by DNA Polymerase POLQ
Matthew J. Yousefzadeh1,2, David W. Wyatt3, Kei-ichi Takata1,2, Yunxiang Mu1, Sean C. Hensley1, Junya Tomida1,2, Go¨ran O. Bylund4, Sylvie Doublie´5, Erik Johansson4, Dale A. Ramsden3,
Kevin M. McBride1,2, Richard D. Wood1,2*
1Department of Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, Texas, United States of America,2The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, Texas, United States of America,3Lineberger Comprehensive Cancer Center, Department of Biochemistry and Biophysics and Curriculum in Genetics and Molecular Biology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, United States of America, 4Department of Medical Biochemistry and Biophysics, Umea˚ University, Umea˚, Sweden,5Department of Microbiology and Molecular Genetics, The University of Vermont, Burlington, Vermont
Abstract
Although a defect in the DNA polymerase POLQ leads to ionizing radiation sensitivity in mammalian cells, the relevant enzymatic pathway has not been identified. Here we define the specific mechanism by which POLQ restricts harmful DNA instability. Our experiments show thatPolq-null murine cells are selectively hypersensitive to DNA strand breaking agents, and that damage resistance requires the DNA polymerase activity of POLQ. Using a DNA break end joining assay in cells, we monitored repair of DNA ends with long 39single-stranded overhangs. End joining events retaining much of the overhang were dependent on POLQ, and independent of Ku70. To analyze the repair function in more detail, we examined immunoglobulin class switch joining between DNA segments in antibody genes. POLQ participates in end joining of a DNA break during immunoglobulin class-switching, producing insertions of base pairs at the joins with homology toIgH switch-region sequences. Biochemical experiments with purified human POLQ protein revealed the mechanism generating the insertions during DNA end joining, relying on the unique ability of POLQ to extend DNA from minimally paired primers. DNA breaks at theIgHlocus can sometimes join with breaks inMyc, creating a chromosome translocation. We found a marked increase inMyc/IgHtranslocations inPolq-defective mice, showing that POLQ suppresses genomic instability and genome rearrangements originating at DNA double-strand breaks. This work clearly defines a role and mechanism for mammalian POLQ in an alternative end joining pathway that suppresses the formation of chromosomal translocations. Our findings depart from the prevailing view that alternative end joining processes are generically translocation-prone.
Citation:Yousefzadeh MJ, Wyatt DW, Takata K-i, Mu Y, Hensley SC, et al. (2014) Mechanism of Suppression of Chromosomal Instability by DNA Polymerase POLQ. PLoS Genet 10(10): e1004654. doi:10.1371/journal.pgen.1004654
Editor:Peter McKinnon, St Jude Children’s Research Hospital, United States of America ReceivedJune 2, 2014;AcceptedAugust 5, 2014;PublishedOctober 2, 2014
Copyright:ß2014 Yousefzadeh et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability:The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding:This research was supported by NIH training grant T32 CA09480 (MJY), NIH grants CA09717 (RDW), CA097096 (DWW, DAR), CA52040 (SD), and grant RP130297 from the Cancer Prevention and Research Institute of Texas (RDW). The M.D. Anderson Research Trust and the Grady F. Saunders Ph.D. Distinguished Research Professorship (RDW), Leukemia SPORE CA100632 (KMM), Swedish Research Council (EJ), the Swedish Cancer Society (EJ), and Insamlingstiftelsen vid den medicinska fakulteten vid Umea˚ Universitet (EJ) also supported this work. We acknowledge NIH Cancer Center Support Grant P30-CA016672 (University of Texas M. D. Anderson Cancer Center). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Competing Interests:The authors have declared that no competing interests exist.
* Email: [email protected]
Introduction
A diverse group of at least 16 DNA polymerases carry out DNA replication, repair, and damage tolerance in the mammalian genome [1,2]. One of these is DNA polymerase theta (POLQ). POLQ homologs are found in multicellular eukaryotes including plants, but an equivalent enzyme is absent from yeast [3]. The large 290 kDa human POLQ protein has an unusual bipartite structure with an N-terminal helicase-like domain and a C-terminal DNA polymerase domain [4]. This domain arrange-ment and the POLQ protein sequence is highly conserved in vertebrates [3].
Several functions have been suggested for POLQ [3] including bypass of template DNA lesions such as abasic sites and thymine
mice showing severe growth retardation [13]. From this observa-tion it was suggested that POLQ has a unique role in maintaining genomic stability that is distinct from the major homologous DNA recombination pathway regulated by ATM [13].
DNA double-strand breaks (DSBs) can be formed in cellular genomes by environmental agents such as ionizing radiation. DSBs also arise during normal cellular duplication cycles, when DNA replication stalls at naturally occurring structures or at sites of internally-generated DNA damage. In diversification steps of the mammalian immune system, DSBs are deliberately formed by regulated enzymatic action, to initiate rearrangement of antibody and receptor segments, and as a means to introduce local variation. Because DSBs are toxic and/or mutagenic if not repaired, organisms have multiple mechanisms for DSB repair [15,16]. The primary strategies are end-joining mechanisms, which process and rejoin the ends of a DSB, and homologous recombination (HR) pathways which employ an undamaged copy of the DNA [17]. End-joining pathways appear to be the first line of defense again DSBs. The most studied pathway is ‘‘classical’’ non-homologous end-joining (cNHEJ), which relies on the DNA-binding Ku70 (XRCC6) and Ku80 (XRCC5) gene products, and the DNA protein kinase (DNA-PK, PRKDC). One or more ‘‘alternative’’ end-joining pathways (altEJ) also exist, which are independent of these factors [18,19]. During immunoglobulin diversification, the regional end-joining process of class switch recombination (CSR) replaces one constant region coding exon for another. This CSR process is known to occur through both cNHEJ and alternative end joining pathways [20]. In mammalian cells, an alternative end-joining repair pathway repair of DSBs is thought to play a role in driving the formation of chromosomal translocations, although the specific enzymology is unclear [21,22].
Here, we report experiments that define a specific function and mechanism of action for POLQ in a pathway for alternative end-joining of DNA double-strand breaks in mammalian cells.
Results
Hypersensitivity to DNA strand-breaking agents in the absence ofPolq
To clarify the cellular role of POLQ in response to DNA damage, we measured the sensitivities of Polq-null and Polq -proficient bone marrow stromal cell (BMSC) lines to various DNA damaging agents. Cells lacking POLQ exhibit hypersensitivity to ionizing radiation (Figure 1) [12,23], and to the double-strand break-inducing chemical bleomycin, as previously reported [12].
We found thatPolq2/2 cells were also hypersensitive to other
agents which directly cause DNA breaks, including the topoisom-erase II inhibitors etoposide and ICRF-193 [24] and camptothe-cin, a topoisomerase I inhibitor. In contrast, loss ofPolqdid not cause hypersensitivity to agents that largely form DNA replication-blocking adducts on one DNA strand including ultraviolet radiation and the methylating agent temozolomide. ThePolq2/2
cells were also not more sensitive than control cells to mitomycin C, cisplatin, and UVA-photoactivated psoralen plus UVA, all of which induce some interstrand DNA crosslinks (ICLs) (Figure 1).
These data indicate that POLQ is most important in a process conferring resistance to direct DNA strand-breaks, particularly double-strand breaks. Cells lackingPolqwere not hypersensitive to the PARP inhibitor olaparib (Figure 1) while control RAD51D-defective cells were hypersensitive (Figure S1A). This suggests that POLQ does not function in the BRCA/homologous recombina-tion pathway [25]. POLQ-proficient cells treated with both olaparib and camptothecin were significantly sensitized compared to camptothecin alone. However, addition of olaparib toPolq-null cells only modestly increased the sensitivity to camptothecin (Figure 1). Consequently, PARP and POLQ may operate within the same subpathway of DNA repair.
Loss ofPolqenhances chromosomal instability in somatic cells
It is important to know whether the elevated level of micronuclei inPolq-defective cells extends to cell types other than peripheral erythrocytes. To answer this question, matched wild-type andPolq2/2 BMSC lines were exposed to etoposide or
x-rays, and the number of cells with micronuclei after 48 h were enumerated (Figure 2A and B).Polq-null cells exhibited a,3 fold
increase in frequency of spontaneous micronuclei formation (Figure 2C). Upon exposure to DNA damaging agents, the percentage of cells with micronuclei increased about 1.5-fold more per unit of damage forPolq2/2cells in comparison toPolq+/
+
cells (Figure 2A and B). This shows that the susceptibility to micronucleus formation inPolq2/2cells is not confined to cells of the hematopoietic lineage, but occurs also in cultured cells, including fibroblast-like BMSCs.
Cells lackingPolqwere analyzed for their ability to proliferate in culture. Two independent BMSC lines devoid ofPolqexpression proliferated at a rate comparable to a pair of wild-type control cells, thePolqBMSCs showing only a 5% increase in population doubling times (Figure 2D and E). We extended this analysis to isogenic immortalized mouse embryonic fibroblast (MEF) cell lines (Figure 2F and G).Polq2/2cells divided at a rate comparable to
Polq-proficient cells. These findings fit with our previous observations that hematopoietic cell counts in irradiatedPolq-null mice recovered at rates comparable to wild-type mice [12]. We have observed no major alterations in growth or development in unchallengedPolqnull or mutant mice, consistent with previous reports [13,14,26]. These observations indicate that despite some increased chromosomal instability, POLQ-defective cells originat-ing from a variety of tissues can proliferate at near-normal rates.
Author Summary
The DNA polymerase activity of POLQ is required to confer resistance to DNA damaging agents
We sought next to investigate which catalytic activities of POLQ are necessary to confer resistance to DNA damaging agents. Lentiviral-delivered expression vectors were constructed to express wild-type or mutant versions of POLQ in immortalized MEFs, in order to test for functional complementation (Figure 3A). A tandem (D2330A,Y2331A) mutation was introduced into the DNA polymerase domain (POL); mutation of the corresponding residues in other DNA polymerases completely inactivates polymerase activity [27]. In a separate construct, a mutation was introduced into the conserved ATP-binding site of the Walker A motif (K121M) in the helicase-like domain (HLD). An equivalent mutation eliminates DNA helicase activity in related enzymes, including HELQ [28]. A third construct (DM) was made harboring mutations in both domains. These vectors expressed full-length recombinant POLQ as tested in transfected 293T cells (Figure 3B and C).
The mutant cDNAs were tested for their ability to genetically complement the bleomycin sensitivity ofPolq-null MEFs. Stable clones with each of the constructs were generated and analyzed for expression of POLQ (Figure 3D). Independent clones of knockout MEFs expressing wild-type recombinant POLQ (WT4 and WT6) were able to rescue bleomycin hypersensitivity (Figure 3E) as an antibody that recognizes endogenous POLQ does not yet exist. Neither the polymerase domain mutant (POL) nor the polymer-ase-helicase double mutant (DM) restored bleomycin sensitivity
(Figure 3E, Figure S1B). Expression of a construct with a mutation only in the helicase-like domain (HLD) was, however, still able to restore resistance to bleomycin. These data indicate that POLQ polymerase activity is essential for conferring resistance to DNA damage, while the ATPase activity of the helicase-like domain is not necessary. Similarly reintroduction of polymerase activity of POLQ intoPolq-deficient MEFs was able to rescue chromosomal instability (micronuclei and DNA DSBs, as measured by 53BP1 andcH2AX colocalization (Figure 3F and 3G, Figure S2).
Mice with an S1932P mutation in Polq (the ‘‘chaos1’’ allele) have an increased spontaneous frequency of micronuclei [13]. We generated a humanPOLQcDNA mimicking thechaos1mutation (S1977P), but attempted expression of POLQ with this mutation in 293T cells did not yield detectable protein (Figure S3). This suggests that the chaos1-encoded mutant protein is unstable, consistent with the finding that chaos1 mice have a phenotype essentially indistinguishable fromPolqknockout mice [13].
POLQ operates in a pathway of altEJ during mouse Ig class-switching
Immunoglobulin class-switch recombination (CSR) uses DNA end joining to exchange one constant region of an antibody gene for another constant region. CSR can occur by both Ku-dependent classical non-homologous end joining and Ku-inde-pendent altEJ [20]. The overall frequencies of CSR are similar in
Polq-defective mice [29] and cultured B cells [30]. To determine whether POLQ is involved in a mechanistically distinct subset of
Figure 1. Hypersensitivity ofPolq2/2bone marrow stromal cells to DNA strand-breaking agents.BMSCs were exposed to x-rays or UVC
at the indicated doses, and to etoposide, ICRF-193, camptothecin, olaparib, temozolomide, mitomycin c, cisplatin, and HMT psoralen+UVA at the indicated concentrations and plated in triplicate. Two isogenic bone marrow stromal cell lines were used of each genotype,Polq+/+orPolq2/2. Colonies were crystal violet stained and counted seven to ten days later. Experiments were repeated three times. Circles,Polq+/+
clone 1; Squares, Polq+/+clone 1; Triangles,Polq2/2clone 1; inverted triangles,Polq2/2clone 3.
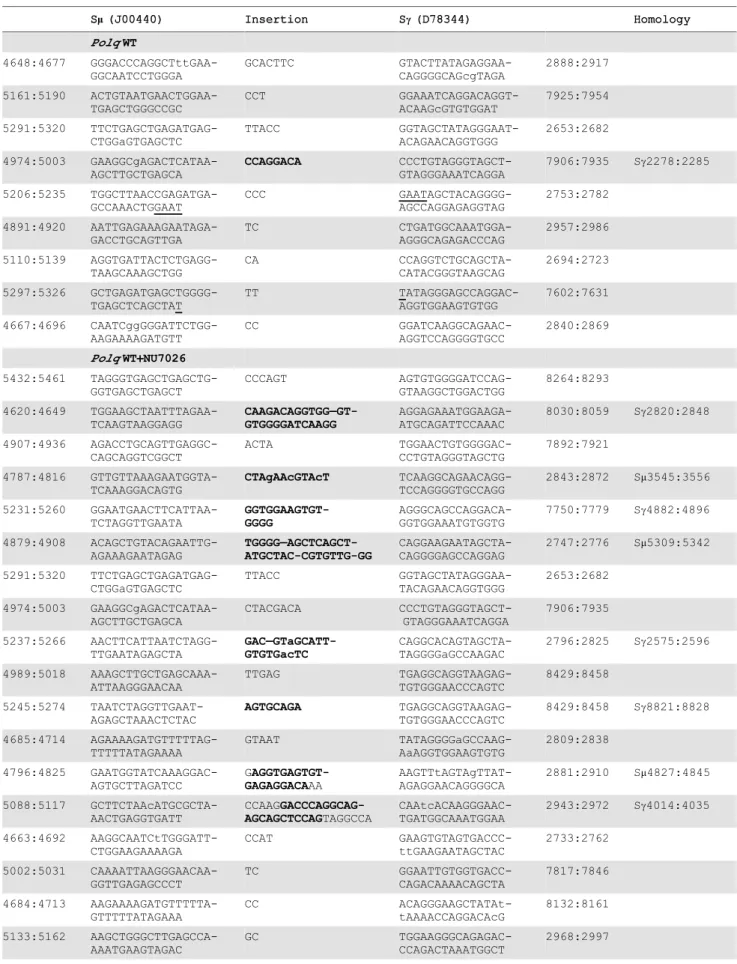
CSR joins, we isolated and analyzed DNA sequences at such joins. Naı¨ve B cells were isolated from the spleens of wild-type andPolq -null mice and stimulated for IgM to IgG class switching, and then the fraction of IgG1-positive B cells was measured by flow cytometry. Parallel B-cell cultures were incubated with NU7026, a DNA-PKcs inhibitor that suppresses cNHEJ [31]. It has been shown that B cells incubated with NU7026 have an increased proportion of CSR junctions with.1 bp insertion at the junction [31]. This suggests that when a pathway of altEJ is used during CSR, it more frequently results in insertion of nucleotides.
We found that B cells fromPolq-proficient and deficient mice had similar overall frequencies of CSR (Figure 4A), and inhibition of DNA-PKcs increased the frequency of CSR in both genotypes by 1.5 to 2 fold (Figure 4B). The Sm-Sc1 junction was then sequenced from 100 clones of each group of IgG1-positive B cells. These data revealed that in wild-type B cells, insertions of.1 bp at Sm-Sc1 junctions, that are thought to be altEJ-dependent, comprised about 9% of total events, and that this increased to
,21% in cells incubated with NU7026 (Figure 4C, Table 1).
Strikingly, in cells lackingPolq, this class of insertions at CSR junctions was absent, even in the presence of NU7026 (Figure 4D, only one insertion of 2 bp observed). Insertion of.1 bp therefore requires POLQ. This class of Polq-dependent joining events
included insertions of between 2 and 35 bp. For longer insertions (greater than,10 bp) homologous sequences were unambiguously
detected up to 2–5 kbp away from the junction site (Table 1), as has been reported for long insertions at Sm-Sc1 junctions in ATM-defective B cells [31]. This suggests that most or all of such insertions are formed in a templated manner during altEJ by POLQ.
Loss ofPolqimpairs an altEJ pathway but not cNHEJ in cultured cells
The most important factor in determining which double-strand break repair pathway is used is whether or not the 59 termini of broken ends are resected [32]. Ends with little or no single stranded overhang are typically rejoined by Ku-dependent cNHEJ. In contrast, CtIP and MRN-dependent resection of 59
termini generates ends with extended single stranded 39 over-hangs; resection is thought to block cNHEJ [33] and enable repair by altEJ [34,35].
To analyze differing requirements for end joining, with or without end resection, we generated two linear DNA substrates with 39 single stranded overhangs; one with a short overhang (6 nt), and one a long overhang (45 nt, a ‘‘pre-resected end’’) (Figure 5A). Both can be aligned with the same 4 nt of terminal complementary sequence. These substrates were then introduced
Figure 2. Loss ofPolqcontributes to chromosomal instability both spontaneously and in the presence of DNA damage.Polq+/+
or Polq2/2bone marrow stromal cells plated in chamber slides were exposed to (A) X-rays or (B) etoposide. 48 hr after damage cells were fixed and stained with DAPI to enumerate cells with micronuclei. Counts represent the average percentage of cells with micronuclei scored in three independent experiments. (Slopes for X-ray and etoposide-induced MN:Polq+/+clone 1 (2.8, 0.73);Polq+/+clone 2 (3.1, 0.85);Polq2/2clone 1 (4.8, 1.2);
Polq2/2clone 3 (6.2, 1.3). The frequency of spontaneous micronuclei for each of the clones in Figure 2A and 2B were combined to generate (C) total spontaneous micronuclei observed for all genotypes. The p-value was determined by Wilcoxon Mann Whitney rank sum test.Polq+/+orPolq2/2(D) bone marrow stromal cells and (F) mouse embryonic fibroblasts were plated in growth medium in triplicate. Cells were counted at the indicated days and cumulative population doublings were recorded. The experiment was repeated three times. (E and G) Absolute quantification ofPolqtranscript numbers in three independent experiments.
into wild-type mouse fibroblasts or fibroblasts harboring deficien-cies inKu70orPolq. Repaired products were recovered from cells and quantified. Repair of the short overhang substrate was, as anticipated, over 10-fold less efficient in cells without Ku70 (Figure 5B) when compared to Ku70-complemented controls. The absence ofPolq2/2had no consequence for repair of this substrate.
End joining with the 45 nt overhang substrate was assessed using qPCR primers located to ensure that at least 10 nt of overhang was included in joined products (Figure 5A). Recovery of these products was no longer dependent on Ku; instead, it was increased 2.8-fold in Ku70-deficient cells (Figure 5C). This is consistent with previous studies arguing Ku suppresses repair by
altEJ. Strikingly, joining of the long overhang substrate inPolq2/2
cells was reduced 10-fold, near background levels of signal observed using this assay. Complementation of the knockout cells with POLQ returned joining to wild-type levels (Figure 5C). These data demonstrate that POLQ participates in some form of alEJ, but cells lacking POLQ maintain proficiency for cNHEJ.
POLQ extends 39DNA ends in a template-dependent manner
Our results demonstrate that POLQ is necessary to form the insertions found in CSR junctions in a process of altEJ. We next
Figure 3. Complementation of the polymerase activity of POLQ rescues DNA damage hypersensitivity in cells lackingPolq.(A) POLQ cDNA was cloned into the pCDH-FH vector containing a FLAG-HA epitope tag on the c-terminus. 293T cells were transiently transfected with pCDH containing either empty-vector control or POLQ cDNA. (B) Crude extracts were immunoblotted with the indicated antibodies to confirm full-length expression of recombinant POLQ or (C) mutant constructs. (D) Stable MEF lines complemented with POLQ expression vectors (or empty vector control) were assayed forPolqexpression by qPCR. WT4 and WT6 are independent clones complemented by wild-type POLQ; POL, mutation in the DNA polymerase domain; HLD, mutation in the DNA helicase domain, DM, mutation in both domains. (E) The complemented MEF lines were treated with bleomycin for 24 hr and cellular ATP levels were measured 72 hr later. (F) Spontaneous micronuclei and (G) DNA double-strand breaks (.2 colocalizedcH2AX and 53BP1 foci per cell), quantified for three independent experiments. The brightness of the entire microscope field was increased to better display the fluorescence for publication, using Adobe Photoshop CS6.
sought to determine the mechanism. Like other DNA polymer-ases, an active polymerase fragment of POLQ [36] can catalyze template-dependent DNA synthesis from an annealed primer (Figure 6A). As is common for family-A DNA polymerases, only a single nucleotide is added to the end of duplex DNA [5]. Unusually, however, POLQ can catalyze extension of single-stranded oligonucleotides [37]. It was unclear whether this reflects a robust terminal deoxynucleotide transferase activity of POLQ on single-stranded DNA, or some form of template-dependent synthesis. For example, POLQ can extend a single-stranded 16-mer oligonucleotide provided without a complementary template (products up to 35 nt long), whileE. colipol I Klenow fragment has no activity on this substrate (Figure 6B). The major 22 nt extension product produced by POLQ on the 16-mer used in Figure 6B may be accounted for by inter- or intra-oligonucleotide pairing (Figure S4C). Neither POLQ nor Klenow fragment could extend an oligonucleotide that was incapable of annealing to itself (Figure S4) [37].
To identify the mechanism of 39single-stranded DNA extension by POLQ, we used a different single-stranded oligonucleotide designed to be unable to form self-complementary base pairs longer than a single nucleotide [37], and sequenced the products of POLQ-mediated extension. Individual extension products of 1 to 30 nt were found (Table S1). Most of the sequenced extension products feature AAC or AAAC sequences that could arise from copying GTTT sequences in the template via inter- or intra-molecular priming and re-priming (Figure 6C) following minimal base pairing at the 39-primer end. These data reveal that POLQ
uniquely extends 39DNA tails through template-dependent DNA synthesis from a primer with minimal base pairing and that the polymerase lacks true TdT-like activity. POLQ indeed has unique biochemical properties compatible with these observations. Unlike other DNA polymerases, POLQ can efficiently extend a DNA chain with a nucleotide incorporated opposite an abasic site [5], or from a mismatched primer-terminus [38]. Further, there is evidence that primers slip on DNA templates with an increased frequency during POLQ-mediated synthesis, as shown by the high frequency of single base pair frameshift mutations generated by purified POLQ [39].
POLQ suppresses chromosomal translocations in B cells Double-strand breaks initiated by AID activity in the immuno-globulin heavy chain (IgH) locus of B cells are necessary to generate immunological diversity, but breaks are sometimes generated at other chromosomal sites, providing an opportunity for dangerous chromosome translocations [21,22,40,41]. For instance the oncogenicMyc/IgHtranslocation that causes Burkitt lymphoma is AID-dependent and requires breaks at both loci, with breaks in theMycgene rate-limiting [42]. An altEJ process is implicated in the formation of oncogenic translocations in lymphoid tissues, including theMyc/IgHtranslocation in murine B cells [21,43,44]. cNHEJ suppresses the formation of such chromosomal translocations [45]. To determine the role of POLQ in chromosomal translocations,Polq+/+
and Polq2/2naı¨ve splenic B cells were stimulated in culture and assayed for the frequency of
Myc/IgH translocations (Figure 7A). Notably, in the absence of
Table 1.Sequence composition of.1 nucleotide CSR insertions.
Sm(J00440) Insertion Sc(D78344) Homology
PolqWT 4648:4677 GGGACCCAGGCTttGAA-GGCAATCCTGGGA GCACTTC GTACTTATAGAGGAA-CAGGGGCAGcgTAGA 2888:2917 5161:5190 ACTGTAATGAACTGGAA-TGAGCTGGGCCGC CCT GGAAATCAGGACAGGT-ACAAGcGTGTGGAT 7925:7954 5291:5320 TTCTGAGCTGAGATGAG-CTGGaGTGAGCTC TTACC GGTAGCTATAGGGAAT-ACAGAACAGGTGGG 2653:2682 4974:5003 GAAGGCgAGACTCATAA-AGCTTGCTGAGCA CCAGGACA CCCTGTAGGGTAGCT-GTAGGGAAATCAGGA
7906:7935 Sc2278:2285
5206:5235 TGGCTTAACCGAGATGA-GCCAAACTGGAAT CCC GAATAGCTACAGGGG-AGCCAGGAGAGGTAG 2753:2782 4891:4920 AATTGAGAAAGAATAGA-GACCTGCAGTTGA TC CTGATGGCAAATGGA-AGGGCAGAGACCCAG 2957:2986 5110:5139 AGGTGATTACTCTGAGG-TAAGCAAAGCTGG CA CCAGGTCTGCAGCTA-CATACGGGTAAGCAG 2694:2723 5297:5326 GCTGAGATGAGCTGGGG-TGAGCTCAGCTAT TT TATAGGGAGCCAGGAC-AGGTGGAAGTGTGG 7602:7631 4667:4696 CAATCggGGGATTCTGG-AAGAAAAGATGTT CC GGATCAAGGCAGAAC-AGGTCCAGGGGTGCC 2840:2869
PolqWT+NU7026
5432:5461 TAGGGTGAGCTGAGCTG-GGTGAGCTGAGCT CCCAGT AGTGTGGGGATCCAG-GTAAGGCTGGACTGG 8264:8293 4620:4649 TGGAAGCTAATTTAGAA-TCAAGTAAGGAGG CAAGACAGGTGG—GT-GTGGGGATCAAGG AGGAGAAATGGAAGA-ATGCAGATTCCAAAC
8030:8059 Sc2820:2848
4907:4936 AGACCTGCAGTTGAGGC-CAGCAGGTCGGCT ACTA TGGAACTGTGGGGAC-CCTGTAGGGTAGCTG 7892:7921 4787:4816 GTTGTTAAAGAATGGTA-TCAAAGGACAGTG CTAgAAcGTAcT TCAAGGCAGAACAGG-TCCAGGGGTGCCAGG
2843:2872 Sm3545:3556
5231:5260 GGAATGAACTTCATTAA-TCTAGGTTGAATA GGTGGAAGTGT-GGGG AGGGCAGCCAGGACA-GGTGGAAATGTGGTG
7750:7779 Sc4882:4896
4879:4908 ACAGCTGTACAGAATTG-AGAAAGAATAGAG TGGGG—AGCTCAGCT-ATGCTAC-CGTGTTG-GG CAGGAAGAATAGCTA-CAGGGGAGCCAGGAG
2747:2776 Sm5309:5342
5291:5320 TTCTGAGCTGAGATGAG-CTGGaGTGAGCTC TTACC GGTAGCTATAGGGAA-TACAGAACAGGTGGG 2653:2682 4974:5003 GAAGGCgAGACTCATAA-AGCTTGCTGAGCA CTACGACA CCCTGTAGGGTAGCT-GTAGGGAAATCAGGA 7906:7935 5237:5266 AACTTCATTAATCTAGG-TTGAATAGAGCTA GAC—GTaGCATT-GTGTGacTC CAGGCACAGTAGCTA-TAGGGGaGCCAAGAC
2796:2825 Sc2575:2596
4989:5018 AAAGCTTGCTGAGCAAA-ATTAAGGGAACAA TTGAG TGAGGCAGGTAAGAG-TGTGGGAACCCAGTC 8429:8458 5245:5274 TAATCTAGGTTGAAT-AGAGCTAAACTCTAC AGTGCAGA TGAGGCAGGTAAGAG-TGTGGGAACCCAGTC
8429:8458 Sc8821:8828
4685:4714 AGAAAAGATGTTTTTAG-TTTTTATAGAAAA GTAAT TATAGGGGaGCCAAG-AaAGGTGGAAGTGTG 2809:2838 4796:4825 GAATGGTATCAAAGGAC-AGTGCTTAGATCC G AGGTGAGTGT-GAGAGGACAAA AAGTTtAGTAgTTAT-AGAGGAACAGGGGCA
2881:2910 Sm4827:4845
5088:5117 GCTTCTAAcATGCGCTA-AACTGAGGTGATT CCAAG GACCCAGGCAG-AGCAGCTCCAGTAGGCCA CAAtcACAAGGGAAC-TGATGGCAAATGGAA
2943:2972 Sc4014:4035
Polq there was a 4-fold increase in translocation frequency (Figure 7B and C). This indicates that mammalian POLQ acts in a subset of altEJ events to suppress chromosomal translocations. Additionally, an increase in intramolecular IgHrearrangements was found in B cells lackingPolq(Figure 7B). Therefore, although POLQ is involved in an altEJ pathway, it prevents rather than promotes chromosomal instability, rearrangements and the formation ofMyc/IgHtranslocations.
Discussion
POLQ suppresses hypersensitivity to direct DNA double-strand breaks
We show that in mammalian cells, POLQ has a specific role in defense against DNA damaging agents that cause direct DNA double-strand breaks, including ionizing radiation, bleomycin, and topoisomerase inhibitors. Our findings indicate that POLQ participates in a novel pathway of alternative-end joining of DSBs, a process that can occur throughout the cell cycle in mammalian cells [17]. The minimal additional sensitization to camptothecin by olaparib inPolq-defective cells suggests that one function of PARP is to participate in a Polq-dependent altEJ pathway. Our experiments indicate that POLQ is an important factor in DNA DSB repair in all cells, not just cells of the hematopoietic lineage. Indeed, Polq is broadly expressed in murine tissues (Figure S5).
Mutants of POLQ homologs in Arabidopsis (TEBICHI), C. elegans (polq-1), and Drosophila (Mus308) are hypersensitive to ICL-inducing agents [3], whereasPolq-defective mammalian cells are not appreciably hypersensitive to such agents (Figure 1). This difference may arise because of differences between organisms in the priority of DNA repair pathway engagement. In proliferating mammalian cells, ICLs are usually dealt with through the Fanconi anemia pathway, which produces enzymatically induced double-strand breaks that are channeled into homologous recombination repair [46]. InDrosophila and some other organisms, an altEJ-dependent pathway may be more important for resolving ICL-associated double-strand breaks. Although Drosophila Mus308
mutants are not hypersensitive to IR, pronounced IR sensitivity occurs in a double mutant when HR is also inactivated [47]. The phenotypic consequences of POLQ-dependent altEJ of double-strand breaks may thus depend on the relative dominance of HR which varies between organisms.
We show here that the DNA polymerase activity of POLQ is necessary to prevent cell death and chromosome breaks (micro-nuclei) caused by a double-strand break-inducing agent. Disrup-tion of the ATPase activity in the helicase-like domain of POLQ did not, however, alter the correcting function of POLQ addition to knockout cells. A previous study with mouse cell lines suggested that disruption of the polymerase domain of the murinePolqgene is less severe than complete disruption ofPolq[30], but the result is difficult to evaluate in the absence of quantitative measurements of expression of the partially deleted form. No activity has yet been shown for the helicase-like domain, other than DNA-dependent ATPase function [4]. It is likely that an additional role remains to be discovered that is dependent on the ATPase function of POLQ.
POLQ aids DNA double-strand break repair through alternative end joining and nucleotide insertions
When double-strand breaks form in mammalian cells, a majority will be repaired through cNHEJ. However, a subset of these breaks will be handled by alternative end-joining pathways in situations where the DNA end is not compatible with processing by Ku-dependent cNHEJ, or if core components of the cNHEJ machinery are absent or unavailable (Figure 7D). In general, altEJ is defined as a means for repair of chromosome breaks that is exclusive of Ku-dependent, classically defined NHEJ [48], and dependent on factors (CtIP, MRN) that resect double-strand breaks to generate extended 39 ssDNA tails [34,35] (Figure 5A). Accordingly, we showed joining of a ‘‘pre-resected’’ extrachromo-somal substrate (substrate with 45 nucleotide 39-ssDNA tails) was stimulated inKu-deficient cells, similar to results using chromo-somal substrates [35]. Joining of this substrate was also dependent on Polq (Figure 5C). Our experiments thus define an altEJ subpathway in mammalian cells that involves POLQ (termed synthesis-dependent end joining, SD-EJ, in Figure 7D), Additional Polq-independent altEJ subpathways may also be operational (Figure 7D). To some extent, different end-joining pathways can be been distinguished from one another by the ligase employed in the pathway, with DNA ligase IV (LIG4) suggested as essential for cNHEJ, and DNA ligase III (LIG3) for altEJ in mammalian cells [21,43,49]. There are caveats, however. For example, some functional redundancy is apparent between LIG1 and LIG3 in altEJ [44,50–52]. Ligase deficiencies may thus not be the best marker for distinguishing different end-joining pathways. For the Table 1.Cont.
Sm(J00440) Insertion Sc(D78344) Homology
5210:5239 TTAACCGAGATGAGCCA-AACTGGAATGAAC
TA
AAAACCAGGACAGGA-GGAAGAATGGGGATC
8148:8177
4816:4845 GCTTAGATCCgAGGTGA-GTGTGAGAGGACA
AA
AAGTTtAGTAgTTATA-GAGGAACAGGGGCA
2881:2910
4897:4926 GAAAGAATAGAGACCTG-CAGTTGAGGCCAG
TT
AGGGTGTGGATCCAG-GCAGGGTAGCTATAG
2634:2663
PolqKO
5360:5389 AGCTACTCTGGAGTAGC-TGAGATGGGGTGA
TT
CAAATGGAAGGGCAG-AGACCCAGACTAAAT
2964:2993
30 bases flanking each side of the insertion are shown. The numbers separated by colons give the position relative to the beginning of the Smand Scgenomic sequences. New mutations (different from the reference sequence) are shown in lower case. For some longer insertions (indicated in bold), homologies were identified in the switch region, at the positions indicated in the right column. Microhomologies at the junction site are underlined. A dash (-) indicates a base deletion. Sequences (graphed in Figure 4E) are fromPolq+/+(WT) and
altEJ subpathway under consideration here, dependence on POLQ is the best available definition.
The biochemical properties of POLQ provide a mechanistic explanation for its contribution to altEJ. POLQ has a unique ability to add nucleotides to the 39ends of single-stranded DNA [37], primed by minimal pairing with other available DNA molecules (Figure 6 and Figure S6). Synthesis by POLQ in this context is consistent with the unusually efficient ability of the polymerase to extend from mismatched DNA termini [5,38], and its tendency towards primer-template slippage [39]. In further biochemical experiments it will be of interest to examine the action of POLQ and DNA ligases at double strand breaks with 39 -single-stranded overhangs that closely mimic the resected ends of a DNA double-strand break. In vivo studies with such substrates, including those that can form hairpins in the single-stranded region, would give insight as to the preferred structures for POLQ-catalyzed extension.
Unique to the POLQ-dependent altEJ process are frequent joins displaying templated DNA insertions. Some form of altEJ has been implicated in resolution of a subset of double-strand break intermediates in CSR, producing templated insertions [20]. Our data support a role for POLQ in generating the CSR products with these templated insertions. These events are consistent with the templated insertions that occur during Mus308-dependent repair of directed double-strand breaks inDrosophila[47,53] and inC. elegans[54]. In the absence of POLQ, the lack of insertion-containing joins is observed, but the global CSR frequency is relatively unchanged (Figure 4). These insertions are best explained by repeated initiation of synthesis by POLQ (Figure 6C) on template sites, ultimately leading to a joined product.
POLQ prevents the formation ofMyc/IgHchromosomal translocations
In the absence of POLQ, we found a,4-fold increase in the
formation of the oncogenic translocationMyc/IgHin mice. This increase is comparable to that seen in B cells that have lostTdrd3,
a regulator of R-loop formation during transcription [55] and
miRNA-155 which regulates AID and suppresses oncogenic translocations [56]. In the absence of Polq there is also an apparent enhancement of rearrangement events in theIgHlocus, consistent with the elevated level of chromosomal instability observed in cells lacking POLQ [57].
altEJ is typically associated with frequent annealing of the DNA ends at existing microhomologies (2–5 bp) and large deletions at repair junctions [19]. Since translocations commonly feature such microhomologies at their breakpoint junctions [58,59] and occur more frequently in cNHEJ defective cells, altEJ is considered the primary mechanism by which translocations occur. Thus, a striking finding of the present work is that the formation ofMyc/ IgHtranslocations is suppressed when the POLQ-dependent altEJ subpathway is operational. It is possible that DNA DSBs persist for a longer time in the absence of POLQ, giving more opportunity for the formation of translocations. Alternatively, the POLQ-dependent pathway may be the most efficient at repairing a structurally distinct class of translocation-prone DNA breaks.
These studies clearly define a role for POLQ in the repair of DNA strand-breaking agents and provide a mechanism of template-dependent extension of DNA ends necessary to repair breaks in a subpathway of altEJ. This distinct altEJ pathway is necessary to prevent the formation of chromosomal translocations as shown by ourin vivoexperiments. It has been suggested that suppression of POLQ may be useful in increasing the efficacy of DNA damaging treatments in cancer [3,23,60]. This promising prospect should be tempered with the knowledge that loss of POLQ may also lead surviving cells to be prone to potentially oncogenic chromosome translocations.
Materials and Methods
Ethics statement
Research mice were handled according to MD Anderson Cancer Center Institutional Animal Care and Use Committee
Figure 5. End joining with extrachromosomal substrates.(A) Substrates were designed to resemble DNA double-strand breaks that are repaired through Ku-dependent NHEJ (6 nt tail with 4 nt of terminal complementary sequence) or alternate end-joining of resected DNA substrates (45 nt tail with 4 nt terminal complementary sequence), introduced into cells, and joining of head-to-tail products assessed by qPCR. (B) qPCR for the classical NHEJ assay uses primers to detect all events having sequences in the duplex immediately flanking the break. Joining efficiencies are expressed as fractions of the mean joining determined for matched wild controls (Polq+/+or Ku70 complemented lines, as appropriate). Three
independent triplicate measurements were made for thePolqcell lines and two independent triplicates for theKucell lines. Error bars represent the standard error of the mean. Joining efficiency was not significantly different, whether cells were deficient inPolq(Polq2/2Empty) or not (Polq+/+, Polq2/2WT4,Polq2/2WT6), but was different when cells expressed Ku (Ku702/2Ku70) when compared toKu702/2Empty cells (t-test, p,0.05) (C) qPCR for the altEJ assay used primers to detect that subset of products including at least 10 nt of each 39overhang. Mean relative joining efficiencies, standard error of the mean, and statistical analysis performed as for panel B. Joining efficiency was significantly different in cells expressingPolq (Polq+/+
,Polq2/2WT4, orPolq2/2WT6) when compared toPolq2/2Empty cells (p,0.05), and in cells expressing Ku (Ku702/2Ku70) when compared to Ku702/2Empty cells (t-test, p,0.05). The background observed in a mock transfected sample was determined to be 0.038,+/20.02 of wild-type controls. p values are represented as: * p,.05, ** p,.01, *** p,.001, ****p,.0001.
policies and protocol 08-08-08732. Mice were euthanized by CO2 euthanasia followed by cervical dislocation.
Cellular proliferation assay Polq+/+
and Polq2/2 bone marrow stromal cells and mouse embryonic fibroblasts were plated in triplicate (200,000 cells per 10 cm dish) with 15 mL of complete media (Dulbecco’s Modified Eagle Medium+Glutamax, 10% FBS, 1% PennStrep). On the indicated days, cells were trypsinized and live cells were counted using trypan blue exclusion (Countess automated cell counter, Life Technologies). Experiments were repeated three times in order to generate standard deviations. Viability was consistently high for all cell lines examined (.95% trypan blue-excluding cells).
Clonogenic assays with bone marrow stromal cells For X-irradiation 56105 cells were plated on a 10 cm plastic culture dish, and exposed the following day at 2 Gy/min, 160 kV peak energy (Rad Source 2000 irradiator, Suwanee, GA). Cells were then trypsinized for replating. For UVC-irradiation (254 nm
peak germicidal lamp) cells were irradiated in 500ml PBSA (105
cells/ml) at 5 J m22min21and then plated. For psoralen-UVA treatment, 56105cells were plated on a 10 cm dish and incubated
in medium with the indicated concentration of HMT-psoralen for 1 h, the dish was irradiated for with 0.9 kJ m22 UVA (365 nm peak, 30 min, 0.5 mJ m22sec21), the psoralen-containing medi-um was removed, and the dish UVA-irradiated in fresh medimedi-um for a further 30 min before replating. Chemicals were added at the indicated concentrations to dishes at the beginning of the experiment. Drugs were solubilized in ethanol (mitomycin c), DMSO (ICRF-193, etoposide, camptothecin, HMT-psoralen, temozolomide, olaparib), or 150 mM NaCl (cisplatin). All chemicals were from Sigma (St. Louis, MO) except ICRF-193 (Enzo LifeScience, Farmingdale, NY), olaparib (AZD2281, Selleck Chemicals, Houston, TX), and mitomycin c (Calbiochem, Darmstadt, Germany). Cells were plated in triplicate in 10 cm dishes and grown for 7–10 days before being fixed and stained with crystal violet. Colonies of 50 or more cells were quantified and experiments were repeated three times to generate standard
Figure 6. Unique template dependent DNA polymerase activity of POLQ.Exonuclease-defectiveE. colipol I Klenow fragment (Kf exo-) or POLQ was incubated at the indicated protein concentrations with (A) a 59-32P-labeled primer 16-mer and 30-mer complementary template, (B) 59-32 P-labeled 16-mer primer and no template. All reaction mixtures included all four deoxynucleotide triphosphates and were incubated at 37uC for 10 min (A) or 20 min (B). The first lane contained no enzyme. The percentage (%) of the primer extended is shown below each lane. (C) Model of intermolecular templating performed by POLQ in the process of extending a different single-stranded oligonucleotide, used to produce the data in Table S1. This model depicts a 12 nt extension product in Table S1. The product can be produced by a series of annealing, extension, slippage and repriming events.
deviations. A clonogenic assay was performed with Rad51D+/+
andRad51D2/2Chinese hamster ovary (CHO) cell lines exposed to varying concentrations of olaparib.
Micronucleus assay
BMSCs were plated at 1.56104cells per well in chambered slides and treated with the indicated amount of x-rays or etoposide the following day. 48 hr later, cells were fixed with 2% para-formalde-hyde, stained with DAPI and coverslipped. Micronuclei were scored by immunofluorescence for 300 cells per group. Experiments were repeated three times to generate standard deviations.
Human cell transfections
293T cells (kindly provided by Dr. Christopher Bakkenist, University of Pittsburgh Medical School) were plated at 150,000
cells in six-well plates and transfected the following day with 2.5mg of either pCDH (System Biosciences, Mountain View, CA) containing empty control, POLQ, K121M, POLQ-D2330A,Y2331A, POLQ-S1977P, or POLQ-DM cDNA using Lipofectamine 2000 (Life Technologies) according to manufac-turer’s specifications. 48 hr after transfection, cells were harvested for RNA isolation (RNeasy, Valencia, CA) or immunoblotting.
Immunoblotting
For immunoblots, cells transfected in six-well dishes were resuspended in 200mL of 26SDS loading buffer (4% SDS, 0.2%
bromophenol blue, 20% glycerol, 100 mM Tris HCl pH 6.8, 12% 2-mercaptoethanol) and heated at 95uC for 5 min. 20mL of extract was separated on a 4–20% polyacrylamide gel, transferred to PVDF membrane, blocked, and blotted with anti-alpha-Tubulin (Abcam, Cambridge, UK) ab4074, 1:10,000), anti-FLAG
Figure 7. POLQ suppresses chromosomal translocation in vivo. (A) Representative schematic for the Myc/IgH translocation assay. PCR amplification primers are represented by black arrows. Closed circles denote centromeric locations on the chromosomes. Naı¨ve B cells from wild-type (WT) orPolq2/2mice were assayed for translocations after 72 hr in culture. (B) Representative agarose gels stained with ethidium bromide and Southern blots withIgHandMycprobes. Each lane contains the DNA content of 16105genomes. Three independent experiments were performed.
(C) Frequency of translocations was plotted and p-values determined using two-tailed Fisher’s exact test. Frequencies were calculated from total translocations (Polq+/+
: 5;Polq2/2: 17) divided by total number of genomes surveyed (9.6
6106). (D) Model of end joining-mediated repair of DNA
double-strand breaks (DSBs). (i) Schematic representing a DSB with existing microhomologies shown in orange. (ii) DSBs are preferentially processed by classical non-homologous end joining (cNHEJ), dependent upon Ku70–80 and Ligase4-XRCC4. This pathway is not thought to promote DNA translocations. In the absence or impairment of critical cNHEJ factors (iii) alternative end joining (altEJ) pathways are utilized. These pathways appear to be suppressed by Ku70–80 and Ligase4-XRCC4. The MMEJ pathway (iv) can orchestrate annealing of ends at pre-existing microhomologies (2– 5 bp) resulting in a net deletion of genomic information. Utilization of this pathway can enhance the formation of chromosomal translocations. In the SD-EJ pathway (v) POLQ can catalyze extension of minimally paired 39single-stranded DNA ends (shown in blue) to facilitate end joining and suppress the formation of chromosomal translocations.
(Sigma F7425, 1:5,000), anti-PCNA (Santa Cruz, Santa Cruz, CA, sc-56, 1:1,000), anti-HA (RW, 1:10,000), or anti-POLQ (MDACC POLQ20, 1:250) antibodies and corresponding secondary anti-bodies (Sigma A0168, A0545; 1:10,000) and visualized with ECL reagent (Pierce, Rockford, IL).
Generation and complementation ofPolqMEFs
Polq-null (Polq2/2) mice [13] were obtained from Jackson
Laboratories and maintained on a C57BL/6J background. Isogenic primary MEFs were generated from 13.5 day pregnant females and cultured in a 2% O2atmosphere. MEFs were then transfected with 1mg of pSV-Tag [61,62] and grown in atmospheric oxygen for six population doublings to allow for immortalization. To generate lentivirus used for transduction, 293T cells were cotransfected with psPAX2 (6mg), pMD2G (6mg), and pCDH (12mg) expression vector (See Text S1 for construction of expression vectors) using Lipofectamine 2000. One day prior to transductionPolq2/2MEFs were seeded into a 10 cm dish at 1.56105 cells with 12 mL complete media. 48 hr post-transfection virus-containing media was harvested, filtered through a 0.45mm syringe filter and used to replace the media on the
plated MEFs. MEFs were incubated in the virus-containing media for 24 hr before being split into T-75 flasks and allowed to grow to 80% confluence before undergoing three weeks of puromycin selection (2.5mg/mL). Following selection, pure clones were
isolated and cultured with complete media containing puromycin (1mg/mL).
Quantitative real-time PCR analysis of complemented MEFs
RNA isolated from the complemented MEF lines were analyzed for quality and purity using RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA). 1mg of total RNA was used to
generate cDNA using the High Capacity cDNA RT kit (Life Technologies). qPCR analysis was performed in triplicate using the ABI Prism 7900 HT thermocycler and the following Taqman Probe set or primer set with iTAQ SYBR Green Supermix with ROX (Bio-Rad, Hercules, CA): MmPolQ_FWD 59 -GGCTC-TGAAGAACTCTTTGCCTTT-39, MmPolQ_REV 59 -GCTG-CTTCCTCTTCTTCATCCA-39, probe 59 -TCCGGGCACT-TTTG-39; HsPOLD1_FWD 59 -CGACCTTCCGTACCTCA-TCTCT-39 HsPOLD1_R 59-ACACGGCCCAGGAAAGG-39, probe 59-CCCTCAAGGTACAAACAT-39; Qexon FWD 59 -TG-CCTTTCAAAAGTGCCCGGAAGGC39, Qexon REV 59 -TG-CCAGTCACCCANATAGTTCNCAT-39. Data were analyzed using the DDCt method. For absolute quantification, titration of pCR-XL-TOPO/MmPolQ and pET/MmPold1 plasmids were used to generate standard curves for expression. Transcript abundance was determined by extrapolation from linear regres-sion analysis of best fit lines from titration experiments. GAPDH was used as an internal control in all experiments.
Bleomycin sensitivity
Complemented MEF lines were plated in triplicate into white 96 well plates at 1250 cells per well and grown overnight using complete media containing puromycin (1mg/mL). The following day, cells are cultured with complete media containing the indicated amounts of bleomycin (dissolved in 150 mM NaCl) for 24 hr before the media was replaced. Cells were allowed to recover for 72 hr before cellular viability was measured using the ATPlite 1Step kit (Perkin Elmer, Waltham, MA) using a Biotek plate reader. Experiments were repeated three times.
Immunofluorescence
Complemented MEF lines were plated at a density of 1.56104 cells per well in 4-well chamber slides and the following day were irradiated with either 0 or 6 Gy of x-rays. Media was changed and cells were allowed to recover for 48 hr after damage before fixation with 2% para-formaldehyde and permeabilized with Triton X-100. Samples were blocked with donkey serum for 30 minutes before being incubated overnight with primary antibodies against 53BP1 (Bethyl, Montgomery, TX, A300-272A, 1:500) andcH2AX (EMD Millipore 05-636, 1:400). Cells were later incubated with AlexaFluor-488 goat-anti-mouse or AlexaFluor-594 goat-anti-rabbit secondary (Life Technologies, 1:1000) and then stained with DAPI before being coverslipped. Cells were scored for DSBs by enumerating the percentage of cells with.2 53BP1 foci and.2cH2AX foci [61,63]. The majority of cells that contained .2 foci for each of the DSB markers, exhibited colocalization of the foci. Cells with pan-staining of
cH2AX were not included in the analysis as they are proposed to represent pre-apoptotic cells [64]. Many of the cells with 53BP1 foci, exhibited enlarged foci that are associated with nuclear OPT (Oct-1, PTF, transcription) domains that sequester damaged DNA in G1 [65,66]. Thus, most of the MEFs that were foci positive contained DSBs [65]. DAPI-stained micronuclei were also scored. For each experiment 250 cells were scored for three independent experiments for a total of 750 cells.
DNA polymerase assays
POLQ was purified as described [36]. Klenow Fragment (39R59 exo-) was purchased from NEB. POLQ was diluted in
buffer containing 30 mM Tris-HCl pH 8.0, 50 mM NaCl, 2 mM DTT, 10% glycerol, 0.01% Triton X-100, and 0.1% BSA. Klenow Fragment (39R59exo-) was diluted in buffer containing
25 mM Tris-HCl pH 7.4, 1 mM DTT, and 0.1 mM EDTA. POLQ reaction mixtures (10ml) contained 20 mM Tris-HCl pH 8.8, 4% glycerol, 2 mM dithiothreitol (DTT), 80mg/ml
bovine serum albumin (BSA), 8 mM MgCl2, 0.1 mM EDTA, 100mM of each dNTP, 30 nM of the primer-template or primer (see Text S1). Klenow Fragment (39R59 exo-) reaction mixtures
(10ml) contained 10 mM Tris-HCl pH 7.9, 50 mM NaCl, 1 mM DTT, 10 mM MgCl2, 100mM of each dNTP, and 30 nM of the primer-template or primer. After incubation at 37uC for 10 min for a 16-1+PA42 substrate or 20 min for 16-1, C20, C19THF substrates, reactions were terminated by adding 10ml of formam-ide stop buffer (98% formamformam-ide, 10 mM EDTA pH 8.0, 0.025% xylene cyanol FF, 0.025% bromophenol blue) and boiling at 95uC for 3 min. Products were electrophoresed on a denaturing 20% polyacrylamide-7 M urea gel, exposed to BioMax MS film, and analyzed with a STORM 860 Phosphor Imager (Molecular Dynamics).
Extrachromosomal substrate assays
-AGTCTGA- GATGGGTGTGAGAGTGAAGATCCTCACCTTCGGAGT-ACTCCTTCTTTTGAGATCTGC-39 to 5-CTCACACCCAT-CTCA-39 (‘‘head’’ linker) and 59 -TGACTATACAGCTAAG- CGATGCTCTCACCGAGCGTATCTGCTGTGTTGTGGAT-GAATTAGATGCAG-39 to 59-CATCGCTTAGCTGTATA-39
(‘‘tail’’ linker). Excess linker was removed by QiaQuick purification and substrate purity validated by polyacrylamide gel electrophoresis. 75 ng of substrate was mixed with 1.1mg of supercoiled pMAX-GFP (Lonza) plasmid carrier and introduced into 26105 cells in a 10ml volume by electroporation with one
30 ms 1350 V pulse (Neon, Invitrogen). Cells were harvested after incubation for 1 hour at 37uC, washed, resuspended in Hank’s buffered saline solution supplemented with 5 mM MgCl2, and extracellular DNA digested by incubation with 6.25 U Benzonase (Novagen) for 15 min at 37uC. Cells were pelleted and DNA purified with the Qiamp kit (Qiagen). Joining efficiency was determined by quantification of head-to-tail junctions by qPCR using primers that either anneal within double-stranded flanks (59- CTTACGTTTGATTTCCCTGAC-TATACAG-39 and 59- GCAGGGTAGCCAGTCTGAGATG-39; 6 nt overhang, Figure 5B) or, for the 45 nt overhang substrate only, which anneal to overhang sequence (59 -TAAGCGATGCTCTCACCGAG and 59- GATGGGTGTGA-GAGTGAAGATC; 45 nt overhang, Figure 5C). Results from electroporated samples were further corrected for differences in transfection and sample processing efficiency using a qPCR specific for substrate (59- GGCACTCTCCAAGGCAAAGA and 59- ACATGTCTAGCCTATTCCCGGCTT).
B cell culture and CSR analysis
B cells were isolated from mouse spleens (n = 6 per genotype) and stimulated for class-switching in culture for 72 hr. Where indicated, cultures were incubated with DNA-PKcs inhibitor 20mM NU7026 (Tocris, Bristol, UK) dissolved in DMSO, or mock-treated. The stimulation procedure and flow-sorting for CSR analysis was as described [31,67]. Prior to this analysis, cells were counted; numbers and viability were similar for all groups. Sm-Sc1 CSR junctions were amplified by PCR using the following conditions for 25 cycles at 95uC (30 s), 55uC (30 s), 68uC (180 s) using the primers (FWD 59 -AATGGATACCTCAGTGGTTTT-TAATGGTGGGTTTA-39; REV 59 CAATTAGCTCCTGCT-CTTCTGTGG-39) andPfuTurbo (Stratagene, La Jolla, CA). To the PCR reaction, 5 U of Taq polymerase (Promega, Madison, WI) was added and incubated at 72uC for 10 min. The resulting product was TOPO TA cloned and transformed into Top10E. coli cells (Life Technologies, Carlsbad, CA) and plasmids were purified and sent for sequencing using M13 FWD and REV primers in addition to the amplification primers for sequencing. 100 clones for each group were analyzed for mutations, deletions, insertions, and sequence overlaps at the junction and both 30 nt upstream and downstream of the junction. p-values were determined by using two-tailed Fisher’s exact test.
Translocation assay
Naı¨ve B cells from three pairs ofPolq+/+
andPolq2/2mice were harvested as above, cultured for 72 hr, and DNA was isolated. 32 separate PCR reactions, each containing the genome from 16105 cells, was performed with primers to amplify Myc/IgH translo-cations and amplified translotranslo-cations were verified by Southern blotting using internal probes to theMycandIgHloci as described [68,69]. Three independent experiments were performed and the p-value was determined using two-tailed Fisher’s exact test. %IgG1 was also measured as an internal control to ensure the B cells from each genotype were switching at a comparable level.
Supporting Information
Figure S1 Cell sensitivity assays. (A) Clonogenic assay of
Rad51D+/+
andRad51D2/2CHO cells treated with the indicated
doses of olaparib, as a positive control [70]. Colonies were crystal violet stained and counted eleven days later. (B)Polq+/+
,Polq2/2,
Polq2/2Empty, and multiple clones of Polq2/2POL and Polq2/
2DM MEF lines were treated with 1mM bleomycin for 24 hr and
cellular ATP levels were measured 72 hr later. (EPS)
Figure S2 Analysis of DNA double-strand breaks and micronu-clei in complemented Polq MEFs. Representative immunofluo-rescence images ofPolq2/2Empty and Polq2/2WT4 MEF lines stained with DAPI and antibodies against 53BP1 andcH2AX. Scale bar represents 25mm. Arrows note micronuclei. Pan staining
of cH2AX (P), OPT domain staining by 53BP1 (OPT), and examples of colocalization (Co) are noted.
(EPS)
Figure S3 Full-lengthchaosImutant protein is poorly expressed. 293T cells were transfected with pCDH constructs that contained either the human POLQ cDNA, ChaosI mutant (S1977P, corresponding to the S1932P mutated residue in mice), or empty control. (A) 48 hr post transfection lysates were made and immunoblotted with antibodies against FLAG and alpha-Tubulin. (B) Total RNA was isolated from cells and qPCR analysis of POLQ transcript levels were performed using theDDCt method. * denotes non-specific band.
(EPS)
Figure S4 POLQ does not extend a single-stranded oligonucle-otide that cannot self-anneal. Increasing amounts of exonuclease-defective E. coli polI Klenow fragment denoted as Kf exo- and POLQ were incubated with (A) 59-32P-labeled 20-mer dC oligonucleotide or (B) a 20-mer dC oligonucleotide with a synthetic abasic site on the 39 end. All reactions include all four deoxynucleotides and were incubated at 37uC for 20 min. The first lane contained no enzyme. The percentage (%) of the product extension from the primer is shown below each lane. * indicates nonspecific band. (C) Model of major 22 nt product formation produced by extension from the primer in Fig. 6B, which has a limited ability to self-anneal.
(EPS)
Figure S5 Absolute quantification of transcript number shows thatPolqis broadly expressed in tissues. Total RNA was isolated from necropsied C57BL/6 mice by Trizol (Life Technologies). Transcript abundance for Polq and DNA polymerase delta catalytic subunitPold1was determined by absolute quantification method as described in the Experimental Procedures.Gapdhwas used as an internal control to normalize samples.
(EPS)
is completed (v) after further DNA synthesis (gray). This process results in an insertion of DNA sequences (blue).
(EPS)
Table S1 POLQ-dependent extension of 39 DNA ends relies upon homology. Primer extension assays were carried out with a defined substrate (top) incubated in the presence of an active polymerase fragment of POLQ. Products were then incubated with terminal deoxynucleotidyl transferase (TdT) in the presence of only dA, dC, dG, or dTTP to create extension products that terminate with homopolymeric runs. The reaction products were then cloned and sequenced for analysis of exten-sion products. Extenexten-sion products are listed above for each homopolymeric run.
(XLSX)
Text S1 Additional materials and methods. Expanded methods, including generation of POLQ antibody and POLQ expression
constructs, PCR primers, oligonucleotide substrates, and sequenc-ing of DNA extended by POLQ.
(DOCX)
Acknowledgments
We thank Sara Lundsten, Kelsey McGahan, Shelley Reh, David Trono and Megan Lowery for technical assistance. We are grateful to our laboratory colleagues for discussion. We acknowledge the support of the M.D. Anderson Molecular Biology Core Facility and Monoclonal Antibodies Facility.
Author Contributions
Conceived and designed the experiments: MJY KT DWW DAR EJ KMM RDW. Performed the experiments: MJY DWW KT YM SCH KMM GOB. Analyzed the data: MJY DWW KMM GOB EJ DAR KT YM RDW. Contributed reagents/materials/analysis tools: JT SD. Wrote the paper: MJY RDW EJ DAR KMM.
References
1. Lange SS, Takata K, Wood RD (2011) DNA polymerases and cancer. Nat Rev Cancer 11: 96–110.
2. Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, et al. (2013) PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol Cell 52: 541–553.
3. Yousefzadeh M, Wood R (2013) DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair 12: 1–9.
4. Seki M, Marini F, Wood RD (2003) POLQ (Polh), a DNA polymerase and
DNA-dependent ATPase in human cells. Nucleic Acids Res 31: 6117–6126. 5. Seki M, Masutani C, Yang LW, Schuffert A, Iwai S, et al. (2004) High-efficiency
bypass of DNA damage by human DNA polymerase Q. EMBO J 23: 4484–4494. 6. Yoon JH, Roy Choudhury J, Park J, Prakash S, Prakash L (2014) A role for DNA polymerase theta in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J Biol Chem 289: 13177–13185.
7. Yoshimura M, Kohzaki M, Nakamura J, Asagoshi K, Sonoda E, et al. (2006) Vertebrate POLQ and POL beta cooperate in base excision repair of oxidative DNA damage. Mol Cell 24: 115–125.
8. Prasad R, Longley MJ, Sharief FS, Hou EW, Copeland WC, et al. (2009) Human DNA polymerase theta possesses 59-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res 37: 1868–1877. 9. Fernandez-Vidal A, Guitton-Sert L, Cadoret JC, Drac M, Schwob E, et al. (2014) A role for DNA polymerase theta in the timing of DNA replication. Nature Communications 5: 4285.
10. Harris PV, Mazina OM, Leonhardt EA, Case RB, Boyd JB, et al. (1996) Molecular cloning ofDrosophila mus308, a gene involved in DNA cross-link repair with homology to prokaryotic DNA polymerase I genes. Mol Cell Biol 16: 5764–5771.
11. Muzzini DM, Plevani P, Boulton SJ, Cassata G, Marini F (2008) Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA interstrand cross-link repair pathways. DNA Repair (Amst) 7: 941–950.
12. Goff JP, Shields DS, Seki M, Choi S, Epperly MW, et al. (2009) Lack of DNA polymerase theta (POLQ) radiosensitizes bone marrow stromal cellsin vitroand increases reticulocyte micronuclei after total-body irradiation. Radiat Res 172: 165–174.
13. Shima N, Munroe RJ, Schimenti JC (2004) The mouse genomic instability mutation chaos1 is an allele ofPolqthat exhibits genetic interaction withAtm. Mol Cell Biol 24: 10381–10389.
14. Shima N, Hartford SA, Duffy T, Wilson LA, Schimenti KJ, et al. (2003) Phenotype-based identification of mouse chromosome instability mutants. Genetics 163: 1031–1040.
15. Kass EM, Jasin M (2010) Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 584: 3703–3708.
16. Rassool FV, Tomkinson AE (2010) Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci 67: 3699–3710.
17. Thompson LH (2012) Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutation Research doi: 10.1016/j.mrrev.2012.06.002. 18. Ramsden DA, Asagoshi K (2012) DNA polymerases in nonhomologous end
joining: are there any benefits to standing out from the crowd? Environmental and molecular mutagenesis 53: 741–751.
19. Decottignies A (2013) Alternative end-joining mechanisms: a historical perspective. Front Genet 4: 48.
20. Boboila C, Alt FW, Schwer B (2012) Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116: 1–49.
21. Simsek D, Jasin M (2010) Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation forma-tion. Nat Struct Mol Biol 17: 410–416.
22. Zhang Y, Jasin M (2011) An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol 18: 80–84.
23. Higgins GS, Prevo R, Lee YF, Helleday T, Muschel RJ, et al. (2010) A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensitization by POLQ knockdown. Cancer Res 70: 2984–2993. 24. Huang KC, Gao H, Yamasaki EF, Grabowski DR, Liu S, et al. (2001)
Topoisomerase II poisoning by ICRF-193. J Biol Chem 276: 44488–44494. 25. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, et al. (2005) Targeting
the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921.
26. Zan H, Shima N, Xu Z, Al-Qahtani A, Evinger Iii AJ, et al. (2005) The
translesion DNA polymerasehplays a dominant role in immunoglobulin gene
somatic hypermutation. EMBO J 24: 3757–3769.
27. Patel PH, Loeb LA (2000) DNA polymerase active site is highly mutable: evolutionary consequences. Proc Natl Acad Sci U S A 97: 5095–5100. 28. Marini F, Wood RD (2002) A human DNA helicase homologous to the DNA
cross-link sensitivity protein Mus308. J Biol Chem 277: 8716–8723. 29. Martomo SA, Saribasak H, Yokoi M, Hanaoka F, Gearhart PJ (2008)
Reevaluation of the role of DNA polymerase theta in somatic hypermutation of immunoglobulin genes. DNA Repair (Amst) 7: 1603–1608.
30. Li Y, Gao X, Wang JY (2011) Comparison of two POLQ mutants reveals that a polymerase-inactive POLQ retains significant function in tolerance to etoposide and gamma-irradiation in mouse B cells. Genes Cells 16: 973–983.
31. Callen E, Jankovic M, Wong N, Zha S, Chen HT, et al. (2009) Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell 34: 285–297.
32. Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annual review of genetics 45: 247–271.
33. Frank-Vaillant M, Marcand S (2002) Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Molecular cell 10: 1189–1199.
34. Lee K, Lee SE (2007) Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 176: 2003–2014.
35. Bennardo N, Cheng A, Huang N, Stark JM (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics 4: e1000110.
36. Hogg M, Seki M, Wood RD, Doublie´ S, Wallace SS (2011) Lesion bypass activity of DNA polymerase theta (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J Mol Biol 405: 642–652. 37. Hogg M, Sauer-Eriksson AE, Johansson E (2012) Promiscuous DNA synthesis
by human DNA polymerase theta. Nucleic Acids Res 40: 2611–2622. 38. Seki M, Wood RD (2008) DNA polymerase theta (POLQ) can extend from
mismatches and from bases opposite a (6-4) photoproduct. DNA Repair (Amst) 7: 119–127.
39. Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA (2008) Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res 36: 3847– 3856.
40. Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, et al. (2011) Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147: 95–106.
41. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, et al. (2011) Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147: 107–119.
43. Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, et al. (2011) DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translo-cation formation. PLoS Genet 7: e1002080.
44. Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, et al. (2012) Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc Natl Acad Sci U S A 109: 2473–2478.
45. Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, et al. (2000) The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci U S A 97: 6630–6633.
46. Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363.
47. Chan SH, Yu AM, McVey M (2010) Dual Roles for DNA Polymerase Theta in Alternative End-Joining Repair of Double-Strand Breaks in Drosophila. PLoS Genet 6: e1001005.
48. Deriano L, Roth DB (2013) Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annual review of genetics 47: 433–455.
49. Frit P, Barboule N, Yuan Y, Gomez D, Calsou P (2014) Alternative end-joining pathway(s): bricolage at DNA breaks. DNA Repair (Amst) 17: 81–97. 50. Arakawa H, Bednar T, Wang M, Paul K, Mladenov E, et al. (2012) Functional
redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res 40: 2599–2610.
51. Simsek D, Furda A, Gao Y, Artus J, Brunet E, et al. (2011) Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471: 245– 248.
52. Han L, Masani S, Hsieh CL, Yu K (2014) DNA ligase I is not essential for mammalian cell viability. Cell Rep 7: 316–320.
53. White TB, Lambowitz AM (2012) The retrohoming of linear group II intron RNAs in Drosophila melanogaster occurs by both DNA ligase 4-dependent and -independent mechanisms. PLoS Genet 8: e1002534.
54. Koole W, van Schendel R, Karambelas AE, van Heteren JT, Okihara KL, et al. (2014) A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun 5: 3216. 55. Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, et al. (2014) Arginine
methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Molecular cell 53: 484–497.
56. Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, et al. (2008) MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 28: 630–638.
57. Roerink SF, van Schendel R, Tijsterman M (2014) Polymerase theta-mediated end joining of replication-associated DNA breaks inC. elegans. Genome Res 24: 954–962.
58. Weinstock DM, Elliott B, Jasin M (2006) A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood 107: 777–780.
59. Bunting SF, Nussenzweig A (2013) End-joining, translocations and cancer. Nat Rev Cancer 13: 443–454.
60. Leme´e F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire M-J, et al. (2010) POLQ up-regulation is associated with poor survival in breast cancer, perturbs DNA replication and promotes genetic instability Proc Natl Acad Sci (USA) 107: 13390–13395.
61. Lange SS, Wittschieben JP, Wood RD (2012) DNA polymerasefis required for proliferation of normal mammalian cells. Nucleic Acids Res 40: 4473–4482. 62. Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, et al. (1996) Requirement
of mammalian DNA polymerasebin base excision repair. Nature 379: 183–186. 63. Ward IM, Minn K, Jorda KG, Chen J (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. The Journal of biological chemistry 278: 19579–19582.
64. Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE (2006) H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A 103: 9891–9896.
65. Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, et al. (2011) Replication stress induces 53BP1-containing OPT domains in G1 cells. The Journal of cell biology 193: 97–108.
66. Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, et al. (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nature cell biology 13: 243–253. 67. Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF,
Nussenz-weig A, et al. (2003) H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med 197: 1767–1778.
68. Kovalchuk AL, Muller JR, Janz S (1997) Deletional remodeling of c-myc-deregulating chromosomal translocations. Oncogene 15: 2369–2377. 69. Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, et al. (2011)
Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol 31: 442–449.