UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
CENTRO DE CIÊNCIAS BIOLÓGICAS
DEPARTAMENTO DE BIOQUÍMICA
MÁRCIA DANIELLE DE ARAÚJO DANTAS
ESTUDO DO GENOMA DO VÍRUS CAUSADOR DA
MIONECROSE INFECCIOSA EM CAMARÕES E
DESENVOLVIMENTO DE MÉTODOS PARA DETECÇÃO DE
POLIMORFISMOS
MÁRCIA DANIELLE DE ARAÚJO DANTAS
ESTUDO DO GENOMA DO VÍRUS CAUSADOR DA
MIONECROSE INFECCIOSA EM CAMARÕES E
DESENVOLVIMENTO DE MÉTODOS PARA DETECÇÃO DE
POLIMORFISMOS
NATAL
2014
Dissertação apresentada ao Programa de Pós-Graduação em Bioquímica da Universidade Federal do Rio Grande do Norte como requisito parcial para obtenção do título de Mestre em Bioquímica.
MÁRCIA DANIELLE DE ARAÚJO DANTAS
ESTUDO DO GENOMA DO VÍRUS CAUSADOR DA MIONECROSE INFECCIOSA EM CAMARÕES E DESENVOLVIMENTO DE MÉTODOS PARA DETECÇÃO DE
POLIMORFISMOS
Dissertação apresentada ao Programa de Pós-Graduação em Bioquímica da Universidade Federal do Rio Grande do Norte como requisito parcial para obtenção do título de Mestre em Bioquímica.
Aprovado em: 01/08/2014
BANCA EXAMINADORA
___________________________________________________ Prof. Dr. Daniel Carlos Ferreira Lanza
Departamento de Bioquímica - UFRN Orientador
____________________________________________________ Prof. Dr.Leonardo Lima Pepino de Macedo
Pesquisador da EMBRAPA - Recursos Genéticos e Biotecnologia Examinador externo
____________________________________________________ Prof. Dr. João Paulo Matos Santos Lima
Dedico esta obra
AGRADECIMENTOS
Agradeço a todos que direta ou indiretamente participaram da realização deste trabalho, em especial agradeço:
A Deus, pai de infinito amor e bondade, pela sua presença constante em minha vida, me abençoando e iluminando meus caminhos.
Aos meus pais, Maria do Socorro de Araújo Dantas e Luis de Vasconcelos Dantas, por todo amor, carinho e dedicação. Vocês são os principais responsáveis pela
pessoa que sou hoje e por todas as minhas conquistas.
Ao meu namorado, Pedro Henrique Sales da Costa, por todo amor, amizade e companheirismo e por sempre estar ao meu lado me apoiando em todos os
momentos.
Ao Programa de Pós-Graduação em Bioquímica, Departamento de Bioquímica da Universidade Federal do Rio Grande do Norte (UFRN) pelos conhecimentos
adquiridos na área.
Aos amigos do Laboratório de Biologia Molecular Aplicada – Laplic, Raffael, Allan, Douglas, Jéssica e Natália, pela amizade e pelos momentos de descontração
durante os trabalhos. Em especial agradeço a Raffael por toda ajuda com as análises de Bioinformática.
Ao professor Dr. João Paulo Matos Santos Lima e seus alunos Diego e Ricardo pela ajuda durante o desenvolvimento deste trabalho.
Ao pessoal do Laboratório de Imunogenética pela disponibilidade de equipamentos extremamente necessários para a realização deste trabalho.
À professora Drª Suely Ferreira Chavante pelo incentivo e apoio em ingressar na pós-graduação.
Aos colegas da minha turma de pós-graduação por compartilhar comigo tantos momentos de alegria e desespero durante esses anos de estudo.
Aos meus grandes amigos da Biologia pelos quatros anos de companheirismo e amizade. Em especial às amigas Sinara Carla, Larissa Maria e Larissa Bahia. Vocês
tornaram os meus momentos mais felizes durante a graduação.
À CAPES pelo fornecimento da bolsa de pesquisa e à FAPERN pelo auxílio financeiro ao projeto de pesquisa.
E claro, o meu muito obrigada ao meu querido orientador Professor Dr. Daniel Carlos Ferreira Lanza pela sua orientação, paciência, profissionalismo e
compromisso com seus orientandos. Obrigada pela sua atuação direta na ampliação dos meus conhecimentos. O senhor, com certeza, é um exemplo de pessoa e de
O cientista não é o homem que fornece as verdadeiras respostas, é quem faz as verdadeiras perguntas.
RESUMO
A carcinicultura é uma das atividades que mais contribui para o crescimento da aquicultura mundial. Entretanto, esta atividade vem sofrendo perdas econômicas significativas devido ao surgimento de doenças virais como a Mionecrose Infecciosa (IMN). A IMN já está disseminada em toda região Nordeste do Brasil e vem causando sérios danos à carcinicultura na Indonésia. O principal sintoma da doença é a mionecrose, que consiste na necrose dos músculos estriados do abdômen e do cefalotórax do camarão. A IMN é causada pelo vírus da mionecrose infecciosa (IMNV), um vírus não envelopado que apresenta protrusões ao longo de seu capsídeo. O genoma viral é formado por uma única molécula de RNA dupla fita e possui duas Open Reading Frames (ORFs). A ORF1 codifica a proteína principal do capsídeo (MCP) e uma possível proteína de ligação a RNA (RBP). A ORF2 codifica uma provável RNA polimerase dependente de RNA (RdRp) e classifica o IMNV dentro da família Totiviridae. Assim, o objetivo desse estudo foi estudar o genoma completo do IMNV e as proteínas codificadas no intuito de desenvolver um sistema que identificasse diferentes isolados do vírus com base na presença de polimorfismos. A relação filogenética entre alguns totivírus foi investigada e mostrou um novo grupo para o IMNV dentro da família Totiviridae. Dois novos genomas foram sequenciados, analisados e comparados a outros dois genomas já depositados no GenBank. Os novos genomas foram mais semelhantes entre si do que com aqueles já descritos. Regiões variáveis e conservadas do genoma foram identificadas através de gráficos de similaridade e alinhamentos utilizando as quatro sequências do IMNV. Esta análise possibilitou o mapeamento de sítios polimórficos e revelou que a região mais variável do genoma se encontra na primeira metade da ORF1 e coincide com as regiões que possivelmente codificam a protrusão viral, enquanto que as regiões mais estáveis se encontraram em domínios conservados de proteínas que interagem com o RNA. Além disso, estruturas secundárias foram preditas para todas as proteínas empregando diversos softwares e modelos estruturais proteicos foram calculados usando modelagens por threading e simulações ab initio. A partir dessas análises foi possível observar que as proteínas do IMNV possuem motivos e formas similares às proteínas de outros totivírus, e novas possíveis funções proteicas foram propostas. O estudo do genoma e das proteínas foi essencial para o desenvolvimento de um sistema de detecção baseado em PCR capaz de discriminar os quatro isolados do IMNV com base na presença de sítios polimórficos.
ABSTRACT
Shrimp farming is one of the activities that contribute most to the growth of global aquaculture. However, this business has undergone significant economic losses due to the onset of viral diseases such as Infectious Myonecrosis (IMN). The IMN is already widespread throughout Northeastern Brazil and has caused serious damage to shrimp farming in Indonesia. The main symptom of disease is myonecrosis, which consists of necrosis of striated muscles of the abdomen and cephalothorax of shrimp. The IMN is caused by infectious myonecrosis virus (IMNV), a non-enveloped virus which has protrusions along its capsid. The viral genome consists of a single molecule of double-stranded RNA and has two Open Reading Frames (ORFs). The ORF1 encodes the major capsid protein (MCP) and a potential RNA binding protein (RBP). ORF2 encodes a probable RNA-dependent RNA polymerase (RdRp) and classifies IMNV in Totiviridae family. Thus, the objective of this research was study the IMNV complete genome and encoded proteins in order to develop a system differentiate virus isolates based on polymorphisms presence. The phylogenetic relationship among some totivirus was investigated and showed a new group to IMNV within Totiviridae family. Two new genomes were sequenced, analyzed and compared to two other genomes already deposited in GenBank. The new genomes were more similar to each other than those already described. Conserved and variable regions of the genome were identified through similarity graphs and alignments using the four IMNV sequences. This analyze allowed mapping of polymorphic sites and revealed that the most variable region of the genome is in the first half of ORF1, which coincides with the regions that possibly encode the viral protrusion, while the most stable regions of the genome were found in conserved domains of proteins that interact with RNA. Moreover, secondary structures were predicted for all proteins using various softwares and protein structural models were calculated using threading and ab initio modeling approaches. From these analyses was possible to observe that the IMNV proteins have motifs and shapes similar to proteins of other totiviruses and new possible protein functions have been proposed. The genome and proteins study was essential for development of a PCR-based detection system able to discriminate the four IMNV isolates based on the presence of polymorphic sites.
LISTA DE FIGURAS
LISTA DE TABELAS
LISTA DE ABREVIATURAS / SIGLAS
Aa Aminoácidos
ABCC Associação Brasileira dos Criadores de Camarão BLAST Basic Local Alignment Search Tool
CDD Conserved Domains Database cDNA DNA complementar
DNA Ácido desoxirribonucleico
dNTP Deoxinucleotídeos trifosfatados DSRM Double-strand RNA binding motif dsRNA RNA dupla fita
EMPARN Empresa Brasileira de Pesquisa Agropecuária do RN ExPASy Expert Protein Analysis System
ha hectare
HIV Human immunodeficiency virus HRMA High-Resolution Melting Analysis IBDV Vírus da doença infecciosa da Bursa
ICTV Comitê Internacional de Taxonomia de Vírus IHHNV Infectious hypodermal and haematopoietic virus IMN Infectious Myonecrosis
IMNV Infectious myonecrosis virus kDa Kilodaltons
Kg Quilogramas
ml mililitros mM milimolar µg microgramas µl microlitros µM micromolar
NCBI National Center for Biotechnology Information nt Nucleotídeo
OIE World Organization for Animal Health ORF Open Reading Frame
PCR Polymerase Chain Reaction PDB Protein Data Bank
PMCV Piscine myocarditis virus pmol Picomole
RBP Proteína de ligação a RNA
RdRp RNA polimerase dependente de RNA RMSD Root-mean-square deviation
RNA Ácido Ribonucleico RNAi RNA de interferência RNAP RNA polimerase RT Transcriptase reversa
RT-LAMP Reverse Transcription Loop Mediated Isothermal Amplification RT-PCR Reverse Transcription Polymerase Chain Reaction
TLR3 receptor Toll-like 3 TM Temperatura de melting TSV Taura syndrome virus U Unidade
UniProt Universal Protein Resource WEEV Western equine encephalitis virus WSSV White spot syndrome virus
SUMÁRIO
1 INTRODUÇÃO 19
1.1 MIONECROSE INFECCIOSA 20
1.2 VÍRUS DA MIONECROSE INFECCIOSA 23
1.2.1 Proteína de ligação a RNA (RBP) 26
1.2.2 Proteína Principal do Capsídeo (MCP) 27
1.2.3 Protrusões virais 30
1.2.4 RNA Polimerase Dependente de RNA (RdRp) 32
1.3 VARIABILIDADE GENÉTICA EM VÍRUS 34
1.4 SISTEMAS DE DETECÇÃO PARA O IMNV 36
2 OBJETIVOS 39
3 MATERIAIS E MÉTODOS 40
3.1 ANÁLISE FILOGENÉTICA 40
3.2 OBTENÇÃO DAS AMOSTRAS DE CAMARÃO 41
3.3 SCREENING INICIAL PARA DETECÇÃO DO IMNV EM DIFERENTES
AMOSTRAS DE CAMARÃO 42
3.3.1 Extração de RNA total 43
3.3.2 RT-PCR 43
3.3.2.1 Síntese de cDNA 43
3.3.2.2 Reação em Cadeia da Polimerase (PCR) 44
3.4 AMPLIFICAÇÃO E SEQUENCIAMENTO DO GENOMA VIRAL 45
3.5 ANÁLISE DAS SEQUÊNCIAS 47
3.6 ANÁLISE ESTRUTURAL DE PROTEÍNAS 48
3.7 MODELAGEM PROTEICA 49
3.8.1 Teste de detecção baseado nas regiões variáveis e conservadas 50 3.8.2 Teste de detecção baseado na análise da curva de melting de fita simples 51
4 RESULTADOS 53
4.1 ANÁLISE FILOGENÉTICA DA FAMÍLIA TOTIVIRIDAE 53
4.2 SCREENING INICIAL PARA DETECÇÃO DO IMNV EM DIFERENTES
AMOSTRAS DE CAMARÃO 54
4.3 AMPLIFICAÇÃO E MONTAGEM DOS NOVOS GENOMAS 55
4.4 CARACTERIZAÇÃO DOS NOVOS GENOMAS 55
4.5 IDENTIFICAÇÃO DE REGIÕES CONSERVADAS E VARIÁVEIS 58 4.6 ANÁLISE DE COMPOSIÇÃO DE AMINOÁCIDOS E PREDIÇÃO DE
ESTRUTURA SECUNDÁRIA 61
4.7 MODELAGEM DE PROTEÍNAS 68
4.8 SISTEMAS DE DETECÇÃO DE POLIMORFISMOS 71
4.8.1 Teste de detecção baseado nas regiões variáveis e conservadas 71 4.8.2 Teste de detecção baseado na análise da curva de melting de fita simples 74
5 DISCUSSÃO 79
6 CONCLUSÃO 91
REFERÊNCIAS 93
1 INTRODUÇÃO
Durante a década de 80, os avanços na produção de camarão proporcionaram um rápido crescimento na carcinicultura (LOTZ, 1997). A produção de crustáceos foi a atividade que mais cresceu na aquicultura mundial nos últimos anos, atingindo 4,5 milhões de toneladas e US$ 17,95 bilhões de dólares em 2006. No mesmo ano a produção de camarão cultivado correspondeu a 17% do valor total dos produtos pesqueiros internacionalmente negociados (FAO, 2008).
No Brasil, as pesquisas com camarão começaram na década de 70 com o Projeto Camarão da Empresa Brasileira de Pesquisa Agropecuária do Rio Grande do Norte S/A EMPARN (BRASIL, 2001). Várias espécies de camarão nativas (Farfantepenaeus subtilis, F. brasiliensis, F. paulensis e Litopenaeus schmitti) foram consideradas como opções de cultivo, porém vários fatores relacionados à produção tornaram estas espécies inviáveis comercialmente (MADRID, 1999). Apenas em meados da década de 80, com a introdução do camarão branco do Pacífico, Litopenaeus vannamei, a carcinicultura brasileira se consolidou (BRASIL, 2001).
Entre os anos de 1996 e 2002, a produção de camarão passou de 2.880 toneladas para mais de 60 mil toneladas e a produtividade, de 900 kg/ha/ano para 5.458 kg/ha/ano (ORMOND, 2004), tonando o Brasil o país líder do hemisfério ocidental, com 90.190 toneladas, e líder mundial em produtividade, com 6.084 kg/ha/ano, no ano de 2003 (ROCHA, 2005a).
Os principais produtores são os estados do Ceará e do Rio Grande do Norte. Existem quase 2.000 produtores no Brasil, a maioria com pequenos cultivos. Atualmente os maiores produtores mundiais de camarão são China, Vietnã, Tailândia, Indonésia, Índia e Equador. O Equador produziu 300.000 toneladas de camarão em 2013 e obteve US$ 1,67 bilhão com a exportação de 215.000 toneladas do produto (Disponível em MPA - Ministério da Pesca e Aquicultura).
Apesar desse crescimento na produtividade, os carcinicultores têm sofrido perdas econômicas significativas nas últimas décadas devido a problemas ambientais relacionados às práticas de cultivo e às doenças virais (MOSS, 2002). Dentre as doenças virais que acometem a carcinicultura mundial, destacam-se aquelas causadas pelo vírus da Mancha Branca (WSSV – White spot syndrome virus), vírus da Cabeça Amarela (YHV – Yellow head virus), vírus da Taura (TSV – Taura syndrome virus), vírus da Necrose Hipodermal e Hematopoiética Infecciosa (IHHNV – Infectious hypodermal and haematopoietic virus) e o vírus da Mionecrose Infecciosa (IMNV – Infectious myonecrosis virus) (LOTZ, 1997; LIU et al., 2009).
1.1 MIONECROSE INFECCIOSA
MARTINS & GESTEIRA, 2004; ANDRADE et al., 2007; SENAPIN et al., 2007; LIGHTNER, 2011). Devido à importância econômica cada vez maior do L. vannamei na região Ásia-Pacífico e ao consequente aumento do transporte dessa espécie entre fronteiras, em janeiro de 2006 a IMN foi adicionada à lista Quartely Aquatic Animal Disease Report com o propósito de aumentar a vigilância e sua detecção (QAAD, 2006).
De acordo com a Associação Brasileira dos Produtores de Camarão, a produção de camarão, no período em que a doença surgiu no Brasil (2002-2005), foi de 150.000 toneladas a menos que o total esperado. Embora essa redução também seja atribuída a variações no cambio e à ação antidumping dos Estados Unidos, estima-se que 60 a 70% das perdas foram causadas por ação da IMN, causando prejuízos próximos a 400 milhões de dólares naquele período (MADRID, 2005a, b; ROCHA, 2005a, b; ANDRADE et al., 2007).
hemoceloma nas brânquias, coração próximo à glândula antenal e cordão nervoso ventral (TANG et al., 2005).
Figura 1: Camarões Litopenaeus vannamei infectados com o vírus da mionecrose infecciosa. (A) O
camarão acima mostra sinais de necrose, evidenciada pela opacidade do sexto segmento abdominal (fase aguda); abaixo se encontra o camarão sadio, sem sinais de infecção (adaptado de POULOS et al., 2006). (B) O camarão apresenta pigmentação avermelhada nos últimos segmentos abdominais –
seta (fase crônica) (adaptado de NUNES, MARTINS & GESTEIRA, 2004).
1.2 VÍRUS DA MIONECROSE INFECCIOSA
Quando comparado a outros vírus que infectam camarão o IMNV apresenta sintomas mais brandos que os vírus da Taura (Taura syndrome virus - TSV), e que os vírus causadores da Mancha Branca (White spot syndrome virus - WSSV) e da Cabeça Amarela (Yellow head virus - YHV). Animais desafiados pelos vírus TSV, WSSV e YHV, em condições de laboratório, apresentam 100% de mortalidade acumulada 10 dias após desafio enquanto os animais desafiados com IMNV só atingem taxas semelhantes 52 dias após o desafio (LU et al., 1994; OVERSTREET et al., 1997; TANG & LIGHTNER, 2000; TANG et al., 2005). A maioria dos vírus patogênicos que acometem camarões pode causar infecções persistentes, mas em baixo nível. Em decorrência disso, geralmente, o cultivo dos camarões infectados por IMNV não é interrompido, permitindo o estabelecimento do vírus em animais saudáveis e gerando grandes prejuízos advindos de gastos com alimentação e manejo.
susceptíveis ao IMNV: L. vannamei, de maior interesse comercial no Brasil, e as espécies L. stylirostris, Farfantepenaeus subtilis e Penaeus monodon (LIGHTNER et al., 2004; TANG et al., 2005).
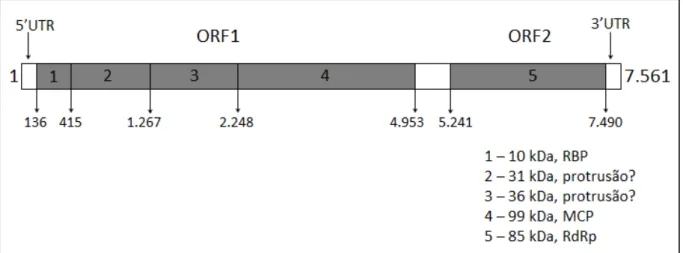
Figura 2: Organização do genoma do IMNV mostrando 5’ e 3’UTR e as duas ORFs não sobrepostas. 1 – Proteína de ligação ao dsRNA (10 kDa); 2 – Protrusão? (31 kDa); 3 – Protrusão? (36 kDa); 4 – Proteína principal do capsídeo (99 kDa); 5 – RNA polimerase dependente de RNA (85 kDa) (adaptado de POULOS et al., 2006; NIBERT, 2007).
Posteriores análises na sequência genômica revelaram a presença de dois motivos 2A-like (GDVESNPGP e GDVEENPGP) na ORF1 do IMNV precedendo a proteína do capsídeo. Os nonapeptídeos GDVESNPGP e GDVEENPGP abrangem os aminoácidos 86-94 e 370-378, respectivamente. A presença desses dois motivos reforça o pressuposto de que a ORF1 é uma poliproteína que posteriormente é clivada gerando fragmentos consecutivos de 93, 284 e 1.228 aa. Esse último fragmento provavelmente também é clivado gerando a MCP (901 aa) e outro fragmento de 327 aa. O fragmento de 93 aa possivelmente corresponde à proteína de ligação a dsRNA (RBP) e os fragmentos de 284 e 327 aa corresponderiam às proteínas de 32kDa e 38 kDa, respectivamente, vistas em gel desnaturante (NIBERT, 2007).
É provável que, em alguns vírus, as duas ORFs estejam sobrepostas em 199 nucleotídeos, sendo traduzidas conjuntamente e gerando uma proteína de 1.734 aminoácidos (196 kDa) que seria, posteriormente, clivada. Isso é possível se ocorrer
frameshifti ribossomal -1 é frequentemente associado a um motivo “shifty heptamer”, XXXYYYZ, onde X é A, C, G ou U, Y é A ou U, e Z é A, C ou U (BEKAERT et al., 2003; JACKS et al., 1988). A sequência GGGUUUU, que se qualifica como um “shifty heptamer” foi encontrada na região de sobreposição das ORFs do IMNV
(NIBERT, 2007). Além disso, imediatamente antes dessa sequência se encontra o dinucleotídeo UC que pode favorecer o frameshifting ribossomal -1 (BEKAERT & ROUSSET, 2005). Esta estratégia de codificação é semelhante aos vírus que infectam Giardia lamblia (WANG et al., 1993; LI, WANG & WANG, 2001) e Saccharomyces cerevisae (DINMAN, ICHO & WICKNER, 1991).
1.2.1 Proteína de ligação a RNA (RBP)
O primeiro produto codificado pela ORF1 (93 aa) apresenta uma região de 60 aminoácidos na extremidade N-terminal que compartilha um domínio conservado de ligação a dsRNA (DSRM) (POULOS et al., 2006). Esses domínios são encontrados em uma variedade de proteínas de ligação a RNA, apresentando diferentes estruturas e funções (BURD & DREYFUSS, 1994).
Hepatite C, a RBP NS5A pode desempenhar um papel significante na regulação da passagem do processo de tradução para replicação do genoma, recrutamento das polimerases e helicases para o RNA viral e aumento nas processividades das mesmas (HUANG et al., 2005). Além do seu papel nos processos de replicação e transcrição, as RBPs virais também parecem aumentar a resposta imune inata iniciada pelo receptor Toll-like 3 (TLR3) (LAI et al., 2011).
1.2.2 Proteína Principal do Capsídeo (MCP)
três subunidades B de decâmeros adjacentes participam de interações assimétricas em torno de cada eixo triplo (Figura 3B). Todos os contatos entre as subunidades A e B são assimétricos. Essas unidades assimétricas são formadas pelo dímero MCP-A/B, totalizando 60 unidades diméricas formando o capsídeo. Com relação ao RNA dupla fita encontrado dentro do capsídeo, este parece se organizar de forma helicoidal, de maneira bastante simétrica (Figura 3A) (TANG et al., 2008).
Figura 3: Reconstrução em imagem 3D do IMNV a uma resolução de 8.0 Å. (A) Secção central com valores de densidade codificados em escala de cinza (quanto mais escuro, mais denso). Os eixos de simetria estão indicados no quadrante superior direito. (B) Visão da superfície do IMNV mostrando a simetria icosaédrica do capsídeo e as subunidades A (azul e ciano) e B (vermelho, rosa e laranja) (adaptado de TANG et al., 2008).
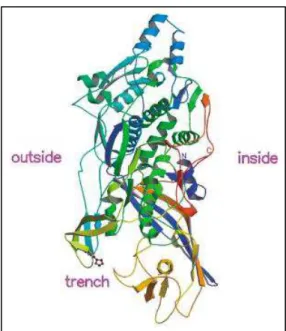
Dados estruturais de alta resolução sobre as proteínas do capsídeo de vírus da família Totiviridae são escassos. Somente uma estrutura em cristal disponível foi resolvida, para a MCP do vírus que infecta Saccharomyces cerevisae L-A (ScV-L-A) (Figura 4) (NAITOW et al., 2002). Como observado no capsídeo do IMNV e de outros totivírus, o capsídeo do ScV-L-A é formado a partir de 60 cópias de pseudo capsômeros, cada um composto de duas moléculas de MCP que possuem sequências de aminoácidos idênticas mas suas estruturas terciárias são diferentes, principalmente nos resíduos localizados na superfície da proteína. Cada molécula de
MCP é formada por duas α-hélices localizadas próximas aos eixos icosaédricos quíntuplos e várias folhas β próximas aos eixos duplos e triplos (NAITOW et al.,
2002).
Figura 4: Modelo em 3D da subunidade A do capsídeo de Saccharomyces cerevisae L-A (ScV-L-A). A
figura mostra o “trench” acessível a partir da superfície da partícula (outside) e o resíduo de His 154.
As extremidades N- e C-terminais estão voltadas para o interior do capsídeo (inside) (adaptado de
NAITOW et al., 2002).
1.2.3 Protrusões virais
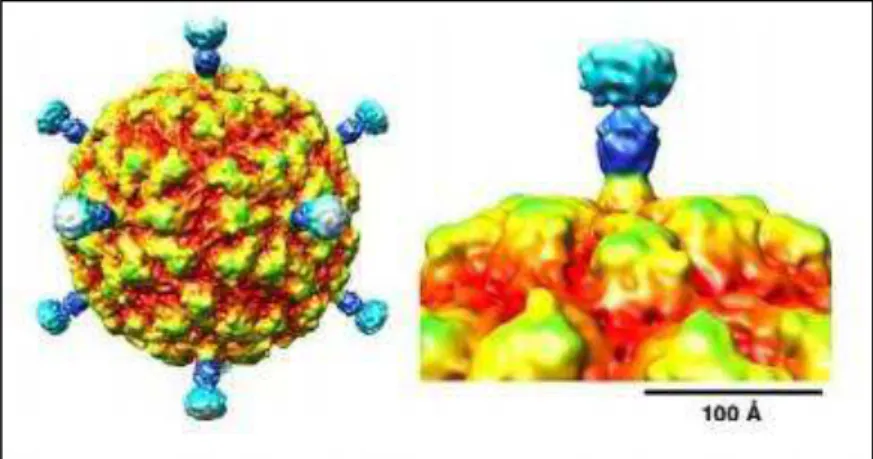
Análises de reconstrução de imagem em 3D mostraram que o capsídeo do IMNV possui fibras complexas que se estendem a aproximadamente 80 Å dos eixos quíntuplos do capsídeo (Figuras 3 e 5). Esses elementos mostraram menos densidade e resolução do que o capsídeo, o que pode ser explicado pela sua flexibilidade lateral e compreendem pelo menos três domínios morfológicos: knob, mais externo; stalk, no meio e; foot mais interno e ancorado ao capsídeo. O comprimento total de cada protrusão, incluindo o domínio foot, é de aproximadamente 100 Å (Figura 5) (TANG et al., 2008).
Figura 5: Estrutura tridimensional prevista para o vírus da mionecrose infecciosa. À direita, um detalhamento da estrutura da protrusão do capsídeo viral (adaptado de TANG et al., 2008).
hospedeiros. Outros membros da família Totiviridae estão associados com infecções avirulentas, enquanto que o IMNV causa uma doença geralmente fatal em camarões peneídos (LIGHTNER et al., 2004; POULOS et al., 2006; GHABRIAL, 2008; TANG et al., 2008). Entretanto, é possível que o totivírus que infecta o salmão do Atlântico, Piscine myocarditis virus (PMCV), também possua essas estruturas. O PMCV causa uma doença fatal em seu hospedeiro e, assim como o IMNV, presume-se que sua transmissão seja extracelular. Além disso, esse vírus possui sequências codificantes extras em seu genoma que podem estar envolvidas nos mecanismos de entrada celular (NIBERT & TAKAGI 2013). O IMNV foi o primeiro vírus da família Totiviridae descoberto como sendo capaz de infectar um hospedeiro diferente de protozoários ou fungos e, até o momento, ele é o único membro dessa família capaz de infectar crustáceos.
Alguns estudos têm mostrado que as regiões que codificam protrusões apresentam grande variabilidade genética em diferentes vírus, permitindo que eles se adaptem a diferentes situações (CAVANAGH, DAVIS & MOCKETT, 1988; LEE et al., 2010; PROMKUNTOD et al., 2014). Já se sabe que proteínas semelhantes a protrusões estão envolvidas na entrada de certos vírus, como os coronavírus, na célula hospedeira, além de desempenhar um papel importante na determinação da gama de hospedeiros (ENJUANES et al., 2006; PERLMAN & NETLAND 2009; BELOUZARD et al., 2012).
1.2.4 RNA Polimerase Dependente de RNA (RdRp)
A ORF2 do genoma do IMNV apresenta um domínio conservado característico de RNA polimerases dependente de RNA. As RdRps desempenham um papel central na replicação dos vírus de dsRNA. Apesar das altas taxas de mutações que ocorrem em vírus de RNA, as RdRps são geralmente bastante conservadas nas famílias virais, especialmente dentro de motivos funcionais, e são bons alvos para estudos filogenéticos (O’REILLY & KAO, 1998). Análises comparativas utilizando a região da RdRp do IMNV o caracterizam como um membro da família Totiviridae, similar ao vírus que infecta o protozoário Giardia lamblia (POULOS et al., 2006; NIBERT, 2007). Estudos sobre estrutura e função de RdRps tem sido relatados para vários vírus como Rhinovírus (LOVE et al., 2004), Picornavírus (KOK & MCMINN, 2009) e o vírus da Hepatite C (WAHEED, BHATTI & ASHRAF, 2013). A primeira estrutura em cristal da RdRp foi resolvida para o poliovírus podendo ser comparada com outras polimerases (Figura 6) (HANSEN, LONG & SCHULTZ, 1997).
As estruturas das RNA polimerases (RNAPs) geralmente apresentam uma arquitetura comum descrita como uma “right hand”, contendo os domínios “thumb”, “fingers” e “palm” (Figura 6). O domínio “palm” parece catalisar a reação de
transferência do grupo fosforila durante o processo de replicação enquanto que o domínio “fingers” está envolvido nas interações envolvendo os trifosfatos de nucleosídeos e pareamento do template. O domínio “thumb” provavelmente desempenha um papel na processividade e translocação da RNAP (STEITZ, 1999). Em RdRps, o domínio “palm” apresenta quatro sequências-motivo encontradas em
adicional E que é única de RdRps e transcriptases reversas (RTs) (Figura 6) (POCH et al., 1989). Outras três sequências-motivo (F1, F2 e F3) também encontradas em RdRps, parecem estar envolvidas na separação da dupla fita do RNA para a transcrição (BRUENN, 2003). Os motivos A e C estão envolvidos na coordenação do magnésio; o motivo A também pode desempenhar um papel na discriminação entre ribose e desoxirribose, assim como o motivo B; o motivo D completa a estrutura do domínio “palm”; e o motivo E está envolvido em interações hidrofóbicas com o domínio “thumb” (O’REILLY & KAO, 1998).
Figura 6: Estrutura da RNA polimerase dependente de RNA do poliovírus. Os domínios “thumb”,
“fingers” e “palm” estão indicados. As sequências-motivo encontradas no domínio “palm” estão
1.3 VARIABILIDADE GENÉTICA EM VÍRUS
O estudo da variabilidade genética é uma abordagem interessante quando se deseja compreender mais profundamente a biologia dos seres vivos. Os vírus de RNA sofrem grande taxa de mutação principalmente devido a falta de atividade de correção de erros da RNA polimerase. O conhecimento de que alterações em regiões específicas do genoma de diferentes vírus influenciam em sua capacidade de infecção e mecanismos de patogenicidade já é amplamente difundido. As primeiras evidências surgiram do estudo de viróides, que tinham sua capacidade de infecção, replicação e patogenicidade afetadas por alterações induzidas em seu genoma de RNA (TABLER & SFINGER, 1985; SANO et al., 1992). Hoje existem inúmeros trabalhos que associam a variabilidade genética de diferentes vírus a sua patogenicidade. Alguns exemplos clássicos em doenças humanas são a correlação da variabilidade observada no gene NS5A do vírus causador da hepatite C e a resistência do vírus ao interferon, a correlação entre polimorfismos no gene que codifica a transcriptase reversa do HIV (Human Immunodeficiency Virus) e alterações na propagação e resistência do vírus, e a presença de polimorfismos associados à infectividade na ORF26 do herpesvírus causador do Sarcoma de Kaposi (ENOMOTO et al., 1996., CHAYAMA et al., 1997; KUROSAKI et al., 1997; ZONG et al., 2007; KEARNEY et al., 2008; BITTAR et al., 2010; PINHO et al., 2010).
estudo desenvolvido por McBridge & Panganiban (1996) relatou-se que estruturas hairpin estão envolvidas no encapsulamento do vírus HIV-1. Foi visto que perturbações em um único hairpin, seja por deleção ou substituição de bases, pode ter um efeito parcial sobre o encapsulamento. Porém, quando dois hairpins são afetados, os efeitos são mais significativos.
O estudo de variabilidade genética em vírus que acometem animais de criação é fundamental para caracterização de cepas virulentas e desenvolvimento de vacinas. Análises de variabilidade genética de proteínas estruturais de diferentes linhagens do vírus causador da encefalite equina (Western equine encephalitis virus - WEEV), uma doença que é problema de saúde pública nos Estados Unidos, possibilitaram definir os principais aminoácidos responsáveis pelos mecanismos de virulência deste vírus (NAGATA et al., 2006). O vírus da doença infecciosa da bursa (IBDV), causador da doença de gumboro, uma das principais doenças para a avicultura brasileira, pode ser classificado de acordo com sua antigenicidade e patogenicidade pela região hipervariável do gene VP2, que codifica uma proteína do capsídeo viral (LANA, BEISEL & SILVA, 1992). A diversidade genética de sete isolados no vírus Sacbrood na China foi estudada e regiões hipervariáveis no gene VP1 já foram sequenciadas e caracterizadas a fim de se obter informações sobre epidemiologia e imunologia do vírus (MINGXIAO et al., 2013).
Indo-Pacífico permitem a diferenciação de pelo menos seis diferentes linhagens do vírus, sendo que, apenas duas dessas linhagens provocam a doença (WIJEGOONAWARDANE et al., 2008).
Existe apenas um trabalho voltado para o estudo da variabilidade genética do IMNV (NAIM, BROWN & NIBERT, 2014), publicado recentemente. Entretanto, até o momento, não foram observados estudos que levem em consideração a variabilidade genética do IMNV no desenvolvimento de métodos diagnósticos capazes de identificar diferentes isolados do vírus.
1.4 SISTEMAS DE DETECÇÃO PARA O IMNV
Os crustáceos parecem não possuir imunidade adaptativa nem memória imunológica e não existem drogas ou vacinas contra as doenças virais que acometem camarões. O sistema de defesa dos camarões depende principalmente da imunidade inata a qual é formada por defesas humorais e celulares (KURTZ, 2004; JIRAVANICHPAISAL, LEE & SÖDERHÄLL, 2006). O monitoramento e o controle da disseminação de doenças na carcinicultura dependem do desenvolvimento de ferramentas rápidas e específicas para detecção e caracterização dos patógenos.
LIU et al., 2013). Recentemente foram desenvolvidas outras ferramentas promissoras para detecção do IMNV, como anticorpos monoclonais para detecção de proteínas virais (KUNANOPPARAT et al., 2011; MELLO et al., 2011; CHAIVISUTHANGKURA et al., 2013) e sistemas de detecção baseados em RT-LAMP (Reverse Transcription Loop Mediated Isothermal Amplification) (ANDRADE & LIGHTNER, 2009; PUTHAWIBOOL et al., 2009; ARUNRUT, SUEBSING & KIATPATHOMCHAI, 2013). Além dessas ferramentas, métodos de imunização baseados em RNA de interferência (RNAi) estão sendo desenvolvidos para o monitoramento e controle do IMNV (LOY et al., 2012; LOY et al., 2013).
Apesar dos avanços recentes nos sistemas de detecção do patógeno, ainda não existem métodos direcionados à identificação da variabilidade genética do IMNV. O monitoramento em campo revela que em algumas amostras dadas como positivas não se observa sintomas nos animais, e geralmente existem diferenças na gradação dos sintomas e evolução da doença em diferentes regiões. Essas diferenças podem estar associadas a variações ambientais e/ou a diferentes variantes do vírus. O monitoramento e o controle de doenças virais que acometem a carcinicultura dependem de ferramentas rápidas e precisas para detecção e caracterização do patógeno.
desses dados é possível inferir como essas variações poderiam afetar a biologia do vírus e desenvolver novos sistemas de detecção mais eficientes.
2 OBJETIVOS
Geral:
Estudar o genoma completo do IMNV e os possíveis produtos gênicos codificados e desenvolver um sistema de detecção baseado em PCR que permita diferenciar isolados do vírus com base na presença de polimorfismos.
Específicos:
Verificar as relações de parentesco entre membros da família Totiviridae utilizando sequências já disponíveis no GenBank;
Identificar a incidência do IMNV em camarões coletados no Nordeste;
Sequenciar e montar o genoma completo de pelo menos dois isolados diferentes do IMNV;
Comparar os genomas obtidos entre si e com genomas completos do IMNV já depositados no GenBank;
Analisar a localização de sítios polimórficos e verificar se esses sítios causam alterações em aminoácidos e/ou em suas características;
Identificar regiões variáveis e conservadas presentes no genoma do vírus;
Estudar in silico a composição de aminoácidos e as estruturas
secundárias das proteínas que compõem o genoma do IMNV;
Modelar in silico a estrutura terciária das proteínas RBP, MCP e RdRp;
Desenhar primers específicos com base nas regiões variáveis e
conservadas identificadas no genoma;
Desenvolver métodos de diagnósticos para diferenciar os isolados
3 MATERIAIS E MÉTODOS
3.1 ANÁLISE FILOGENÉTICA
indels e sítios não informativos (missing data) no alinhamento foram tratados como deleções parciais, com um corte de 75%, para permitir potenciais regiões ambíguas nas topologias, resultado em 530 sítios de parcimônia informativos no dataset. A inferência Bayesiana foi conduzida em três corridas (runs) independentes, com modelos fixos LG ou WAG, taxas de distribuição gama entre os sítios e frequências de aminoácidos fixas. Cada Cadeia de Markov foi iniciada com uma árvore aleatória e rodada por 106 gerações, com dados amostrados a cada 100 gerações, e a árvore consenso foi estimada usando um “burn in” de 1.000.000 árvores. A convergência das corridas foi avaliada usando a ferramenta Tracer versão 1.5 (DRUMMOND & RAMBAUT, 2007), a fim de avaliar estatisticamente a robustez da análise Bayesiana. As árvores geradas pelos programas foram editadas no programa FigTree versão 1.4.1 (DRUMMOND & RAMBAUT, 2007).
3.2 OBTENÇÃO DAS AMOSTRAS DE CAMARÃO
3.3 SCREENING INICIAL PARA DETECÇÃO DO IMNV EM DIFERENTES AMOSTRAS DE CAMARÃO
Inicialmente, para a detecção dos vírus nas amostras de camarão, foram utilizadas duas estratégias: (i) PCR-Nested, utilizando primers desenvolvidos por Senapin et al. (2007), especificamente os pares de primers F13/R13 e F13N/R13N e; (ii) o método de identificação tradicional que utiliza os primers desenvolvidos pela World Organization for Animal Health (OIE). Após as análises iniciais, a identificação dos vírus foi realizada empregando o par de primers F14/R14. As sequências dos primers usados em cada análise estão listadas na Tabela 1.
As amostras de camarões, possivelmente infectadas foram analisadas como descrito a seguir.
Tabela 1: Lista dos primers usados em cada análise para identificar o IMNV nas amostras de
camarões Litopenaeus vannamei.
Primer Sequência (5’ →3’) Referência
F13 TTTATACACCGCAAGAATTGGCCAA
Senapin et al., 2007
R13 AGATTTGGGAGATTGGGTCGTATCC
F13N TGTTTATGCTTGGGATGGAA
R13N TCGAAAGTTGTTGGCTGATG
4587F CGACGCTGCTAACCATACAA
OIE
4914R ACTCGGCTGTTCGATCAAGT
4725NF GGCACATGCTCAGAGACA
4863NR AGCGCTGAGTCCAGTCTTG
F14 GAGTACCATCAGGAGTGAGAATAAC
Senapin et al., 2007
3.3.1 Extração de RNA total
A extração de RNA total foi realizada utilizando o KIT NUCLEOSPIN®RNA II, seguindo as instruções do fabricante. Aproximadamente, 50 µg de tecido foram retirados do sexto segmento abdominal do camarão. Em seguida, o tecido foi macerado, vortexado e submetido às etapas de centrifugação descritas no manual do kit de extração. Para as amostras de lisados de células de camarão conservadas congeladas ou em etanol 70%, foram utilizados 30 µl e 60 µl de amostra, respectivamente.
3.3.2 RT-PCR
3.3.2.1 Síntese de cDNA
A síntese de cDNA foi realizada seguindo o protocolo da enzima
3.3.2.2 Reação em Cadeia da Polimerase (PCR)
A reação em cadeia da polimerase foi realizada de acordo com o protocolo da Taq Biotools, com algumas modificações. As amplificações foram conduzidas em volume final de 20 µl contendo 10,8 µl de água Milliq, 2 µl de Reaction Buffer 10X, 1 µl de MgCl2 50 mM, 0,8 µl de dNTP mix 5 mM cada, 0,4 µl de Taq polimerase (5 U/µl), 4 µl de cDNA e 1 µl de primer. Para a amostra conservada em etanol 70%, as quantidades de água e cDNA foram 7,8 µl e 7 µl, respectivamente.
Os ciclos térmicos empregados nas análises variaram conforme os primers usados:
(i) Desnaturação inicial a 94 ºC por 2 minutos, seguido por 30 ciclos de desnaturação a 94 ºC por 40 segundos, anelamento a 45 ºC por 40 segundos, extensão a 72 ºC por 40 segundos, e extensão final a 72 ºC por cinco minutos. Na reação nested, o ciclo correspondia a uma desnaturação inicial a 94 ºC por 5 minutos, seguido por 25 ciclos de desnaturação a 94 ºC por 45 segundos, anelamento a 50 ºC por 45 segundos, extensão a 72 ºC por 30 segundos, e extensão final a 72 ºC por cinco minutos (primers F13/R13 e F13N/R13N desenvolvidos por Senapin et al., 2007).
(iii) Desnaturação inicial a 94 ºC por 2 minutos, seguido por 30 ciclos de desnaturação a 94 ºC por 30 segundos, anelamento a 55 ºC por 45 segundos, extensão a 72 ºC por 45 segundos, e extensão final a 72 ºC por cinco minutos (primers F14/R14 desenvolvidos por Senapin et al., 2007).
Os produtos da amplificação foram visualizados em gel de agarose 1%, na presença de brometo de etídio. Os tamanhos dos fragmentos foram determinados através de um marcador de peso molecular DNA Marker Hae III (Sigma) e visualizados em transluminador UV.
3.4 AMPLIFICAÇÃO E SEQUENCIAMENTO DO GENOMA VIRAL
Para a amplificação do genoma viral completo, as etapas de extração de RNA total, síntese de cDNA e técnica de PCR procederam conforme descrito anteriormente. O ciclo térmico empregado foi o mesmo daquele utilizado para os primers F14/R14.
Figura 7: Estratégia empregada para amplificar o genoma completo do IMNV (adaptado de SENAPIN
et al., 2007).
Tabela 2: Lista de primers usados para amplificar o genoma completo do IMNV (SENAPIN et al.,
2007).
Região Nome do primer foward /Sequência Nome do primer reverse/Sequência
1 F1/GGCAATTTCAACCTAATTCTAAAAC R1/TGAAAAATAAGCTGTGCCCCATGTT
2 F2/AATACTACATCATCCCCGGGTAGAC R2/GACTTTCTTCCCAAGATGGAGTCTC
3 F3/GAAGTTAAAGATGTAACACTTGCCT R3/ATACTCCTTCTCCAAAGGGTGTACG
4 F4/GATCCAGTTCTAACTAGAGAAGATA R4/TCCAGATACAATTACCATGCTGGTT
5 F5/GCAGCTTGGCTAAACAACAGACCAT R5/ATTCAATCCACGAATTTGTCTTGGT
6 F6/CTTCGTGATAATGACTCTATTAGGG R6/CTGTGGAACAGATTGTAAAGTAAGA
7 F7/ATAAATAATGGTGTTAATATATTTG R7/ACTAATTGGCAGTGTTGTTTTCATT
8 F8/GTTGGTGTGGCCCTGCCAACTGTAA R8/ACTACCTTGCATTGAACTCCACGAA
9 F9/GTTTGGTATTCAACAAGACGTATTT R9/AACATTAATACAACTCTCATCATGA
10 F10/GAGACAGGCAATGTATTCAGACCAT R10/CTCTTGCTGACTCGGCTGTTCGATC
11 F11/TCGGGTTTTATGAATGCCCGTTCCA R11/TTGATAACTGTTTTGCAATTTCAAT
12 F12/TTGTACAAAACATTTGTATCTATAT R12/CTTCGATGTTAGATGCCACAGCAAG
13 F13/TTTATACACCGCAAGAATTGGCCAA R13/AGATTTGGGAGATTGGGTCGTATCC
14 F14/GAGTACCATCAGGAGTGAGAATAAC R14/GATGTATGTCCTCTACGTTAACCAA
15 F15/AATATCTAGAATTGCCAAAACGACT R15/CATGGCTGGCCACAAAACCCAACTG
Os produtos de PCR foram enviados à empresa Hellixa para serem purificados e submetidos ao sequenciamento usando a plataforma da Applied Biosystems® 3500 Genetic Analyzer, de acordo com as especificações do fabricante. As concentrações dos produtos da amplificação foram mensuradas previamente a fim de se verificar se os mesmos estavam na concentração adequada para o sequenciamento. Os fragmentos foram sequenciados através do método de sequenciamento Sanger, utilizando os primers descritos na Tabela 2 na concentração de 5 µM cada primer. A reação de sequenciamento ocorreu em ambas às direções, foward e reverse, para cada região. As regiões do genoma foram sequenciadas no mínimo duas vezes (forward + reverse) e no máximo seis vezes até a obtenção de uma sequência confiável. As análises dos eletroferogramas e a montagem dos genomas foram realizadas usando parâmetros pré-definidos no programa Geneious versão 6.1.6 (Disponível em Biomatters) e inspeção visual cuidadosa.
3.5 ANÁLISE DAS SEQUÊNCIAS
As sequências genômicas completas obtidas foram analisadas no Basic Local
(LARKIN et al., 2007), com parâmetros default, usando o software Jalview versão 2.8 (WATERHOUSE et al., 2009) para identificação dos sítios polimórficos e regiões conservadas. Diferenças no nível de similaridade entre as regiões genômicas foram plotadas pelo programa SimPlot versão 3.5.1 (LOLE et al., 1999), com os seguintes parâmetros: modelo de árvore Neighbor joining, bootstrap igual a 1000 e modelo de distância Kimura (2-parameter). Adicionalmente, a fim de investigar a similaridade entre os genomas, uma análise de agrupamento, utilizando o método de Neighbor joining foi feita no software MEGA versão 6.06 (TAMURA et al., 2013), usando o modelo de substituição nucleotídica de Tamura-Nei e 1000 repetições de bootstrap, como teste de confiança na topologia. Para esta análise, além das sequências completas obtidas no presente estudo, foram usadas as seguintes sequências disponíveis no GenBank: números de acesso JN391187.1, AB555544.1, NC_013499.1, NC_014609.1, AY570982, EF061744.1.
3.6 ANÁLISE ESTRUTURAL DE PROTEÍNAS
A predição de regiões estruturadas e não estruturadas foi realizada através do software Foldindex© que prediz se uma sequência de proteínas é intrinsecamente desdobrada implementando o algorítimo de Uversky e colaboradores, o qual é baseado na hidrofobicidade média dos resíduos e carga líquida das sequências (PRILUSKY et al., 2005). Para a predição de coiled coils foi usado o programa Coils (LUPAS, VAN DYKE & STOCK, 1991) com windows 14, 21 e 28. O programa
predizer estruturas em α-hélices e folhas β, assim como regiões da proteína expostas ou não a solvente.
3.7 MODELAGEM PROTEICA
A predição de modelos estruturais foi realizada com auxílio dos programas I-TASSER (servidor online) (ZHANG, 2008; ROY et al,. 2010) e Phyre2 (KELLEY & STERNBERG, 2009). A metodologia do I-TASSER é baseada na predição por homologia de estrutura e alinhamento por threading das sequências alvo com base nas estruturas disponíveis na biblioteca do Protein Data Bank – PDB (http://www.rcsb.org/pdb/home/home.do), complementando com simulações ab initio para as cadeias laterais (ZHANG, 2007). O programa I-TASSER gera modelos classificados pelo Z-Score, RMSD, densidade do cluster e, principalmente pelo C-score. O C-score corresponde ao escore de confiança que estima a qualidade do modelo gerado pelo I-TASSER cujo valor pode ser de -5 a 2 (quanto maior o valor do C-score, maior será a confiança do modelo). Com relação ao programa Phyre2, este realiza modelagem basicamente por homologia (KELLEY & STERNBERG, 2009).
Os modelos foram avaliados usando os softwares MOLPROBITY (CHEN et al., 2010) e SWISS-MODEL (ARNOLD et al., 2006). Para a visualização dos modelos, foram usados os softwares UFSC Chimera (PETTERSEN et al., 2004) e PyMOL versão 1.5.0.4 (http://www.pymol.org/).
3.8 SISTEMAS DE DETECÇÃO DE POLIMORFISMOS
3.8.1 Teste de detecção baseado nas regiões variáveis e conservadas
Pares de primers foram desenhados com base em uma região conservada e uma região variável do genoma. Todos os primers foram analisados nos programas PrimerBlast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) para verificar o TM (temperatura de melting) e anelamento inespecífico, e AutoDimer (VALLONE & BUTLER, 2004) para verificar a formação de hairpins e dímeros de primers.
Um método de PCR convencional foi desenvolvido para identificar as duas regiões escolhidas. Após a síntese de cDNA, foi realizada a PCR como descrito a seguir: os primers (5 µM cada) foram usados em 20 µl de reação contendo 10,8 µl de água Milliq, 2 µl de Reaction buffer 10X, 1 µl de MgCl2 50 mM, 0,8 µl de dNTP mix 5 mM cada, 0,4 µl de Taq polimerase 5 U/µl e 4 µl de cDNA. A reação foi submetida à desnaturação inicial a 94 ºC por 2 minutos, seguido por 30 ciclos de 94 ºC por 30 segundos, 58 ºC por 45 segundos, 72 ºC por 45 segundos, e extensão final a 72 ºC por 5 minutos. Os amplicons gerados foram de 200 pb para a região conservada e 456 pb para a região variável. Os produtos da PCR foram analisados em gel de agarose 1,5% na presença de brometo de etídio, e visualizados em transluminador.
3.8.2 Teste de detecção baseado na análise da curva de melting de fita simples
Seis regiões no genoma (cada uma com 200 pb) foram selecionadas como candidatas para o sistema de identificação de polimorfismos através da análise da curva de melting de fita simples. Essas regiões foram submetidas à análise in silico no programa RNAfold (http://rna.tbi.univie.ac.at) para predição da estruturas secundárias de RNA e DNA, e nos softwares MELTSIM (BLAKE et al., 1999) e RNAheat (HOFACKER et al., 1994) para a geração de curvas de melting de DNA fita dupla e RNA fita simples, respectivamente. Os seis pares de primers (sequências não mostradas) foram desenhados e analisados usando os programas PrimerBlast e AutoDimer.
As análises in vitro procederam da seguinte maneira, em duas etapas:
(I) Em um volume final de 20 µl foram adicionados 10,8 µl de água Milliq, 2 µl de Reaction Buffer 10X, 1 µl de MgCl2 50 mM, 0,8 µl de dNTP mix 5 mM cada, 0,4 µl de Taq polimerase (5 U/µl), 4 µl de cDNA e 1 µl de primer (5 µM cada). O protocolo de termociclagem compreendia um passo inicial a 94 ºC por 2 minutos, seguido por 30 ciclos de desnaturação a 94 ºC por 30 segundos, anelamento a 55 ºC por 45 segundos, extensão a 72 ºC por 45 segundos, e extensão final a 72 ºC por cinco minutos.
(II) 4 µl do produto da primeira reação foi utilizado como template em um volume final de 20 µl contendo 10 µl de mix SYBR green, 1 µl de primer
extensão final a 72 ºC por cinco minutos. Os parâmetros da curva de dissociação foram padrões.
4 RESULTADOS
4.1 ANÁLISE FILOGENÉTICA DA FAMÍLIA TOTIVIRIDAE
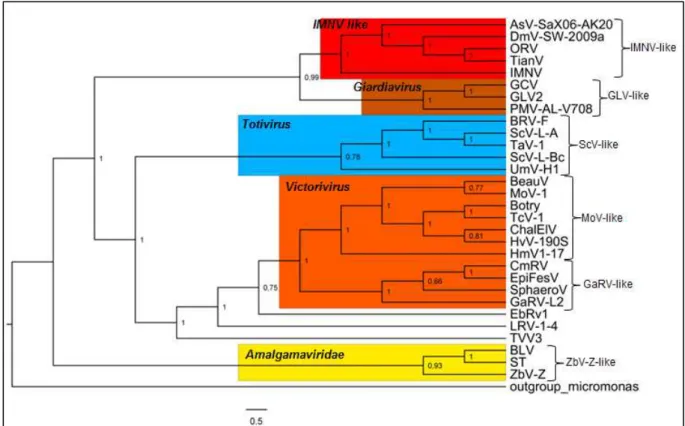
Figura 8: Análise filogenética de membros representativos da família Totiviridae. A árvore foi calculada a partir do alinhamento da sequência de aminoácidos da RdRp de 30 membros representativos da família Totiviridae, usando Inferência Bayesiana. As IDs das sequências estão
indicadas nas Tabelas Suplementares 1 e 2. Os números indicam a consistência topológica e a confiança de cada ramo. As chaves indicam os grupos identificados neste estudo e nomeados de acordo com Liu et al. (2012) e as cores representam os gêneros de acordo com a ICTV.
4.2 SCREENING INICIAL PARA DETECÇÃO DO IMNV EM DIFERENTES AMOSTRAS DE CAMARÃO
4.3 AMPLIFICAÇÃO E MONTAGEM DOS NOVOS GENOMAS
Dois novos genomas de isolados diferentes do IMNV foram amplificados e sequenciados no presente estudo. Os isolados foram nomeados IMNVgen1 (coletado em 2009 no estado de Pernambuco/BR) e IMNVgen2 (coletado em 2013 no estado do Rio Grande do Norte/BR). As sequências dos genomas completos estão mostradas nas Figuras Suplementares 2 e 3.
A estratégia empregada para obtenção da sequência nucleotídica completa de IMNVgen1 e IMNVgen2 foi aquela desenvolvida para o genoma proveniente da Indonésia descrita por Senapin et al. (2007). Exceto pelos últimos 43 nucleotídeos em um dos genomas, a qualidade das bases foi alta, em torno de 50 (> 20), equivalente ao escore Phred (EWING & GREEN, 1998), indicando que a sequência genômica foi confiável para os dois isolados.
4.4 CARACTERIZAÇÃO DOS NOVOS GENOMAS
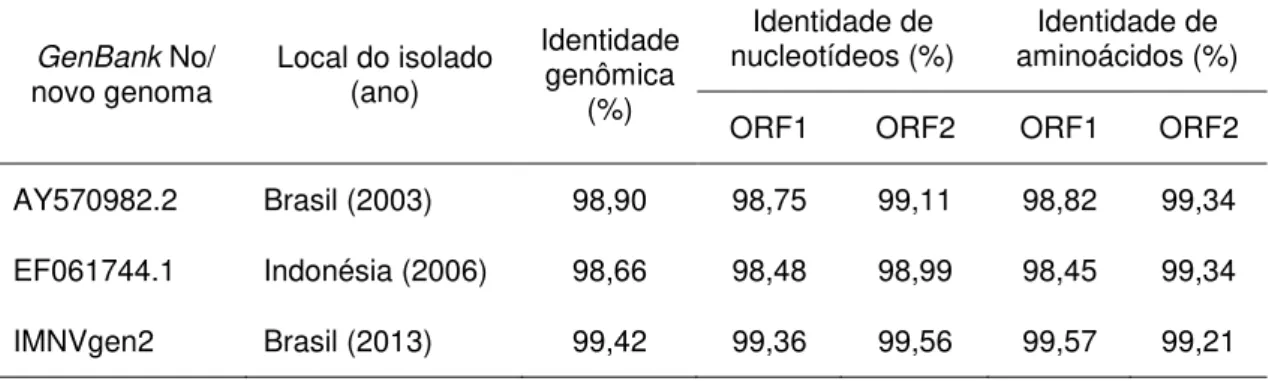
A, 25,3% de T, 20,0% de G e 17,9% de C. O conteúdo G/C e A/T do genoma foi calculado para ser 1,12% e 1,45%, respectivamente. Como mostrado na Tabela 3, IMNVgen1 é muito similar a IMNVgen2 (99,42% de identidade), assim como é muito parecido com os isolados do Brasil e Indonésia. Valores de identidade semelhantes foram observados quando IMNVgen2 foi comparado com esses mesmos isolados (Tabela 4).
Tabela 3: Comparação das sequências de nucleotídeos e aminoácidos do IMNVgen1 com outras sequências disponíveis no GenBank e IMNVgen2.
GenBank No/
novo genoma Local do isolado (ano)
Identidade genômica
(%)
Identidade de
nucleotídeos (%) aminoácidos (%) Identidade de
ORF1 ORF2 ORF1 ORF2
AY570982.2 Brasil (2003) 98,90 98,75 99,11 98,82 99,34
EF061744.1 Indonésia (2006) 98,66 98,48 98,99 98,45 99,34
IMNVgen2 Brasil (2013) 99,42 99,36 99,56 99,57 99,21
Tabela 4: Comparação das sequências de nucleotídeos e aminoácidos do IMNVgen2 com outras sequências disponíveis no GenBank e IMNVgen1.
GenBank No/
novo genoma Local do isolado (ano)
Identidade genômica
(%)
Identidade de
nucleotídeos (%) aminoácidos (%) Identidade de
ORF1 ORF2 ORF1 ORF2
AY570982.2 Brasil (2003) 98,90 98,75 99,11 98,82 99,21
EF061744.1 Indonésia (2006) 98,67 98,48 98,99 98,38 99,21
As sequências codificantes de IMNVgen1 e IMNVgen2 estão organizadas em duas ORFs, como esperado para um típico totivírus e de acordo com os genomas já publicados (POULOS et al., 2006; SENAPIN et al., 2007). Em ambos, a ORF1 apresentou 4.818 nucleotídeos, começando no nucleotídeo 136, e a ORF2 apresentou 2.250 nucleotídeos, começando no nucleotídeo 5.241, estando as ORFs em diferentes frames. A sequência aminoacídica da ORF1 dos novos genomas foi pelo menos 98,38% similar as sequências dos isolados do Brasil e Indonésia (Tabela 4). Com relação à sequência aminoacídica da ORF2, IMNVgen1 mostrou 99,34% de similaridade com os genomas já publicados, enquanto que IMNVgen2 mostrou 99,21% de similaridade.
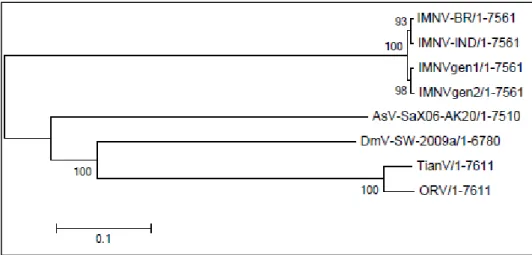
A análise de agrupamento por Neighbor joining usando os quatro genomas completos dentro do grupo IMNV-like gerou um dendograma com dois grupos bem definidos para o IMNV: um grupo composto pelos vírus sequenciados neste estudo e outro compreendendo os genomas previamente descritos (Figura 9).
Figura 9: Análise Neighbor joining do grupo IMNV-like incluindo os quatro genomas completos do
IMNV. O dendograma foi calculado baseado no alinhamento da sequência de nucleotídeos, usando o modelo Tamura-Nei e 1000 replicações de Bootstrap. As análises foram realizadas no programa
MEGA 6.06. IMNV - Penaeid shrimp infectious myonecrosis virus; AsV - Armigeres subalbatus virus;
4.5 IDENTIFICAÇÃO DE REGIÕES CONSERVADAS E VARIÁVEIS
Figura 10: Representação esquemática do genoma do IMNV e análise de variabilidade. Os plots de
similaridade gerados no Simplot comparam IMNVgen1 e IMNVgen2 com os genomas disponíveis no
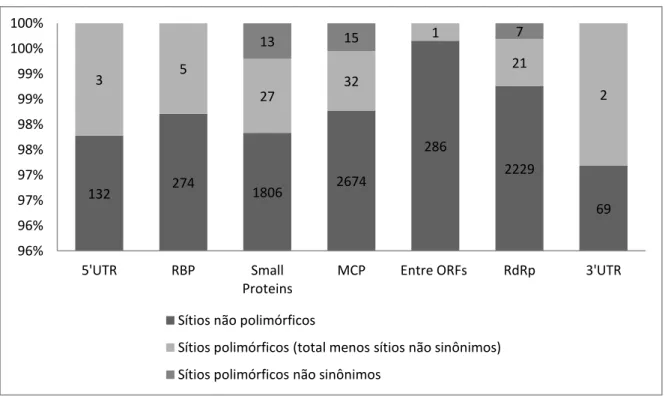
Os resultados obtidos no alinhamento das sequências nucleotídicas e aminoacídicas mostrou a presença de 126 sítios polimórficos, dos quais 5 estão na proteína de ligação a dsRNA (RBP), 40 nas regiões das Small Proteins (SP1 e SP2), 47 na proteína do capsídeo e 28 na RdRp. Desses 126 sítios polimórficos, 37 causam alteração em aminoácidos, estando 13 nas regiões de SP1 e SP2, 15 na proteína do capsídeo e 7 na RdRp (Figura 11 e Tabela Suplementar 4). Levando em consideração a região codificante do genoma, verificamos que, proporcionalmente, a maior densidade de sítios polimórficos, inclusive aqueles que alteram aminoácidos, se encontram nas regiões da SP1 e SP2 (~1 polimorfismo a cada 46 nt), seguido pela proteína do capsídeo (~1 polimorfismo a cada 57 nt) e pela RdRp (~1 polimorfismo a cada 80 nt). Os polimorfismos encontrados na região da proteína de ligação da dsRNA não alteram o quadro de leitura dos aminoácidos, constituindo mutações silenciosas.
Figura 11: Sítios polimórficos identificados no genoma do IMNV. O número total de sítios polimórficos compreende os sítios polimórficos sinônimos e não sinônimos mostrados proporcionalmente em cada região do genoma.
132 274 1806 2674
286
2229
69
3 5
27
32
1
21
2
13 15 7
96% 96% 97% 97% 98% 98% 99% 99% 100% 100%
5'UTR RBP Small Proteins
MCP Entre ORFs RdRp 3'UTR
Sítios não polimórficos
Sítios polimórficos (total menos sítios não sinônimos)
Com base nos resultados obtidos no Simplot e no alinhamento das quatro sequências, foi possível mapear os sítios polimórficos ao longo do genoma e, assim identificar quais regiões eram mais conservadas e quais eram mais variáveis. Os dados indicam que as regiões previstas para codificarem a protrusão do vírus (SP1 e SP2) variaram mais em relação às outras regiões, enquanto que a região da RdRp apresentou-se mais conservada. Nesse sentido, podemos dizer que as proteínas que são candidatas a formarem a protrusão do vírus são mais susceptíveis a sofrerem mudanças do que as outras proteínas do IMNV e, provavelmente apresentam uma região hipervariável.
4.6 ANÁLISE DE COMPOSIÇÃO DE AMINOÁCIDOS E PREDIÇÃO DE ESTRUTURA SECUNDÁRIA
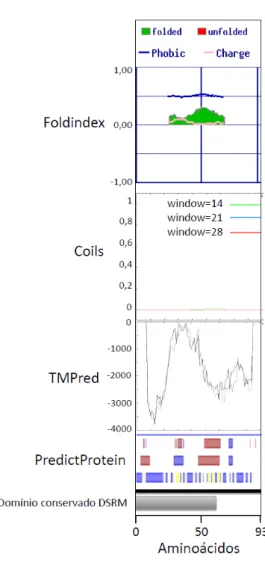
Figura 12: Análise de composição de aminoácidos e predição de estrutura secundária para RBP. A predição de regiões estruturadas (verde) e desestruturadas (vermelho), coiled coils e hélices
transmembrana foram realizadas usando os programas Foldindex, Coils e TMPred, respectivamente.
O software PredictProtein foi usado para predizer a presença de α-hélices (caixas rosas) e folhas β
(caixas azuis) (primeira e segunda linha), e exposição a solvente (caixas azuis indicam exposição a solvente e caixas amarelas indicam não exposição a solvente). Abaixo, uma representação esquemática mostrando o domínio conservado DSRM (em cinza claro). O eixo X representa a posição dos aminoácidos.
mostrou-se uma proteína quase totalmente desestruturada, com alta quantidade de aminoácidos carregados, alta hidrofilicidade e uma pequena região estruturada na porção C-terminal. Além disso, a região desestruturada que apresentou o maior pico coincidiu com uma região não exposta a solvente. SP2 também mostrou uma região de coiled coil com um escore significante na porção N-terminal, que foi predito em diferentes reading windows (Figura 13 e Figura Suplementar 4C).
Figura 13: Análise de composição de aminoácidos e predição de estrutura secundária para SP1 e SP2. A predição de regiões estruturadas (verde) e desestruturadas (vermelho), coiled coils e hélices
transmembrana foram realizadas usando os programas Foldindex, Coils e TMPred, respectivamente.
O software PredictProtein foi usado para predizer a presença de α-hélices (caixas rosas) e folhas β
A proteína principal do capsídeo (MCP) apresentou um número considerável de sítios polimórficos, sendo superada apenas pela região das proteínas SP. De acordo com o esperado para uma proteína globular, ela contém uma alta proporção de aminoácidos neutros (31%) e hidrofóbicos (50,5%). Esta proteína, formada por 901 aminoácidos, apresentou alto grau de regiões estruturadas e duas regiões desestruturadas mais evidentes na sua segunda metade. A região desestruturada entre os aminoácidos 804 a 846 contém alto conteúdo de α-hélices predito pelos programas Coils e PredictProtein (Figura 14 e Fig. Suplementar 4D). As análises in silico também revelaram cinco picos hidrofóbicos preditos pelo TMPred. A comparação entre os quatro genomas do IMNV mostrou que a região correspondente a MCP possui 15 sítios onde ocorrem mudanças de aminoácidos. Esses sítios estão distribuídos ao longo da proteína e alguns coincidem com os picos hidrofóbicos.
Figura 14: Análise de composição de aminoácidos e predição de estrutura secundária para MCP. A predição de regiões estruturadas (verde) e desestruturadas (vermelho), coiled coils e hélices
transmembrana foram realizadas usando os programas Foldindex, Coils e TMPred, respectivamente.
O software PredictProtein foi usado para predizer a presença de α-hélices (caixas rosas) e folhas β
A RNA polimerase dependente de RNA mostrou-se uma proteína muito conservada entre os quatro isolados apresentando apenas sete sítios polimórficos não-sinônimos. As análises in silico revelaram que a RdRp possui características de uma proteína estruturada com pequenas regiões desestruturadas interpassadas, lembrando uma proteína com domínios conectados por pontes móveis (Figura 15 e Figura Suplementar 4E). Duas hélices hidrofóbicas preditas pelo TMPred estão localizadas em domínios estruturados e provavelmente estão escondidas na estrutura globular. A predição de estruturas secundárias revelou a existência de um alto conteúdo de α-hélices e dois motivos coiled coil com alta probabilidade de
ocorrência compreendendo os aminoácidos 243 a 256 e 636 a 649. Em ambos os motivos não foram detectadas mudanças de aminoácidos (Figura 15). A Figura 15 mostra que as sequências-motivo conservadas em RNA polimerases estão localizadas em regiões estruturadas e, exceto pela sequência-motivo F1, todas coincidem com a região do domínio conservado da RdRp.
Figura 15: Análise de composição de aminoácidos e predição de estrutura secundária para RdRp. A predição de regiões estruturadas (verde) e desestruturadas (vermelho), coiled coils e hélices
transmembrana foram realizadas usando os programas Foldindex, Coils e TMPred, respectivamente.
O software PredictProtein foi usado para predizer a presença de α-hélices (caixas rosas) e folhas β
4.7 MODELAGEM DE PROTEÍNAS
As estruturas terciárias das proteínas RBP, MCP e RdRp potencialmente codificadas pelo isolado da Indonésia foram preditas utilizando os programas I-TASSER e Phyre2. Os valores para o escores e parâmetros de avaliação dos modelos são mostrados na Tabela Suplementar 5. Não foi possível obter modelos com escores de confiabilidade significantes para as proteínas SP1 e SP2. Os modelos da MCP e RdRp também foram preditos para os três isolados do Brasil (Figuras Suplementares 5 e 6). Não foram gerados modelos da RBP para os isolados do Brasil devido à sequência de aminoácidos da proteína ser idêntica para os quatro isolados.
Figura 16: Modelo estrutural para a proteína RBP do IMNV gerado pelo I-TASSER. O domínio conservado DSRM está destacado em cinza claro. (MolProbity score 4,03, ZScore 0,627, Cscore -2,26).
Figura 17: Modelo estrutural para a MCP do IMNV gerado pelo I-TASSER. O core conservado da MCP está destacado em cinza claro. Os sítios polimórficos não sinônimos e suas posições estão indicados em vermelho. (MolProbity score 3,47, Z-Score -6,341, C-score -2,27).
Figura 18: Modelo estrutural para a RdRp do IMNV gerado pelo I-TASSER. O domínio conservado da RdRp está destacado em cinza claro. Os sítios polimórficos não sinônimos e suas posições estão indicados em vermelho. Os domínios “thumb”, “fingers”, e “palm” estão mostrados, assim como as
sequências-motivo conservadas em RdRps estão destacadas como segue: A – azul escuro; B – azul claro; C – rosa; D – verde; E – amarelo; F1, F2 e F3 – laranja. (MolProbity score 3,96, Z-Score -5,14, C-score 0,23).
4.8 SISTEMAS DE DETECÇÃO DE POLIMORFISMOS
4.8.1 Teste de detecção baseado nas regiões variáveis e conservadas
para ser usado como controle positivo da reação e amplifica um fragmento de 600 pb presente no gene do fator de elongação-1α do camarão.
Tabela 5: Lista de primers empregados no sistema de identificação do IMNV.
Nome do primer Sequência (5’→3’) TM (ºC)
SP-F CATCAAGTGTACTCGGAAG 52,85
SP-R CTTCTCTAGTTAGAACTGGATC 52,90
RdRp-F ACGTAGAGGACATACATCC 53,05
RdRp-R GATTGGTATTGTCTGATTTCTAC 53,02
EF1α-F GCGCAAGAGCGACAACTATG 59,97
EF1α-R ACATAGGCTTGCTGGGAACC 60,03
Figura 19: Alinhamentos de nucleotídeos dos fragmentos de 456 pb e 200 pb referentes às regiões variável e conservada, respectivamente. Esses fragmentos foram empregados no sistema de detecção do IMNV. Os sítios polimórficos de cada região estão mostrados em caixas.
Figura 20: Sistema de detecção do IMNV. (A) Gel de agarose mostrando os resultados do sistema de identificação do IMNV utilizando os novos marcadores para as regiões variável e conservada do genoma. O sistema foi capaz de identificar a presença do IMNV em duas amostras diferentes do camarão coletadas em diferentes locais do Brasil. 1 – marcador de peso molecular; 2 e 6 – controle positivo (EF-1α); 3 e 7 – controle negativo; 4 e 8 – detecção usando os primers da região mais
variável; 5 e 9 – detecção usando primers da região conservada. Os asteriscos indicam as bandas de
456 pb e 200 pb esperadas. (B) Análise Neighbor joining baseado no fragmento de 456 pb da região
mais variável. O dendograma foi calculado baseado no alinhamento da sequência de nucleotídeos, usando o modelo Tamura-Nei e 1000 replicações de Bootstrap. As análises foram realizadas no
programa MEGA 6.06.
4.8.2 Teste de detecção baseado na análise da curva de melting de fita simples