobre
oCNT) eaglomeradosde prataou obre. Apósosresultados, serão apresentadas as
prin-ipais onlusões sobre os trabalhos expostos e em seguida as perspetivas de pesquisas
Metodologia
Pois se foi permitido ao homem tantas oisas onheer,
é melhor que todos saibam o que pode aonteer
Cássia Eller(Gilberto Gil)- Queremos saber
Neste apítuloserãoapresentados os métodos utilizadosneste trabalho. As três seções
seguintes tratamdeonheimentospreliminares. NaquartaseçãofalaremossobreaTeoria
doFunionalda Densidade,queé ametodologiaapliadapara os álulospresentes nesta
tese. A quinta, sexta e sétima seções tratam sobre assuntos independentes da Teoria do
FunionaldaDensidade,mas seu onteúdonormalmenteaompanhaesse assuntoeos
mé-todos apresentados nessas seções são frequentemente utilizadosjuntoom um formalismo
de primeiros prinípios. A última seção é dediada ao programa que foi utilizado, onde
apresentaremosalgumas araterístiasdo mesmo.
2.1 O sistema de unidades atmio
Em Físia da matéria ondensada, os átomos têm uma posição de destaque, já que
são osformadoresdos objetosde estudostantonoâmbito experimentalquantonoteório.
quântio do sistema. Para isso, vamos supor que nosso sistema seja onstituído de
M
núleos (indexados porletras maiúsulas) eN
elétrons(indexados porletras minúsulas), todos interagindo entre si. Considerando que não haja poteniaisexternose queosefeitosrelativístios são desprezíveis, todos os possíveis termos do hamiltoniano estão listadas a
seguir:
•
Energia inétiados núleos→
T
ˆ
n
=
X
I
ˆ
P
2
I
2M
I
•
Energia inétiados elétrons→
T
ˆ
e
=
X
i
ˆ
P
2
i
2m
•
Interação núleo-núleo→
V
ˆ
nn
=
1
2
X
I
6
=J
e
2
4πǫ
0
Z
I
Z
J
|
R
I
−
R
J
|
•
Interação núleo-elétron→
V
ˆ
ne
=
−
X
i,I
e
2
4πǫ
0
Z
I
|
r
i
−
R
I
|
•
Interação elétron-elétron→
V
ˆ
ee
=
1
2
X
i
6
=j
e
2
4πǫ
0
1
|
r
i
−
r
j
|
,ouseja, podemosesrever ohamiltoniano totaldo sistemaomo sendo:
ˆ
H
tot
= ˆ
T
n
+ ˆ
T
e
+ ˆ
V
ee
+ ˆ
V
ne
+ ˆ
V
nn
.
(2.1)Todos os termos do hamiltoniano estão repletos de onstantes que podemos eliminar
adotando o sistema de unidades atmias, que será utilizado daqui em diante. Nesse
sistema,asunidadessãoesaladasapartirdeonstantesfísiasfundamentais. Porexemplo:
massas são dadas em unidades da massa do elétron (
m
), omprimentos em unidades do raio de Bohr (a
0
= 4πǫ
0
~
2
/me
2
), energias em unidades da energia de Hartree (
E
H
=
me
4
/4πǫ
0
~
2
,que orresponde ametade daenergiade ionizaçãodoestado fundamentaldoque
a
0
=
e
=
~
=
m
= 4πǫ
0
= 1
emtodas as equações. Adotando-o, o hamiltonianopassa a ser esrito omo:ˆ
H
tot
=
−
X
I
1
2M
I
∇
2
I
−
1
2
X
i
∇
2
i
+
1
2
X
i
6
=j
1
|
r
i
−
r
j
|
−
X
i,I
Z
I
|
r
i
−
R
I
|
+
1
2
X
I
6
=J
Z
I
Z
J
|
R
I
−
R
J
|
.
(2.2)Adotar um sistema de unidades apenas ajuda a enxugar as equações, não tendo
im-pliação nenhuma na resolução do mesmo. Como o problema é muito omplexo, já que
envolve
4(M
+
N
)
variáveis (ada núleo ou elétron tem três variáveis posiionais e uma de spin), poderíamos busar uma maneira de reduzir a quantidade de variáveis a seremaluladas. Tal aproximação hama-se aproximação de Born-Oppenheimer e será o tema
dapróxima seção.
2.2 Aproximação de Born-Oppenheimer
A aproximação de Born-Oppenheimer (BO) [15℄ é oloada tradiionalmante omo
sendo a anulação de
h
T
ˆ
n
i
eh
V
ˆ
nn
i
se tornar onstante devido ao argumento das massas nuleares serem de três a ino ordens de grandeza maiores que a massa eletrnia. Issoquer dizerqueadistribuiçãoespaialdos elétronsseadequaquase queinstantaneamentea
qualquermudançanadistribuiçãoespaialnulear,permitindoonsiderarosnúleos omo
sendo xos.
Esse argumento intuitivo para a aproximação de BO é bastante razoável, mas
rigoro-samente não é nisso emque elase baseia. A dedução matemátia daaproximação de BO
segue nas próximaslinhas [16℄.
Vamos esrevera equação de Shrödinger para ohamiltoniano desritoem (2.1):
ˆ
onde
r
= (
r
1
,
r
2
, . . . ,
r
N
)
eR
= (
R
1
,
R
2
, . . . ,
R
M
)
representam o onjuto de oordenadas eletrnias e nuleares, respetivamente1
. Essa onvenção será usada apenas nesta seção.
Considerando as massas nuleares muito maiores que a massa eletrnia, vamos
des-prezara enegiainétianulear,ouseja, quando
M
→ ∞
entãoh
T
ˆ
n
i
=
T
n
→
0
,o quenos permite denir ohamiltonianode núleos xosH
ˆ
f ix
daseguintemaneira:ˆ
H
f ix
= ˆ
T
e
+ ˆ
V
ee
+ ˆ
V
ne
+
V
nn
.
(2.4)Note que o termo
V
nn
tornou-se uma onstante. Denindo agora o hamiltoniano ele-trnioH
ˆ
de formaqueˆ
H
= ˆ
T
e
+ ˆ
V
ee
+ ˆ
V
ne
,
(2.5)veriamos queele omuta om asoordenadas nuleares:
[ ˆ
H
,
R
] = 0.
(2.6)oquequerdizerqueexistemautofunçõessimultâneaspara
H
ˆ
eR
. Issonospermiteesrever uma equação de Shrödinger daseguinteforma:ˆ
H
Ψ
n
(
r
;
R
) =
ε
n
(
R
)Ψ
n
(
r
;
R
),
(2.7)onde
Ψ
n
(
r
;
R
)
é a autofunção eletrnia eε
n
(
R
)
a orrespondente autoenergia, ambas dependendo parametriamente das oordenadas nuleares. O autovalor do hamiltonianode núleosxos,
E
n
(
R
)
, a então dado porE
n
(
R
) =
ε
n
(
R
) +
1
2
X
I
6
=J
Z
I
Z
J
|
R
I
−
R
J
|
.
(2.8)1
A introdução dasoordenadasde spin apenas aumentaria aomplexidade da notação, podendo ser
Como os
Ψ(
r
;
R
)
formam um onjunto ompleto de funções, podemos usá-los para expandir afunção de onda doproblema iniial, dado naequação (2.3):Ψ
tot
(
r
,
R
) =
X
m
φ
m
(
R
)Ψ
m
(
r
;
R
),
(2.9)onde
φ
m
(
R
)
são os oeientes de expansão. Substituindo essa expansão em(2.3), temosX
m
−
X
I
1
2M
I
∇
2
I
+
E
m
(
R
)
!
φ
m
(
R
)Ψ
m
(
r
;
R
) =
E
tot
X
m
φ
m
(
R
)Ψ
m
(
r
;
R
),
(2.10)onde foramutilizadasasequações(2.7) e (2.8). Para evitar uma sobrearga nas notações,
vamos omitiradependêniaexplíita de
Ψ
emr
eR
. Resolvendo olaplaianonoprimeiro membro, enontramosX
m
−
X
I
1
2M
I
∇
2
I
φ
m
(
R
) +
E
m
(
R
)φ
m
(
R
)
!
Ψ
m
=
E
tot
X
m
φ
m
(
R
)Ψ
m
+
+
X
m
X
I
1
M
I
∇
I
φ
m
(
R
)
· ∇
I
Ψ
m
+
1
2
φ
m
(
R
)
∇
2
I
Ψ
m
.
(2.11)
Após multipliar por
Ψ
∗
n
, integrar sobre as oordenadas eletrniasr
, usar aortonor-malidadedos
Ψ
's eisolarφ
m
(
R
)
, temos aseguinteexpressão:−
X
I
1
2M
I
∇
2
I
φ
n
(
R
) +
E
n
(
R
)φ
n
(
R
) =
E
tot
φ
n
(
R
)+
+
X
m
"
X
I
1
M
I
Z
Ψ
∗
n
∇
I
Ψ
m
· ∇
I
+
1
2
Ψ
∗
n
∇
2
I
Ψ
m
d
3
r
#
φ
m
(
R
).
(2.12)
Denindo
C
nm
(
R
,
∇
)
≡
X
I
1
M
I
Z
Ψ
∗
n
∇
I
Ψ
m
· ∇
I
+
1
2
Ψ
∗
n
∇
2
I
Ψ
m
podemosesrever aequação para
φ
de maneiramais ompata:−
X
I
1
2M
I
∇
2
I
φ
n
(
R
) +
E
n
(
R
)φ
n
(
R
) =
E
tot
φ
n
(
R
) +
X
m
C
nm
(
R
,
∇
)φ
m
(
R
).
(2.14)Chegamos a um resultado bastante interessante: se todos os oeientes
C
nm
forem nulos,teremosumaequaçãodeShrödingerparaφ
omE
n
(
R
)
atuandoomoumpotenial efetivo (sendo hamado de superfíie de energia de BO) e ujo autovalor é exatamentea energia total do problema iniial. A aproximação de BO onsiste em fazer todos os
oeientes
C
nm
desprezíveis 2, oque nos leva aesrever:
ˆ
H
nuc
φ
n
(
R
) =
−
X
I
1
2M
I
∇
2
I
φ
n
(
R
) +
E
n
(
R
)φ
n
(
R
) =
E
tot
φ
n
(
R
),
(2.15)Por onstrução, omo os
C
nm
aoplam diferentes estados eletrnios, a aproximação de BO onsiste naverdade emimpor que osestados eletrnios estão desaoplados. Comisso, obviamente o suesso dessa aproximação não depende do fato do núleo atmio ser
muito mais massivo que o elétron, mas sim se há um frao aoplamento entre os estados
eletrnios.
Comessaaproximação,épossívelfailmenteveriarqueaseparaçãodoshamiltonianos
eletrnio e nulearresultou emresolver oproblema eletrnio para uma distribuição xa
denúleos. Essadistribuiçãonulearxaatuaomoum potenialexternoparaoselétrons,
de maneira que podemos generalizar o hamiltoniano eletrnio esrevendo-o da seguinte
forma:
ˆ
H
= ˆ
T
e
+ ˆ
V
ee
+ ˆ
V
ext
,
(2.16)om
V
ˆ
ext
sendo agora o operadorque representaqualquer potenialesalar.Portanto, para se resolver o problema de muitos orpos utilizando-se a aproximação
2
Existetambémahamadaaproximaçãoadiabátia,quedesprezaapenas os
C
nm
forada diagonal,onuleariniial,enontrandoosautovaloresqueservirãodepotenialefetivoparaoproblema
nulear, de onde se tirarão novas distribuições nuleares e assim o ilo reiniia até a
onvergênia. Esse esquema é retratado nodiagramamostrado na gura.(2.1)
Iníio
R
Distribuiçãoiniialˆ
H
= ˆ
T
e
+ ˆ
V
ee
+ ˆ
V
ext
Hamiltonianoeletrnioˆ
H
Ψ
n
(
r
;
R
) =
ε
n
(
R
)Ψ
n
(
r
;
R
)
ProblemaeletrnioE
n
(
R
) =
ε
n
(
R
) +
1
2
X
I
6
=J
Z
I
Z
J
|
R
I
−
R
J
|
Potenialefetivonulear−
X
I
1
2M
I
∇
2
I
φ
n
(
R
) +
E
n
(
R
)φ
n
(
R
) =
E
tot
φ
n
(
R
)
ProblemanulearR
DistribuiçãonalConvergênia? Critério de onvergênia
Fim Sim Não
Figura2.1: Cilo de autoonsistênia para a aproximação de Born-Oppenheimer.
Essa aproximação põe toda a importâniano hamiltonianoeletrnio. Portanto,para
simpliar, daqui em diante quando for menionado hamiltoniano, está implíito que se
trata dohamiltoniano doproblema eletrnio.
oordenadas eletrnias (ontando om o spin). Para
N
>
2
, o problema não admite solução analítia e a outra maneira de se resolver a equação, que seria numeriamente,torna-seinviávelamedidaqueonúmerodeelétronsrese. Paraontornaressadiuldade,
devemos busar uma alternativa à solução do problema eletrnio. Felizmente existem
maneirasde seresolveresseproblemasempreisardeterminarafunçãode ondaeletrnia.
Dentre essas maneiras, destaa-se a Teoria do Funional da Densidade, que substitui o
problema envolvendo a função de onda por outro equivalente mas que usa uma variável
mais simples: a densidade eletrnia. A denição e algumas propriedadesimportantes da
densidade eletrnia serão disutidas napróxima seção.
2.3 A densidade eletrnia
O operadordensidade eletrnia
ˆ
n(
r
)
de um sistemaonstituído porN
elétronsé de-nido daseguinteforma:ˆ
n(
r
)
≡
N
X
i
δ(
r
−
r
i
).
(2.17)Ao alularo valor esperado desse operador, enontramos a densidade eletrnia. Sua
expressãoemtermosdafunçãode ondaeletrnia
Ψ
é(novamenteomitiremosospin para não arregar muita notação):n(
r
) =
Z
|
Ψ(
r
1
,
r
2
, . . . ,
r
N
)
|
2
N
X
i
δ(
r
−
r
i
)d
3
r
1
d
3
r
2
· · ·
d
3
r
N
.
(2.18)Apliando as propriedades de (anti)simetria da função de onda, podemos failmente
veriarque
n(
r
) =
N
Z
Ψ
∗
(
r
,
r
funçõesde onda normalizadas,temos exatamenteisso:
Z
n(
r
)d
3
r
=
N
(2.20)Vamos agora ver omo esrever o valoresperado de um operador que dependa apenas
das oordenadas eletrnias de um únio elétron emtermos da densidade eletrnia. Seja
esse operador
O
ˆ
,uja expressão pode ser esrita generiamenteomo:ˆ
O
=
X
i
ˆ
O(
r
i
).
(2.21)O valoresperado desse operador édado usualmentepelafunção de onda:
h
Oi
ˆ
=
X
i
Z
Ψ
∗
(
r
1
,
r
2
, . . . ,
r
N
) ˆ
O(
r
i
)Ψ(
r
1
,
r
2
, . . . ,
r
N
)d
3
r
1
d
3
r
2
· · ·
d
3
r
N
.
(2.22)Usando a (anti)simetriadafunção de onda,podemos esrever
h
Oi
ˆ
=
N
Z
Ψ
∗
(
r
1
,
r
2
, . . . ,
r
N
)O(
r
1
)Ψ(
r
1
,
r
2
, . . . ,
r
N
)d
3
r
1
d
3
r
2
· · ·
d
3
r
N
.
(2.23)Podemos manipular oresultado aima da seguinte maneira:
h
Oi
ˆ
=
Z
O(
r
1
)
N
Z
Ψ
∗
(
r
1
,
r
2
, . . . ,
r
N
)Ψ(
r
1
,
r
2
, . . . ,
r
N
)d
3
r
2
· · ·
d
3
r
N
d
3
r
1
.
(2.24)A integral dentro do parênteses é justamente a densidade eletrnia avaliada em
r
1
.Fazendo umamudançade variáveis naintegral em
r
1
,amos nalmenteomh
Oi
ˆ
=
Z
que atuenas oordenadaseletrnias pode ser failmenteesritoomo funionalda
densi-dadeeletrnia. Essa propriedadeépartiularmenteútilparadeterminaro valoresperado
dopotenialexterno:
ˆ
V
ext
=
X
i
v
ext
(
r
i
),
(2.26)andoo valoresperado esrito omo:
h
V
ˆ
ext
i
=
Z
v
ext
(
r
)n(
r
)d
3
r.
(2.27)2.4 A Teoria do Funional da Densidade
Desde o surgimento daTeoria do FunionaldaDensidade (DFT) emmeados dos anos
60, vários trabalhos vêm sendo desenvolvidos seja apliando-a ou melhorando-a.
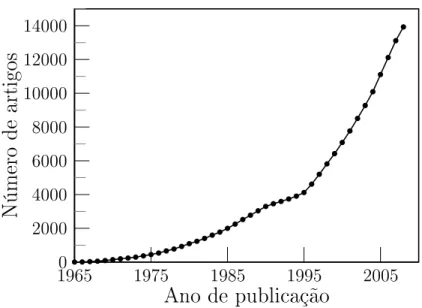
Atual-mente a DFT é o método mais largamente utilizado para lidar om problemas na esala
nanométria. Pode-se ter uma ideia da quantidade de trabalhos publiados utilizando-se
essa ténia através dagura (2.2).
Em 1998, a omunidade ientía mundial reonheeu que a DFT representou um
avançobastantesigniativoparaodesenvolvimentodaiênia. NaqueleanoWalterKohn,
o prinipalarquitetoda DFT, foi laureadoom o prêmionobel emQuímia.
Dois trabalhosmaramoiníiodaDFT: oprimeirofundamentaosprinípiosdateoria
em dois teoremas simples e foi publiado em 1964 por Hohenberg e Kohn (HK) [17℄. O
segundo, publiado no ano seguinte por Kohn e Sham (KS) [18℄, apresenta um esquema
autoonsistenteparaaresoluçãodoproblemaeletrnioatravésdasoluçãodeumaequação
baseadanoansatz demapearosistemainteragenteemum sistemaauxiliarnãointeragente
utilizando a densidade eletrnia omo variável do problema. Os teoremas e o esquema
0
2000
4000
6000
8000
10000
12000
14000
1965
1975
1985
1995
2005
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
b
Ano de publiação
Número
de
artigos
Figura2.2: Número de artigospubliados por ano utilizando-sea DFT.
2.4.1 Os teoremas da DFT
OsteoremasquefundamentamaDFT[17℄estabeleemquetodasaspropriedadesdeum
sistemademuitosorposdesritaspelafunçãodeondapodemseresritasomofunionais
dadensidadeeletrnia. Comisso, adensidade eletrniapassa adesempenharopapelde
variávelfundamentalnoproblema de muitos orpos.
O primeiro teorema garante que em um sistema de partíulas interagentes, o
poten-ial externo
v
ext
(
r
)
que age sobre o sistema é determinado uniamente (exeto por uma onstante aditiva)peladensidade eletrnia doestado fundamental.O segundo teorema arma que a energia pode ser esrita omo um funional
univer-sal válido para qualquer potenial externo. A energia do estado fundamental é obtida
minimizando-seesse funionalom respeito àdensidade eletrnia, sendoque adensidade
que produz esse mínimoé a densidade exata doestado fundamental.
Demonstração do 1 teorema
Para demostrar esse teorema, vamos apliar o método de redução ao absurdo.
Supo-nhamos que existam dois poteniais externos
v
ext
ev
′
ext
que diferem por mais que umaonstante aditiva porém gerem a mesma densidade do estado fundamental
n
0
(
r
)
. Esses poteniais produzemhamiltonianosH
ˆ
eH
ˆ
′
distintos entre si e funçõesde onda do estado
fundamental
Ψ
eΨ
′
tambémdistintas. Partindo do prinípiode que o valor esperado do
hamiltonianoavaliadopara qualquer função de onda quenão seja exatamenteado estado
fundamentalserá sempremaior queo valordaenergiapara oestado fundamentalorreto,
teremos:
E
0
<
h
Ψ
′
|
H|
ˆ
Ψ
′
i
E
′
0
<
h
Ψ
|
H
ˆ
′
|
Ψ
i
,
onde
E
0
eE
′
0
sãoasenergiasdoestadofundamentaldeH
ˆ
eH
ˆ
′
,respetivamente. Somando
e subtraindo
H
ˆ
′
aprimeira expressão e
H
ˆ
na segunda, temos
E
0
<
h
Ψ
′
| H
+
H
′
− H
′
|
Ψ
′
i
=
h
Ψ
′
| H
′
|
Ψ
′
i
+
h
Ψ
′
| H − H
′
|
Ψ
′
i
E
′
0
<
h
Ψ
| H
′
+
H − H |
Ψ
i
=
h
Ψ
| H |
Ψ
i
+
h
Ψ
| H
′
− H |
Ψ
i
.
Umavez que oshamiltonianosdiferem apenasnos poteniaisexternos,pode-se failmente
mostrar que
E
0
< E
0
′
+
Z
[v
ext
(
r
)
−
v
ext
′
(
r
)]
n
0
(
r
)
d
3
r
E
0
′
< E
0
+
Z
[v
ext
′
(
r
)
−
v
ext
(
r
)]
n
0
(
r
)
d
3
onde usamos
E
0
=
h
Ψ
|
H|
ˆ
Ψ
i
,E
′
0
=
h
Ψ
′
|
H
ˆ
′
|
Ψ
′
i
e a equação (2.27). Somando essas duas expressões, hegamos ao absurdoE
0
−
E
′
0
< E
0
−
E
0
′
, ou seja, dois poteniais distintos não podem geraramesmadensidade doestadofundamental,o queimpliaemdizer queadensidade determinauniamenteopotenialexterno. Comoorolário,pode-se dizerque o
hamiltonianoé uniamentedeterminado,exeto por umaonstante, peladensidade do
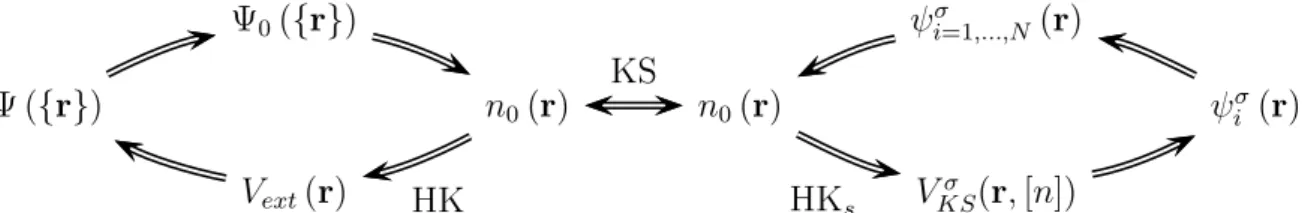
es-tadofundamental. Esseteoremapodeser resumidopelodiagramamostradonagura(2.3)
v
ext
(
r
)
n
0
(
r
)
Ψ (
{
r
}
)
Ψ
0
(
{
r
}
)
HK
Figura2.3: Representação esquemátia doteoremade Hohenberg-Kohn. As setasindiam
oaminhodasoluçãodaequaçãodeShrödinger. AindiadaporHK,refere-seaoteorema
de Hohenberg-Kohn.
Demonstração do 2 o
teorema
Suponha umhamiltoniano
H
ˆ
ujafunção de ondae densidadeeletrnia de um estado qualquer sejamΨ
en(
r
)
, respetivamente. O primeiro teorema nos permite esrever a energiapara esse estadoomo funional dadensidade, alulada da seguintemaneira:E[n] =
h
Ψ
|
T
ˆ
e
+ ˆ
V
ee
+ ˆ
V
ext
|
Ψ
i
(2.28)E[n] =
F
[n] +
h
Ψ
|
V
ˆ
ext
|
Ψ
i
,
(2.29)onde
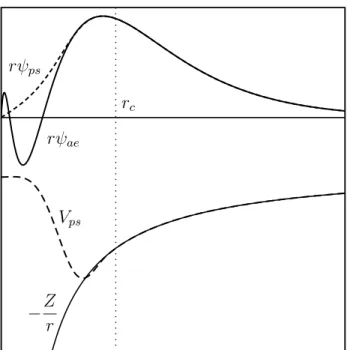
F
[n]
é um funional ompletamenteuniversal válido para sistemas oulombianos,já que depende apenas deT
ˆ
e
eV
ˆ
ee
. A expressão orrespondente para a energia do estado fundamentalé:Podemosapliaroteoremavariaionaldamaneiraonvenional,ouseja,om
E
esrito omo funional deΨ
:E[Ψ
0
]
6
E[Ψ]
(2.31)h
Ψ
0
|
T
ˆ
e
+ ˆ
V
ee
+ ˆ
V
ext
|
Ψ
0
i
6
h
Ψ
|
T
ˆ
e
+ ˆ
V
ee
+ ˆ
V
ext
|
Ψ
i
(2.32)F
[n
0
] +
h
Ψ
0
|
V
ˆ
ext
|
Ψ
0
i
6
F
[n] +
h
Ψ
|
V
ˆ
ext
|
Ψ
i
(2.33)E[n
0
]
6
E[n].
(2.34)Comisso, podemosapliaroprinípiovariaionalminimizando
E[n]
omrelaçãoan(
r
)
que hegaremos ao estado fundamental produzido pela densidade do estado fundamentalexata.
Ao demostrar esses dois teoremas, Hohenberg e Kohn garantem que é possível obter
o estado fundamental de um sistema sem que se tenha onheimento da função de onda.
Falta ainda riar uma maneira de se enontrar essa densidade do estado fundamental, o
que implia em desenvolver equações que permitam o álulo da densidade. Isso foi feito
noano seguinteà demostração desses teoremas eserá oobjetivo dapróxima subseção.
2.4.2 As equações básias da DFT
Vimos que os teoremas de HK garantem que a densidade eletrnia do estado
funda-mentalpodesubstituirafunçãodeondanaobtençãodosobserváveis. Issosedáesrevendo
ovaloresperadodosoperadoresomofunionaisdadensidade. Dentretodososobserváveis
físios, a energia total talvez seja o mais importante deles. De aordo om HK, elapode
ser esrita daseguintemaneira:
esrita omo funionaldadensidade, dada na equação (2.27):
h
V
ˆ
ext
i
=
V
ext
[n] =
Z
v
ext
(
r
)n(
r
)d
3
r.
(2.36)Faltam portanto
T
e
[n]
eV
ee
[n]
para enontrarmos a energia. Sabemos que o operador energiainétia esrito noespaço de onguraçõesé proporionalao operador∇
2
e que a
interação elétron-elétron édo tipo
1/r
. Como então podemos esrever derivadas einverso da distânia omo funionais den(
r
)
? Infelizmente ninguém até agora desobriu omo fazê-lo.Para ontornar essa diuldade, em vez de prourar uma formaexplíita para os
fun-ionais
T
e
[n]
eV
ee
[n]
, Kohn e Sham [18℄ propuseram um método que onsegue enontrar a densidade eletrnia do estado de maneira (em prinípio) exata. Eles se basearam emduas suposições:
1. A densidade exata do estado fundamental pode ser representada pela densidade de
um sistema auxiliarde partíulas não-interagentes.
2. Ohamiltonianoauxiliar(tíio)éesolhido de formaquepossua umaenergia
iné-tia esritadaformausual e umpotenialefetivo loalatuando sobre um elétronde
spin
σ
naoordenadar
.De aordo om essas suposições, o hamiltoniano tíio é esrito omo um hamiltoniano
de um elétron:
ˆ
H
σ
aux
=
−
1
2
∇
2
+
V
σ
(
r
)
.
(2.37)
Com isso, troamos um problema de vários elétrons interagindo entre si para vários
problemas de um elétron, já que esses são não-interagentes. Para um sistema de
N
=
N
↑
+
N
fundamental é obtido aloando-se um únio elétron em ada um dos
N
σ
orbitais
ψ
σ
i
(
r
)
de mais baixa energia. A densidade eletrnia para esse sistema é dada pela soma dos
módulos quadrados do orbital
ψ
σ
i
(
r
)
:n
(
r
) =
X
σ
n
(
r
, σ) =
X
σ
N
σ
X
i=1
|
ψ
i
σ
(
r
)
|
2
,
(2.38)que émuito mais simplesque a formaesritana equação (2.19).
A energia inétiapara esse hamiltonianotíioassume a seguinte formafunional:
T
s
=
−
1
2
X
σ
N
σ
X
i=1
Z
ψ
i
σ
∗
(
r
)
∇
2
ψ
i
σ
(
r
)
d
3
r,
(2.39)onde o índie
s
signia single partile, denotando que essa energia inétia refere-se a partíulas não-interagentes3
. A energia
T
s
é diferente da energia inétia realT
e
por um termo quenos édesonheido mas que será levado emonta mais adiante.Para a interação elétron-elétron, vamos proeder de maneira diferente. Imaginemos
que essa interação é omposta por dois termos: o primeiro dado pela solução lássia de
uma distribuiçãoxade argas eoutroque ompensa oerro ausado peloprimeirotermo,
dessaformamantemosaindaaexatidãodasolução. Essasoluçãolássiaéonheidaomo
energiade Hartree
E
H
:E
H
=
1
2
Z
n(
r
)n(
r
′
)
|
r
−
r
′
|
d
3
rd
3
r
′
.
(2.40)
Comessasanálises,perebemosqueopotenialefetivototalmostradonaequação(2.37)
deve gerar três ontribuições paraa energia total:
•
Aenergiadevido aopotenialexternoV
ˆ
ext
,aluladopreviamentenaequação(2.27);•
A energia devido ao potenial de Hartree,E
H
, solução doproblema de distribuição 3Note que
T
s
não éumfunional explíito den
, massimdeψ
. Aimportâniadessa formafunional•
Umaenergia potenialque ompense oserros ometidos nas avaliaçõesdeT
s
eE
H
.O quedifereniaaenergia inétiaparaelétronsnão-interagentes ea energiapotenial
de uma distribuição xa de argas de um sistema real são os efeitos de muitos orpos,
onheidos omo troa e orrelação. O efeito de troa está relaionado ao prinípio de
exlusão apliado a elétrons om spins paralelos, enquanto que os efeitos de orrelação
estão assoiados ao fato de elétrons om spins antiparalelos terem seus movimentos
or-relaionados. Devido a esses efeitos, o tereiro termo do potenial efetivo total mostrado
na equação (2.37) é onheido omo potenial de troa-orrelação(XC). Com todos esses
argumentos, a energiaa então dada por
E[n] =
T
s
[n] +
1
2
Z
n(
r
)n(
r
′
)
|
r
−
r
′
|
d
3
rd
3
r
′
+
Z
v
ext
(
r
)n(
r
)d
3
r
+
E
xc
σ
(
r
,
[n]),
(2.41)om
T
s
[n]
dado naequação (2.39) eE
σ
xc
(
r
,
[n])
éa energiade XC.Agora, omo determinar a densidade eletrnia do estado fundamental? O segundo
teorema da DFT nos permite fazer uma minimização na energia variando-se
n(
r
)
e a densidade que gera esse mínimo de energia é a densidade que prouramos. Fazendo essaminimizaçãoapliando-se a téniados multipliadoresde Lagrangeonde o vínulo se faz
sobre osorbitais
ψ
σ
i
,que devemser normalizados:δ
δn
E[n]
−
ε
σ
i
Z
|
ψ
σ
i
(
r
)
|
2
d
3
r
−
1
= 0,
(2.42)onde
ε
σ
i
é o multipliador de Lagrange para o vínulo da funçãoψ
σ
i
. Com o auxílio daequação (2.41),a derivada funional
E[n]
pode ser alulada daseguintemaneira:δE
[n]
δn
=
δT
s
[n]
δn
+
Z
n(
r
′
)
|
r
−
r
′
|
d
3
r
′
+
v
ext
(
r
) +
δE
σ
xc
(
r
,
[n])
Para alulara derivada funionalde
T
s
[n]
, devemos apliara regra daadeia:δT
s
[n]
δn
=
δT
s
[n]
δψ
σ
∗
i
δψ
σ
∗
i
δn
,
(2.44)onde om o auxíliodas equações(2.39) e (2.19),enontramos failmente
δT
s
[n]
δn
=
−
1
2
∇
2
ψ
σ
i
ψ
σ
i
.
(2.45)Substituindo esse resultado naequação (2.43), temos que a ondição de extremodada
pela equação(2.42) implia emesrever
−
1
2
∇
2
+
Z
n(
r
′
)
|
r
−
r
′
|
d
3
r
′
+
v
ext
(
r
) +
δE
σ
xc
(
r
,
[n])
δn
ψ
σ
i
(
r
) =
ε
σ
i
ψ
σ
i
(
r
).
(2.46)Otermoentreolhetesnoprimeiromembroéexatamenteohamiltonianotíio
men-ionadoanteriormente, agora om o potenialefetivoexpliitado emtrês termosjá
disu-tidos. Denindo esse hamiltonianoomo hamiltonianode KS, temos
ˆ
H
σ
KS
=
−
1
2
∇
2
+
Z
n(
r
′
)
|
r
−
r
′
|
d
3
r
′
+
v
ext
(
r
) +
V
xc
σ
(
r
,
[n]),
(2.47)om
V
xc
σ
(
r
,
[n])
≡
δE
σ
xc
(
r
,
[n])
δn
(2.48)sendo o potenial de XC. O potenial efetivo total de antes a agora sendo hamado de
potenial efetivo de KS:
V
KS
σ
(
r
,
[n])
≡
Z
n(
r
′
)
|
r
−
r
′
|
d
3
−
1
2
∇
2
ψ
σ
i
+
V
σ
KS
(
r
,
[n])ψ
σ
i
=
ε
σ
i
ψ
σ
i
(
r
),
(2.50)que deve ser resolvida para ada elétron do sistema. As autofunções da equação de KS,
ψ
σ
i
(
r
)
, são onheidas omo orbitais de KS. A densidade do estado fundamental é então determinadapormeio daequação (2.38).Graças aos teoremas demonstrados por Hohenberg e Kohn eà equação onstruída por
Kohn e Sham, podemos resolver o ompliado problema real em um problema tíio
onsideravelmente mais simples. Quem faz essa onexão entre o real e o tíio é
justa-mente a densidade eletrnia do estado fundamental. A gura (2.4) resume as prinipais
araterístiasdesses dois pontos.
Ψ
0
(
{
r
}
)
ψ
i=1,...,N
σ
(
r
)
Ψ (
{
r
}
)
n
0
(
r
)
n
0
(
r
)
ψ
i
σ
(
r
)
V
ext
(
r
)
HKV
KS
σ
(
r
,
[n])
HK
s
KSFigura 2.4: Diagrama resumindo as prinipais impliações dos teoremas de
Hohenberg-KohneométododeKohn-Sham. AssetasindexadasporHKeHK
s
referem-seaosteoremasdeHohenberg-Kohnapliadosaossistemasrealetíio,respetivamente. Asetaindexada
por KS india o ansatz de Kohn-Sham em onetar o sistema real om o tíio através
dadensidade eletrnia do estadofundamental.
Agora, parase esrevero potenialde KS,queé opontohavedaequação,preisamos
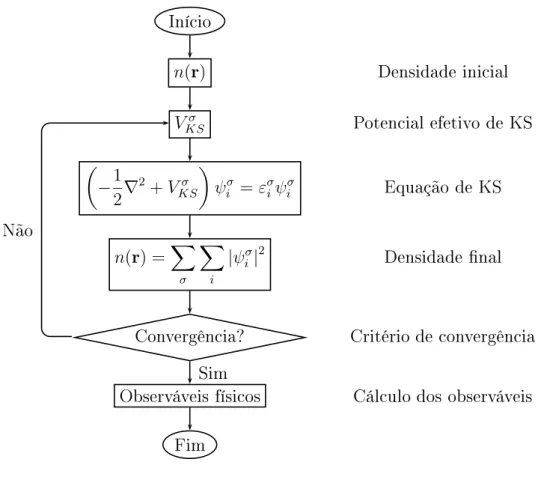
primeiramenteonheeradensidadeeletrnia,jáqueopotenialdependefunionalmente
dela. Amaneiramaisadequadadeseresolveresse tipode problemaédemaneiraiterativa,
ou seja: primeiramente se propõe uma densidade eletrnia iniial para que o potenial
seja onstruído e assim montada a equação de KS. Após resolvê-la e enontrar uma nova
densidade,ompara-seestaomainiial. Seadiferençaentreelasatenderaumerto