Abra˜ao Cefas Torres Dias
Transic¸˜oes de Fase Induzidas por Altas Press˜oes no
Molibdato de ´Itrio.
Fortaleza – CE
Transic¸˜oes de Fase Induzidas por Altas Press˜oes
Hidrost´aticas no Molibdato de ´Itrio.
Dissertac¸˜ao apresentada ao departamento de F´ısica da Universidade Federal do Cear´a, como parte dos requisitos para a obtenc¸˜ao do t´ıtulo de Mestre em F´ısica.
Orientador:
Prof. Dr. Antˆonio Gomes de Souza Filho
Co-orientador:
Dr. Cleˆanio da Luz Lima
MESTRADO EM F´ISICA DEPARTAMENTO DE F´ISICA
CENTRO DE CIENCIASˆ
UNIVERSIDADEFEDERAL DO CEARA´
Fortaleza – CE
Dissertac¸˜ao de Mestrado sob o t´ıtulo Transic¸˜oes de Fase Induzidas por Altas Press˜oes
Hidrost´aticas no Molibdato de ´Itrio., defendida por Abra˜ao Cefas Torres Dias e aprovada em 20
de Setembro de 2011, em Fortaleza, Cear´a, pela banca examinadora constitu´ıda pelos doutores:
Prof. Dr. Antˆonio Gomes de Souza Filho
Departamento de F´ısica - Universidade Federal do Cear´a Orientador
Prof. Dr. Paulo de Tarso Cavalcante Freire
Departamento de F´ısica - Universidade Federal do Cear´a
Prof. Dr. Jerias Alves Batista
Departamento de F´ısica - Universidade Federal do Maranh˜ao
Dr. Cleˆanio da Luz Lima
Departamento de F´ısica - Universidade Federal do Piau´ı Co-orientador
Agradecimentos
Ao professor Gomes, pelas oportunidades e orientac¸˜ao deste trabalho.
Ao Cleˆanio, pela co-orientac¸˜ao deste trabalho.
Ao professor Josu´e, pela motivac¸˜ao constante.
Ao professor Carlos Alberto, pela iniciac¸˜ao cient´ıfica e amizade.
Ao professor Paulo de Tarso, pela ajuda com os experimentos.
Ao professor Waldeci Paraguassu, pelos calculos de dinˆamica de rede.
Ao professor Bojan Marinkovic, pelas amostras.
Ao professor Humberto Terrones, pelas dicuss˜oes sobre este trabalho.
Ao professor Milton, pelos excelentes cursos de Mecˆanica Quˆantica.
Ao professor Ramos, pelos cursos de F´ısica Matem´atica e Mecˆanica Estat´ıstica.
Ao professor Evangelista, pelos v´arios cursos ministrados na graduac¸˜ao.
`
A Katiane, pela amizade e ajuda nos experimentos.
Ao Acr´ısio, pela amizade e pelas in´umeras pequenas ajudas ao longo do mestrado.
Ao Eduardo, pela amizade e discuss˜oes sobre diversos assuntos da F´ısica.
Ao Rafael, pela amizade e pelas excelentes discuss˜oes sobre F´ısica do Estado S´olido.
Ao Bruno, pelas discuss˜oes sobre F´ısica de Altas Press˜oes e aux´ılio no laborat´orio.
Ao colegas Paschoal, Saulo, Ernerson, Pablo, J.J., Ana, S´ergio e Neves, pelo aprendizado.
`
A CAPES, ao CNPQ e `a FUNCAP pelo financiamento das nossas pesquisas e bolsas.
Resumo
Neste trabalho investigamos as propriedades vibracionais do molibdato de ´ıtrio hidratado e
anidro em func¸˜ao da press˜ao hidrost´atica. A an´alise dos modos vibracionais mostra que esse
material experimenta uma transic¸˜ao de fase da estrutura ortorrˆombica para uma fase de mais
baixa simetria para valores de press˜ao em torno de 0,3 GPa, provavelmente monocl´ınica, antes
de se transformar em uma estrutura com alto grau de desordem a partir de 2,4 GPa. A transic¸˜ao
estrutural da fase ordenada para a fase desordenada n˜ao ´e revers´ıvel, indicando que o material
provavelmente entrou em um estado de amorfizac¸˜ao. O comportamento dos modos vibracionais
Conte ´udo
Resumo p. vi
Abstract p. vii
Lista de Figuras p. x
Lista de Tabelas p. xii
Introduc¸˜ao p. 1
1 Propriedades F´ısicas dos Molibdatos p. 3
1.1 Molibdatos e Tungstatos . . . p. 3
1.2 Efeitos observados nos molibdatos sob altas press˜oes . . . p. 5
1.3 O Molibdato de ´Itrio . . . p. 6
2 M´etodos p. 8
2.1 Fundamentos do espalhamento Raman . . . p. 8
2.2 Dinˆamica de Rede . . . p. 13
2.3 T´ecnica para obter altas press˜oes hidrost´aticas . . . p. 14
2.4 Detalhes do experimento . . . p. 16
2.5 Tratamento dos dados experimentais . . . p. 17
3 Resultados e Discuss˜oes p. 18
3.1 An´alise de Teoria de Grupos . . . p. 18
4 Conclus˜oes e Perspectivas p. 37
Bibliografia p. 39
Lista de Figuras
1.1 Estrutura cristalina do Y2(MoO4)3 (Pbcn). Tetraedros escuros representam
MoO4e octaedros claros representam YO6. Adaptac¸˜ao do original [33]. . . . p. 7
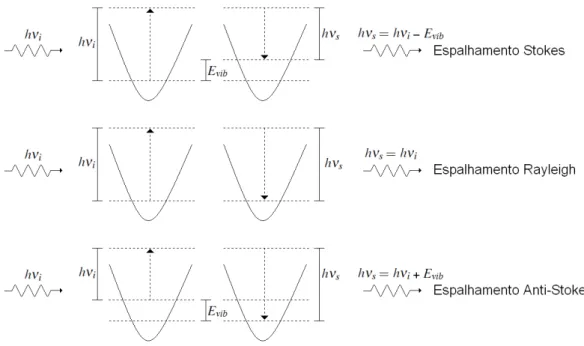
2.1 Diagramas de n´ıveis ilustrando os diferentes tipos de espalhamento de luz
n˜ao ressonantes. Respectivamente, hνi, hνs e Evib representam as energias
dos f´otons incidentes, espalhados e as energias das vibrac¸˜oes. . . p. 9
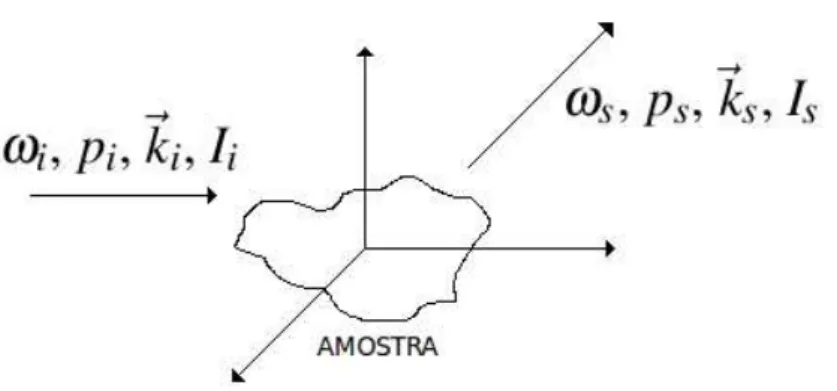
2.2 Modelo cl´assico da interac¸˜ao entre radiac¸˜ao eletromagn´etica e mat´eria. Os
´ındices ‘i’ e ‘s’ est˜ao para incidente e espalhado, respectivamente. ω, p e ~ks˜ao frequˆencia, polarizac¸˜ao e vetor de propagac¸˜ao, respectivamente. Ii ´e a
intensidade da luz incidente eIs ´e a intensidade da luz espalhada. . . p. 10
2.3 Detalhe da bigorna de diamante, gaxeta e amostra. Adaptac¸˜ao do original
[41]. . . p. 14
2.4 Detalhe da gaxeta. Adaptac¸˜ao do original [41]. . . p. 15
2.5 C´elula de press˜ao tipo NBS. Adaptac¸˜ao do original [41]. . . p. 15
2.6 Deslocamento do rubi em relac¸˜ao `a energia do laser (2,41 eV) utilizado na
excitac¸˜ao. . . p. 16
3.1 Valores calculados para as constantes de forc¸a em func¸˜ao da distˆancia da
ligac¸˜ao entre os diferentes ´atomos do Y2(MoO4)3. . . p. 21
3.2 Autovetores e autovalores para os modos de baixa energia dos tetraedros
MoO4para a representac¸˜ao irredut´ıvel Ag. . . p. 22
3.3 Autovetores e autovalores para os modos de alta energia dos tetraedros MoO4
para a representac¸˜ao irredut´ıvel Ag. . . p. 22
3.4 Espectros Raman do molibdato de ´ıtrio nas fases anidra (Y2(MoO4)3) e hidratada
(Y2(MoO4)3·3H2O) medidos em condic¸˜oes de temperatura e press˜ao ambiente. p. 23
3.5 Espectro Raman do Y2(MoO4)3 logo ap´os ser transferido para o interior da
GPa e 0,3 GPa). . . p. 28
3.8 Comparac¸˜ao dos espectros tomados em 2,4 GPa (regime de compress˜ao), 9,6
GPa (m´axima press˜ao atingida em nosso experimento) e 0,2 GPa (regime de
descompress˜ao). . . p. 29
3.9 Variac¸˜ao da largura dos modos de flex˜ao com a press˜ao. . . p. 30
3.10 Variac¸˜ao da largura dos modos de estiramento assim´etrico com a press˜ao. . . p. 30
3.11 Variac¸˜ao da largura dos modos de estiramento sim´etrico com a press˜ao. . . . p. 31
3.12 Variac¸˜ao da largura dos modos remanescentes durante a descompress˜ao. . . . p. 31
3.13 Variac¸˜ao da posic¸˜ao dos modos do Y2(MoO4)3durante a compress˜ao. . . p. 32
3.14 Espectro Raman do Y2(MoO4)3·xH2O (x< 3) ap´os ser transferido para a
c´elula de press˜ao. . . p. 33
3.15 Espectros do Y2(MoO4)3·xH2O (x<3) em func¸˜ao da press˜ao hidrost´atica. . . p. 34
3.16 Dependˆencia da frequˆencia dos modos do Y2(MoO4)3·xH2O (x<3) em func¸˜ao
da press˜ao. . . p. 35
3.17 Dependˆencia da largura de linha dos modos do Y2(MoO4)3·xH2O (x<3) em
func¸˜ao da press˜ao. . . p. 36
Lista de Tabelas
3.1 Tabela de correlac¸˜ao dos modos para 8MoO4ocupando s´ıtio C1 na simetria
D2h. . . p. 19
3.2 Tabela de correlac¸˜ao dos modos para 4MoO4ocupando s´ıtio C2 na simetria
D2h. . . p. 19
3.3 Valores dos parˆametros utilizados no potencial de Buckingham. . . p. 20
3.4 Frequˆencias e identificac¸˜ao dos modos vibracionais dos tetraedros MoO4
cal-culados usando dinˆamica de rede. . . p. 21
3.5 Identificac¸˜ao do modos do Y2(MoO4)3observados experimentalmente. . . . p. 24
3.6 Valores deα (cm−1GPa−1) eω0(cm−1) determinados para: Fase 1 (entre 0,3
e 1,1 GPa) e Fase 2 (entre 2,4 GPa e 9,6 GPa). . . p. 32
3.7 Valores de ω0 (cm−1) e α (cm−1GPa−1) obtidos ajustando as frequˆencias
Introduc¸˜ao
As t´ecnicas de Altas Press˜oes est˜ao presentes em muitos estudos nas diversas disciplinas
do conhecimento na literatura cient´ıfica, constituindo uma importante ferramenta de pesquisa
interdisciplinar. Do ponto de vista da Geologia, essas t´ecnicas reproduzem em laborat´orio
as condic¸˜oes extremas de altas press˜oes encontradas nas camadas interiores da Terra, onde a
press˜ao ´e uma vari´avel determinante para a s´ıntese natural de alguns materiais (o diamante ´e
um exemplo cl´assico). Do ponto de vista da F´ısica de estado s´olido, oferece um meio eficaz
de investigar a estrutura da mat´eria, interac¸˜oes interatˆomicas, propriedades eletromagn´eticas e
mecˆanicas.
Podemos dizer que a F´ısica de Altas Press˜oes consiste em aliar modelos te´oricos e t´ecnicas
experimentais para descrever e aplicar press˜oes elevad´ıssimas (da ordem de gigapascais, 1atm
= 105Pa) e observar os efeitos induzidos na mat´eria utilizando em geral difrac¸˜ao e absorc¸˜ao de raios-X, espalhamento Raman (usada neste trabalho), absorc¸˜ao no infravermelho e, em alguns
casos, medidas el´etricas. Realizar um experimento de espectroscopia Raman em altas press˜oes
significa acessar a resposta vibracional de um material com o aumento da press˜ao sobre ele
apli-cada e determinar a partir dos resultados a influˆencia da press˜ao na estrutura e nas propriedades
do material estudado. Essa resposta ´e intimamente ligada ao arranjo molecular e as mudanc¸as
estruturais s˜ao observadas atrav´es das mudanc¸as no espectro vibracional.
Na mat´eria, as distˆancias interatˆomicas s˜ao reguladas pelas interac¸˜oes de curto e longo
al-cance entre os ´atomos. Podemos interferir nas distˆancias interatˆomicas de um material variando
a temperatura ou a press˜ao. Variac¸˜oes de temperatura provocam mudanc¸as na populac¸˜ao de
fˆonons, o que n˜ao ocorre se utilizarmos como parˆametro a press˜ao mantendo a temperatura
aproximadamente constante. A press˜ao hidrost´atica em n´ıveis elevados induz variac¸˜oes mais
dr´asticas nas distˆancias interatˆomicas, tornando-a mais eficiente do que a temperatura para esta
finalidade. De modo geral, a press˜ao hidrost´atica aplicada sobre um material tem como
con-sequˆencia a reduc¸˜ao das distˆancias entre os ´atomos que o comp˜oe, bem como a reduc¸˜ao do
volume, e ´e, portanto, uma ferramenta fundamental para investigar o delicado balanc¸o entre
as forc¸as de longo e curto alcance. Reduzir as distˆancias interatˆomicas e detectar as mudanc¸as
ocorridas nas propriedades estruturais de um material pode fornecer pistas sobre os mecanismos
2
ferramenta para as ciˆencias e engenharia dos materiais.
Nos s´olidos cristalinos (cristal, por simplicidade), o balanc¸o entre as interac¸˜oes de curto e
longo alcance ´e respons´avel pela estabilidade da estrutura cristalina e pela emergˆencia de
diver-sos fenˆomenos coletivos, tais como a ferroeletricidade. Quando um ´atomo ´e deslocado da sua
posic¸˜ao de equil´ıbrio dentro de um cristal, ele sofre ac¸˜ao de uma forc¸a restauradora devido as
interac¸˜oes interatˆomicas e portanto vibra em determinadas frequˆencias caracter´ısticas,
determi-nadas pela dinˆamica dos fˆonons da rede cristalina [1]. Essas vibrac¸˜oes s˜ao fortemente
depen-dentes da estrutura da rede e influenciam diretamente as propriedades ´oticas e de transporte do
material [2]. Ao realizar um experimento de espectroscopia Raman, acessamos as frequˆencias
vibracionais dos fˆonons ´oticos. Como a press˜ao influi na dinˆamica dos fˆonons, experimentos
de espectroscopia Raman em altas press˜oes permitem estudar a mudanc¸a do comportamento
dos fˆonons com a variac¸˜ao da press˜ao e consequentemente obter informac¸˜oes sobre as
propri-edades do cristal em estudo. A espectroscopia Raman em altas press˜oes tamb´em ´e ´util para
detectar transic¸˜oes de fase estruturais induzidas por essa vari´avel. Determinar para quais
valo-res de pvalo-ress˜ao hidrost´atica um material transita de uma fase cristalina para outra fase cristalina
ou ainda para uma fase desordenada de alta press˜ao contribui com a construc¸˜ao de diagramas de
fases que s˜ao essenciais para a descric¸˜ao dos aspectos termodinˆamicos do material. Os dados
obtidos num experimento de altas press˜oes para um material ‘bulk’ s˜ao importantes para o
apri-moramento de modelos te´oricos que descrevem o material. Os dados obtidos em altas press˜oes
para o material ‘bulk’ podem ser usados para a caracterizac¸˜ao do n´ıvel de ‘strain’ efetivo quando
esse material ´e usado na forma de filmes finos sobre substratos e nanocristais.
Esta dissertac¸˜ao descreve o estudo do molibdato de ´ıtrio em condic¸˜oes extremas de altas
press˜oes hidrost´aticas atrav´es da espectroscopia Raman. A redac¸˜ao est´a dividida em quatro
cap´ıtulos. No cap´ıtulo 1 comentamos os principais resultados fornecidos pela F´ısica de Altas
Press˜oes para os molibdatos e tungstatos, as propriedades de interesse cient´ıfico e tecnol´ogico
desses materiais e a motivac¸˜ao pela escolha do molibdato de ´ıtrio como material de estudo.
No cap´ıtulo 2 revisamos brevemente a teoria do espalhamento Raman, as t´ecnicas empregadas
para atingir altas press˜oes hidrost´aticas, o modelo te´orico utilizado para efetuar c´alculos de
dinˆamica de rede e alguns detalhes do experimento realizado. No cap´ıtulo 3 descrevemos em
detalhes os resultados dos nossos experimentos de espectroscopia Raman. Como suporte ao
trabalho experimental, inclu´ımos an´alise de Teoria de Grupos e c´alculos de dinˆamica de rede
aplicados ao molibdato de ´ıtrio. No cap´ıtulo 4 conclu´ımos o trabalho enfatizando as implicac¸˜oes
dos resultados obtidos para o conhecimento cient´ıfico sobre os molibdatos e tungstatos e as
1
Propriedades F´ısicas dos Molibdatos
Neste cap´ıtulo fazemos um resumo do conhecimento estabelecido na literatura sobre os
molibdatos de diversos elementos qu´ımicos. Comentamos sobre as propriedades e os
resulta-dos experimentais obtiresulta-dos utilizando as t´ecnicas da F´ısica de Altas Press˜oes. Em particular,
destacamos o molibdato de ´ıtrio, Y2(MoO4)3, cujas propriedades de expans˜ao t´ermica negativa
motivaram nosso estudo de espectroscopia Raman em condic¸˜oes de altas press˜oes para este
material.
1.1
Molibdatos e Tungstatos
Chamamos de molibdatos os materiais formados pela ligac¸˜ao do ´ıon molibdato MoO24− a c´ations. Podemos definir os tungstatos de maneira an´aloga, pela ligac¸˜ao com ´ıon tungstato
WO24−. Esses ´ıons tem forma tetra´edrica com um ´atomo de Mo/W no centro e quatro ´atomos de oxigˆenio nos v´ertices. Na literatura, muitos dos molibdatos (e tungstatos) s˜ao
represen-tados pela f´ormula qu´ımica B2(XO4)3, onde B ´e um c´ation trivalente (do Al ao Dy) e X =
W, Mo [3]. Entre outros molibdatos e tungstatos de interesse atual na literatura, podemos
citar: Ln2Mo4O15 (Ln = lantan´ıdeos), Bi2MoO6, K3Fe(MoO4)2(Mo2O7), ABW2O9 (A=K, Rb
e B=Nb, Ta), Cs4W11O35, Rb4W11O35, Bi2WO6, Bi2W2O9. Destacam-se tamb´em os
compos-tos AB(XO4)2, com A=metal alcalino, B=Sc, Y, Al, Fe, Bi, Ln e X=Mo/W. De modo geral,
os c´ations trivalentes s˜ao elementos qu´ımicos esperados para formar molibdatos e tungstatos
B2(XO4)3, nos quais o tamanho desses c´ations tem influˆencia direta nos coeficientes de
ex-pans˜ao t´ermica desses materiais [4]. De modo geral, a fam´ılia B2(XO4)3 desperta interesse
cient´ıfico por suas propriedades ´oticas, ferroel´etricas, ferroel´asticas e termodinˆamicas.
Em nosso trabalho, focalizamos nosso interesse na fam´ılia B2(XO4)3, visto que esses
mate-riais ganharam atenc¸˜ao da comunidade cient´ıfica porque alguns deles possuem coeficientes de
expans˜ao t´ermica negativos, ou seja, reduzem de volume com o aumento da temperatura, em
contraste com a maior parte dos materiais conhecidos, que expandem o volume com o aumento
4
apresentam expans˜ao t´ermica negativa (NTE), sendo alguns deles: Y2(MoO4)3 [7], Y2(WO4)3
[8], Sc2(MoO4)3 [9], Sc2(WO4)3 [10], Er2(MoO4)3 [3], Er2(WO4)3 [11], Yb2(MoO4)3 [4],
Yb2(WO4)3 [11], Lu2(MoO4)3 [4], Lu2(WO4)3 [4]. Estes exemplos tamb´em ilustram que o
comportamento de expans˜ao t´ermica entre molibdatos e tungstatos de um mesmo elemento
qu´ımico podem apresentar semelhanc¸as [4]. O grande interesse da comunidade cient´ıfica pela
NTE surgiu ap´os a descoberta dessa propriedade no ZrW2O8 [12]. Embora antes relatada para
outros materiais, a NTE no ZrW2O8 ´e intensa e aproximadamente isotr´opica num intervalo de
temperatura apreci´avel, tornando-o um sistema modelo para o estudo da propriedade de
ex-pans˜ao t´ermica negativa e consequentemente alvo de outros estudos [13, 14].
Os materiais que possuem NTE tornaram-se candidatos para o desenvolvimento de ligas
met´alicas com expans˜ao t´ermica controlada, dispositivos para placas de circuito impresso,
com-ponentes para instrumentos ´oticos de precis˜ao e outras aplicac¸˜oes [5]. Vale salientar que a NTE
ocorre apenas em determinanos intervalos de temperatura e press˜ao, sendo necess´ario observar
os limites dessas grandezas para tirar bom proveito da propriedade de NTE em aplicac¸˜oes.
Al-guns trabalhos atribuem a expans˜ao t´ermica negativa aos modos vibracionais de baixa energia
(chamados de modos das unidades r´ıgidas) e as vibrac¸˜oes anarmˆonicas transversais [15]. Evans
et al.descrevem algumas condic¸˜oes para a ocorrˆencia de NTE num material [5]:
1. A presenc¸a de ligac¸˜oes M-O que naturalmente apresentam baixa expans˜ao;
2. ´Atomos de oxigˆenio de coordenac¸˜ao dupla que permitem vibrac¸˜oes transversais;
3. A estrutura deve suportar modos vibracionais transversais de baixa energia;
4. Ausˆencia de c´ations intersticiais na estrutura;
5. Falta ou supress˜ao de transic¸˜oes para fases de mais baixa simetria;
A literatura relata que certos materiais B2(XO4)3com estrutura ortorrˆombica apresentam o
fenˆomeno da NTE [8, 9, 16]. Evanset al. tamb´em sugerem que uma dopagem qu´ımica
ade-quada pode suprimir transic¸˜oes de fases nesses compostos e isto pode ser utilizado como
meca-nismo de controle dos coeficientes de expans˜ao t´ermica, bastante interessante para aplicac¸˜oes
[5], pois fazendo uma engenharia apropriada de compostos com expans˜ao t´ermica negativa com
outros de expans˜ao t´ermica positiva ´e poss´ıvel obter um material com expans˜ao t´ermica nula.
Alguns materiais da fam´ılia B2(XO4)3que apresentam NTE amorfizam em altas press˜oes,
lev-antando a hip´otese da existˆencia de alguma correlac¸˜ao entre amorfizac¸˜ao e NTE [17]. Outra
pista para a investigac¸˜ao desse fenˆomeno ´e dada por Sikkaet al., que sugere uma relac¸˜ao entre
O primeiro estudo de um material do tipo B2(XO4)3 em altas press˜oes foi publicado em
1972 [18], relatando o fenˆomeno de amorfizac¸˜ao induzida por press˜ao (Pressure Induced
Amor-phization, PIA) para o Gd2(MoO4)3. Neste mesmo ano um trabalho relata as dimens˜oes das
c´elulas unit´arias dos compostos La2(MoO4)3, Ce2(MoO4)3, Pr2(MoO4)3 e Nd2(MoO4)3 em
func¸˜ao da press˜ao [19]. Em 1993 foi observado para o Gd2(MoO4)3 uma transic¸˜ao de fase
estrutural em torno de 2,0 GPa que antecede o processo de amorfizac¸˜ao induzida pela press˜ao
[20]. Posteriormente, mais experimentos de altas press˜oes (inclu´ındo difrac¸˜ao de raios-X e
es-pectroscopia Raman) mostraram diversas transic¸˜oes de fases estruturais (ordem e
ordem-desordem) ocorridas nos molibdatos e tungstatos B2(XO4)3, al´em de amorfizac¸˜ao em alguns
casos.
Alguns compostos B2(XO4)3mostram comportamentos semelhantes em altas press˜oes. Por
exemplo, o Sc2(MoO4)3 e o Al2(WO4)3 transitam da fase ortorrˆombica para monocl´ınica em
torno de 0,29 GPa e 0,28 GPa, respectivamente [21, 22]. Press˜oes mais elevadas induzem
uma desordem na estrutura de ambos, exibindo comportamento similar ao j´a citado para o
Gd2(MoO4)3. J´a os compostos Y2(WO4)3 e Lu2(WO4)3 transitam diretamente para a fase
amorfa, sem passar por uma fase cristalina intermedi´aria (pelo menos quando essa rota ´e
in-vestigada `a temperatura ambiente) [23, 24]. Dentre os compostos B2(XO4)3 que apresentam
amorfizac¸˜ao induzida por press˜ao, citamos: Sc2(MoO4)3[21], Sm2(MoO4)3[25], Eu2(MoO4)3
[26], Gd2(MoO4)3 [20], Tb2(MoO4)3 [27], Nd2(MoO4)3 [28], Y2(WO4)3 [23], Sc2(WO4)3
[29], e Lu2(WO4)3[24].
O fenˆomeno de amorfizac¸˜ao induzida por press˜ao ´e atribu´ıdo a trˆes fatores: deformac¸˜ao
mecˆanica, decomposic¸˜ao qu´ımica e proibic¸˜ao cin´etica (kinetic hindrance) da transic¸˜ao de uma
fase cristalina para outra. Para explicar a amorfizac¸˜ao dos compostos B2(XO4)3 citados, a
deformac¸˜ao mecˆanica pode ser descartada, visto que para esses compostos a amorfizac¸˜ao ´e
obtida sob condic¸˜oes de altas press˜oes hidrost´aticas e mostrou-se irrevers´ıvel quando ´e realizada
a descompress˜ao do sistema [25]. A amorfizac¸˜ao nos compostos tipo B2(XO4)3 ´e relacionada `a
decomposic¸˜ao qu´ımica, mas de forma geral, a proibic¸˜ao cin´etica para atingir outra fase cristalina
precisa ser considerada, pois a impossibilidade de atingir uma fase cristalina leva `a amorfizac¸˜ao.
Nesse caso, a fase amorfa induzida por press˜ao ´e cineticamente preferida [17, 30]. Prop˜oe-se
que o modelo estrutural para amorfizac¸˜ao induzida por press˜ao nos compostos do tipo B2(XO4)3
´e similar ao modelo adotado para oα-ZrW2O8e para oα-ZrMo2O8[17, 31].
6
se inclinem, reduzindo o valor m´edio dos ˆangulos B-O-X e das distˆancias entre os oxigˆenios
n˜ao ligantes. Quando as distˆancias entre os oxigˆenios n˜ao ligantes tornam-se muito curtas, elas
passam a contribuir de forma repulsiva para a energia livre e limitam espacialmente a estrutura,
ocasionando a mudanc¸a da fase de press˜ao ambiente original para uma fase metaest´avel de alta
press˜ao. Uma compress˜ao maior forc¸a a perda da correlac¸˜ao de longo alcance (aumentando a
entropia). O processo de amorfizac¸˜ao nesses compostos pode ser atribu´ıdo aos v´ınculos
espa-ciais (‘steric constrains’) e `a impossibilidade do sistema superar a barreira de energia para uma
fase ordenada de alta press˜ao [17].
1.3
O Molibdato de ´Itrio
Centramos agora nossa atenc¸˜ao no Y2(MoO4)3, que ´e o nosso sistema de estudo.
Trata-se de um dos compostos inorgˆanicos com f´ormula B2(XO4)3 que apresenta a propriedade de
NTE, possu´ındo os coeficientes de expans˜ao t´ermica (nas direc¸˜oes cristalogr´aficasa,bec)αa
=-11,19x10−6K−1,αb=-6,57x10−6K−1eαc=-10,04x10−6K−1, destacando-se como os mais
isotr´opicos entre os molibdatos e tungstatos de terras raras [32]. Essa caracter´ıstica de expans˜ao
isotr´opica ´e desej´avel para aplicac¸˜oes que podem tirar proveito da NTE [7].
Em condic¸˜oes de temperatura e press˜ao ambiente (n˜ao exposto `a umidade), o Y2(MoO4)3
´e ortorrˆombico, pertencendo ao grupo espacial Pbcn [7, 15]. Sua estrutura ´e formada por
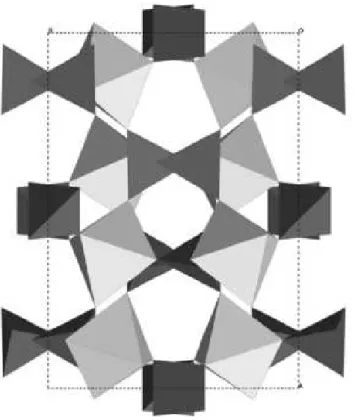
oc-taedros YO6 e tetraedros MoO4ligados por seus v´ertices, num arranjo onde todos os poliedros
compartilham todos os seus v´ertices [7]. Como ´e mostrado na Figura 1.1, cada octaedro YO6
compartilha seus seis v´ertices com os tetraedros MoO4 e cada um destes compartilha seus
v´ertices com quatro octaedros YO6[33, 34]. C´ations de tamanho iˆonico grande como o Y3+e a
flexibilidade dos poliedros YO6e MoO4s˜ao vistos como fatores chave para ocorrˆencia de NTE
nesse material [7]. Devemos lembrar que a NTE nos compostos B2(XO4)3 ´e mais acentuada
para c´ations B3+ com maiores raios iˆonicos [35].
Uma dificuldade experimental para lidar com o Y2(MoO4)3 ´e o fato dele ser altamente
higrosc´opico. Quando exposto `a umidade ambiente, assume a forma tri-hidratada est´avel,
com f´ormula Y2(MoO4)3·3H2O, acumulando mol´eculas d’´agua nos microcanais existentes na
direc¸˜ao cristalogr´afica ‘c’ de sua estrutura [7]. Sabe-se que as mol´eculas de ´agua limitam os
movimentos dos poliedros MoO4 e por consequencia disso, o fenˆomeno de NTE n˜ao ocorre
na fase hidratada desse material. Um tratamento t´ermico pode ser efetuado para “expulsar” as
mol´eculas d’´agua e a amostra deve ser prontamente isolada em ambiente seco para que seja
Figura 1.1: Estrutura cristalina do Y2(MoO4)3(Pbcn). Tetraedros escuros representam MoO4e
octaedros claros representam YO6. Adaptac¸˜ao do original [33].
Uma consulta na literatura sobre o Y2(MoO4)3mostra-o isoestrutural em relac¸˜ao aos
com-postos Y2(WO4)3, Sc2(MoO4)3 e Sc2(WO4)3 em condic¸˜oes de temperatura e press˜ao
ambi-ente. Sob press˜ao, estes trˆes compostos amorfizam respectivamente em torno de 4 GPa, 6 GPa
e 11 GPa [21, 23, 29]. Isto nos leva a conjecturar sobre o comportamento do Y2(MoO4)3 em
condic¸˜oes an´alogas. Recordando o que j´a comentamos sobre a relac¸˜ao entre a fase ortorrˆombica
e NTE nesses compostos, percebemos que estudar as poss´ıveis transic¸˜oes de fase do Y2(MoO4)3
pode fornecer mais informac¸˜oes sobre os fenˆomenos de NTE e amorfizac¸˜ao para os compostos
B2(XO4)3.
At´e a presente data, n˜ao encontramos publicac¸˜oes de estudos em condic¸˜oes de altas press˜oes
para o Y2(MoO4)3. Propomos ent˜ao realizar experimentos de espectroscopia Raman em altas
press˜oes com amostras policristalinas de Y2(MoO4)3. Nosso objetivo ´e estudar o
comporta-mento dos fˆonons ´oticos deste material com a variac¸˜ao da press˜ao. Com os resultados obtidos,
podemos tanto detectar transformac¸˜oes estruturais como estimar algumas propriedades desse
8
2
M´etodos
Neste cap´ıtulo descrevemos a metolologia utilizada para estudar o molibdato de ´ıtrio em
condic¸˜oes de altas press˜oes hidrost´aticas. Comentamos de forma sucinta alguns fundamentos da
teoria do espalhamento Raman e o princ´ıpio de funcionamento da c´elula de press˜ao a extremos
de diamante. Alguns detalhes t´ecnicos dos equipamentos e m´etodos experimentais utilizados
s˜ao tamb´em descritos.
2.1
Fundamentos do espalhamento Raman
O fenˆomeno de espalhamento de luz pode ser explicado como um processo de absorc¸˜ao de
um f´oton (incidente) e emiss˜ao instantˆanea de outro f´oton (espalhado). Devemos diferenci´a-lo
do fenˆomeno de absorc¸˜ao, quando uma amostra (cristal ou mol´ecula) ´e excitada para um dos
seus estados de mais alta energia ao absorver a energia do f´oton incidente. No espalhamento
n˜ao ressonante, os el´etrons s˜ao excitados para um estado virtual. Esse estado virtual ´e assim
chamado por n˜ao corresponder a um dos estados eletrˆonicos reais do material [37]. A radiac¸˜ao
espalhada prov´em dos momentos de multipolo el´etrico e magn´etico oscilantes induzidos no
material pelo campo eletromagn´etico da radiac¸˜ao incidente.
O espalhamento pode ser el´astico ou inel´astico. No espalhamento el´astico (chamado de
espalhamento Rayleigh) o f´oton espalhado e o f´oton incidente possuem energias iguais, n˜ao
havendo mudanc¸a nos estados vibracionais. No espalhamento inel´astico (chamado de
espa-lhamento Raman) o f´oton espalhado possui energia diferente do f´oton incidente, indicando a
ocorrˆencia de alguma mudanc¸a entre dois estados do sistema. O efeito Raman ´e justamente
o fenˆomeno de espalhamento inel´astico devido `a interac¸˜ao da radiac¸˜ao eletromagn´etica com a
mat´eria, havendo mudanc¸a de energia (ou frequˆencia) entre radiac¸˜ao incidente e espalhada.
De-vemos sempre lembrar que o espalhamento Raman ´e incoerente, n˜ao havendo correlac¸˜ao entre
as fases das radiac¸˜oes incidente e espalhada.
Es=hω¯ s=hνs (2.1)
Ei=hω¯ i=hνi. (2.2)
Se Es<Ei (νs <νi), temos o espalhamento chamado de Stokes. O f´oton incidente ´e
ab-sorvido (virtualmente) e espalhado pela amostra, que passa do seu estado fundamental para um
estado virtual de energia mais elevada, deca´ındo em seguida para um estado excitado de menor
energia, cujo valor ´e a diferenc¸a entre a energia do f´oton espalhado em relac¸˜ao ao incidente. A
diferenc¸a de energia ´e exatamente igual a energia de um modo vibracional.
Se Es >Ei (νs >νi), temos o espalhamento chamado de anti-Stokes. O f´oton incidente
´e absorvido (virtualmente) e espalhado pela amostra, que passa de um de seus estados
exci-tados para um estado virtual de energia mais elevada, deca´ındo em seguida para seu estado
fundamental. A diferenc¸a de energia ´e associada a um quantum de energia vibracional que foi
aniquilado.
Na Figura 2.1 ilustramos os espalhamentos Rayleigh, Stokes e anti-Stokes em um diagrama
de n´ıveis.
Figura 2.1: Diagramas de n´ıveis ilustrando os diferentes tipos de espalhamento de luz n˜ao ressonantes. Respectivamente, hνi, hνs e Evib representam as energias dos f´otons incidentes,
espalhados e as energias das vibrac¸˜oes.
10
Figura 2.2: Modelo cl´assico da interac¸˜ao entre radiac¸˜ao eletromagn´etica e mat´eria. Os ´ındices ‘i’ e ‘s’ est˜ao para incidente e espalhado, respectivamente.ω, pe~ks˜ao frequˆencia, polarizac¸˜ao e vetor de propagac¸˜ao, respectivamente. Ii ´e a intensidade da luz incidente eIs ´e a intensidade
da luz espalhada.
momento de dipolo induzido~Pno sistema pelo campo de radiac¸˜ao como:
~Pmn = (αi j)mn~E, (2.3)
ondeα ´e o tensor polarizabilidade, cujas componentes s˜ao dadas por:
Px=αxxEx+αxyEy+αxzEz (2.4)
Py=αyxEx+αyyEy+αyzEz (2.5)
Pz=αzxEx+αzyEy+αzzEz. (2.6)
Sendo a polarizabilidade func¸˜ao do movimento vibracional, podemos ent˜ao expandir as
componentes do tensorα em s´erie de Taylor na coordenada normal q (modo vibracional) do
sistema:
α =αo+
dα
dq
0
q+1
2
d2α dq2
0
q2+... (2.7)
onde o zero subscrito indica que a derivada em relac¸˜ao `a coordenada normal ´e calculada em
torno da posic¸˜ao de equil´ıbrio.
Podemos escrever o campo el´etrico da radiac¸˜ao incidente na forma:
~
E=~E0cos(ωit), (2.8)
ondeωi´e frequˆencia da radiac¸˜ao incidente. Supondo que as vibrac¸˜oes s˜ao harmˆonicas, podemos
escrever a coordenada vibracional como:
~
P=αo~E0cos(ωit) +
dα
dq
0
q0cos(ωqt)E~0cos(ωit), (2.10)
onde consideramos apenas os dois primeiros termos da s´erie. Na equac¸˜ao 2.10 temos a
de-pendˆencia do momento de dipolo el´etrico em termos do campo el´etrico da radiac¸˜ao incidente e
da coordenada normal de vibrac¸˜ao do sistema. Usando a identidade trigonom´etrica
cos(ωqt)cos(ωit) =
1
2cos(ωit+ωqt) + 1
2cos(ωit−ωqt) (2.11)
e aplicando-a na equac¸˜ao 2.10, obtemos o momento de dipolo el´etrico na forma
~P=αo~E0cos(ωit) +1 2 dα dq 0
q0~E0
1
2[cos(ωit+ωqt) +cos(ωit−ωqt)]. (2.12)
Se(dα/dq)0=0, a equac¸˜ao 2.12 se reduz a
~
P=αo~E0cos(ωit) (2.13)
e temos um dipolo oscilando com frequˆenciaωi(portanto emitindo radiac¸˜ao) igual a frequˆencia
do f´oton incidente, caracterizando o espalhamento Rayleigh. Se(dα/dq)06=0, al´em do
espa-lhamento Rayleigh, temos um termo onde a radiac¸˜ao espalhada aumenta a frequˆencia e ganha
energia (ωs =ωi+ωq), o que caracteriza o espalhamento Raman anti-Stokes, e outro termo
onde a radiac¸˜ao espalhada diminui de frequˆencia e perde energia (ωs =ωi−ωq),
caracteri-zando o espalhamento Raman Stokes.
A condic¸˜ao para existˆencia do espalhamento Raman ´e (dα/dq)06=0, ou seja, a
polariza-bilidade deve variar com uma pequena mudanc¸a na coordenada normal em torno da posic¸˜ao
de equil´ıbrio, do contr´ario, n˜ao temos espalhamento Raman. Podemos fazer uma descric¸˜ao do
fenˆomeno utilizando as func¸˜oes de onda dos estados final e inicial no processo de espalhamento
Raman reescrevendo a expans˜ao do tensor polarizabilidade como
(αi j) = (αi j)o+
dα
i j dq
0
q+1
2
d2αi j dq2
0
q2+... (2.14)
As componentes do tensor ser˜ao dadas por:
(αi j)mn= Z
ψm(αi j)ψndτ (2.15)
(αi j)mn= Z
ψm(αi j)0ψndτ+ Z ψm dα i j dq 0
12
(αi j)mn= (αi j)0
Z
ψmψndτ+
dα i j dq 0 Z
ψmqψndτ. (2.17)
Se o estado inicial for diferente do final, temosm6=ne consequentemente
Z
ψmψndτ =0. (2.18)
Se m = n, temos espalhamento Rayleigh. Se m 6=n, ent˜ao (αi j)mn 6= 0 se e somente se
(dαi j/dq)0 6= 0. Ent˜ao, m6=n implica que h´a variac¸˜ao em ao menos um dos componentes
do tensor polarizabilidade com a vibrac¸˜ao em torno da posic¸˜ao de equil´ıbrio, ocorrendo o
es-palhamento Raman. Outra propriedade importante a ser discutida ´e a paridade das func¸˜oes de
onda envolvidas no processo de espalhamento. Se tivermos
Z
ψmqψndτ6=0, (2.19)
ent˜aoψmqψn´e uma func¸˜ao par. Como o operadorq(posic¸˜ao) ´e uma func¸˜ao ´ımpar, ent˜aoψmψn
´e uma func¸˜ao ´ımpar, ou seja, ψm e ψn possuem diferentes paridades, e isso tem implicac¸˜oes
importantes para as regras de selec¸˜ao.
Fizemos uma ilustrac¸˜ao simples, com uma ´unica variac¸˜ao em torno da posic¸˜ao de equil´ıbrio
mas, de forma geral, as componentes do tensor polarizabilidade s˜ao dadas por [37]
(αi j)mn=
1
h
∑
r
hψn|µi|ψrihψr|µj|ψmi νrm−ν0+iΓr
+hψn|µj|ψrihψr|µi|ψmi
νrm+ν0+iΓr
. (2.20)
Os kets|ψmi,|ψnie|ψrirepresentam os estados inicial, final e virtual, com ´ındicesm,ner. µi eµj s˜ao as componentes do operador momento de dipolo el´etrico. Os ´ındicesie jrecebem
os valoresx, y e z dos eixos coordenados, νrm ´e a frequˆencia de transic¸˜ao eletrˆonica, ν0 ´e a
frequˆencia da radiac¸˜ao incidente eΓr ´e o fator de amortecimento relacionado ao tempo de vida
do estado virtual|ψri, que para o espalhamento Raman ´e da ordem de 10−15s. Os estados|ψmi, |ψni e|ψris˜ao mistos, envolvendo parte eletrˆonica e parte vibracional. Para o efeito Raman,
as transic¸˜oes entre os estados acontecem em dois passos: do estado inicial|ψmipara o estado
virtual|ψri, representado pelo brackethψr|µj|ψmiseguida da transic¸˜ao do estado virtual|ψri
para o estado final|ψni, representado pelo brackethψn|µi|ψri.
A intensidade Raman depende do quadrado do m´odulo do tensor de polarizabilidade, sendo
dada por
Imn=
16π2
9c4 Iiν
4
s
∑
i∑
j|(αi j)mn|2, (2.21)
pr´oximo da frequˆencia de excitac¸˜ao.
2.2
Dinˆamica de Rede
Quando um ´atomo pertencente a um cristal ´e desclocado da sua posic¸˜ao de equil´ıbrio, ele
sofre ac¸˜ao de forc¸as restauradoras e consequentemente vibra numa determinada frequˆencia
ca-racter´ıstica determinada pela dinˆamica dos fˆonons da rede cristalina. Devemos lembrar que
a rede cristalina ´e formada por uma infinidade de ´atomos unidos num arranjo peri´odico. As
vibrac¸˜oes desses ´atomos dependem do arranjo atˆomico e influenciam diretamente as
proprie-dades ´oticas e de transporte do cristal. Podemos ent˜ao estudar as proprieproprie-dades de um cristal
atrav´es do comportamento dos seus fˆonons. Num cristal h´a fˆonons ac´usticos e ´oticos, que
po-dem ser transversais ou longitudinais. A espectroscopia Raman ´e capaz de acessar os fˆonons
´oticos e os modos transversais tˆem relevˆancia especial para o nosso estudo pela sua relac¸˜ao ao
fenˆomeno de expans˜ao t´ermica negativa [38].
Nem sempre todas as propriedades vibracionais de um material (um s´olido cristalino em
nosso caso) podem ser facilmente acessadas num experimento. Uma das maneiras para
con-tornar essa dificuldade ´e discutir os resultados experimentais sob a luz de um modelo te´orico.
Entre as ferramentas te´oricas utilizadas para esta finalidade, quando se trata de propriedades
vibracionais, temos o C´alculo da Dinˆamica de Rede. Uma modelagen te´orica bastante usada
nesse tipo de c´alculo computacional ´e Modelo do ´Ion R´ıgido, que para alguns sistemas fornece
resultados satisfat´orios. Este modelo cosidera o ´atomo como uma esfera r´ıgida representando
o n´ucleo atˆomico e a carga eletrˆonica, tratando o material como um conjunto dessas esferas
in-teragindo entre si atrav´es de potenciais cl´assicos de curto e longo alcance. Para sistemas como
o nosso, a literatura frequentemente utiliza o chamado potencial inter-atˆomico de
Bucking-ham, que obtemos adicionando, respectivamente, o potencial de Coulomb; respons´avel pelas
interac¸˜oes de longo alcance; o termo de Born-Meyer, que representa as forc¸as repulsivas de
curto alcance e o termo de van der Waals; devido `as interac¸˜oes atrativas de curto alcance do tipo
dipolo-dipolo, resultando na equac¸˜ao:
Ui j(ri j) = zizj
ri j
e2+bi je−ri j/ρi j− ci j ri j6
(2.22)
onde zi e zj s˜ao as cargas efetivas dos ´ıons i e j separados pela distˆancia ri j. O raio iˆonico
14
modelo n˜ao descreve satisfatoriamente as interac¸˜oes covalentes fortes do tipo M-O (M = Mo,
W, V, etc). Quando este ´e o caso, ´e necess´ario incluir correc¸˜oes no modelo [38].
C´alculos de dinˆamica de rede utilizando o modelo do ´ıon r´ıgido tem mostrado bons
resulta-dos na previs˜ao das propriedades vibracionais de molibdatos e tungstatos, auxiliando inclusive a
compreens˜ao dos fenˆomenos de polimorfismo e amorfizac¸˜ao induzidos por press˜ao hidrost´atica
observados nesses compostos, [38–40].
2.3
T´ecnica para obter altas press˜oes hidrost´aticas
O princ´ıpio de funcionamento da c´elula do tipo bigorna de diamante (DAC - Diamond Anvil
Cell) consiste em comprimir uma amostra entre as faces de dois diamantes diretamente opostos
empurrados um contra o outro por uma ac¸˜ao externa, como ilustrado na Figura 2.3 [41]. O
volume de amostra utilizado ´e pequeno, resultando numa superf´ıcie de contato extremamente
reduzida entre o compartimento da amostra e os diamantes. Como a press˜ao ´e inversamente
proporcional `a ´area na qual a forc¸a ´e aplicada, ´e poss´ıvel induzir altas press˜oes dentro da c´elula
de press˜ao sem a necessidade de aplicar forc¸as demasiadamente intensas. As c´elulas atuais
permitem alcanc¸ar press˜oes da ordem de centenas de gigapascais. Lembrando que 1atm = 105 Pa, ´e poss´ıvel obter press˜oes maiores que um milh˜ao de vezes a press˜ao atmosf´erica.
Figura 2.3: Detalhe da bigorna de diamante, gaxeta e amostra. Adaptac¸˜ao do original [41].
Em geral, as montagens utilizadas para aplicar forc¸as nos diamantes definem os tipos de
c´elulas. Em nosso experimento utilizamos o tipo NBS [41], cujo esquema ´e mostrado na Figura
2.5. O princ´ıpio de funcionamento de uma c´elula NBS ´e bastante simples: ao girarmos o
Figura 2.4: Detalhe da gaxeta. Adaptac¸˜ao do original [41].
Figura 2.5: C´elula de press˜ao tipo NBS. Adaptac¸˜ao do original [41].
A gaxeta ´e uma folha met´alica resistente que tem por func¸˜ao limitar o volume da amostra.
A gaxeta possui uma identac¸˜ao para acomodar os diamantes de modo que eles mantenham-se
alinhados quando submetidos a forc¸as externas. Essa condic¸˜ao de alinhamento ´e crucial para
garantir a seguranc¸a da c´elula quanto `a quebra dos diamantes.
O compartimento da amostra ´e simplesmente um furo aberto na gaxeta com profundidade
e diˆametro da ordem de algumas dezenas de micrˆometros (200µm em nosso equipamento).
O compartimento armazena um fragmento do material a ser estudado e um fragmento de rubi
imersos em um fluido. O rubi ´e utilizado para medir a press˜ao interna e o fluido serve como
meio transmissor da press˜ao.
O fluido deve preencher a c´elula e distribuir a press˜ao aplicada na superf´ıcie da amostra de
maneira uniforme. Isto exige que a press˜ao aplicada n˜ao exceda o limite de press˜ao para o qual
o fluido deixa de responder hidrostaticamente (limite hidrost´atico do fluido), caso contr´ario, a
press˜ao no compartimento da amostra n˜ao ser´a a mesma em todos os pontos. Para este fim h´a
v´arios compostos e misturas conhecidas na literatura, como ´oleos de parafina, etanol e misturas
metanol:etanol [41].
Para medir a press˜ao dentro da c´elula de diamante utilizamos o deslocamento de frequˆencia
das linhas R1 e R2 do rubi sob variac¸˜ao da press˜ao. O deslocamento com a press˜ao das linhas
R1e R2 ´e linear entre 0 e 10 GPa e dentro desse limite podemos utilizar a equac¸˜ao abaixo para
16
P= ωRi−ω
0
Ri
0,7535 , (2.23)
sendo a press˜ao dada em GPa,ωRieωRi0 em cm−1, sendoω medido em relac¸˜ao `a excitac¸˜ao do
laser. A Figura 2.6 ilustra o deslocamento das linhas R1 e R2 dos espectros rubi em press˜oes
diferentes. Os valores das frequˆencias das linhas R1substitu´ıdos na equac¸˜ao 2.23 resultam 0 e
2,4 GPa.
Figura 2.6: Deslocamento do rubi em relac¸˜ao `a energia do laser (2,41 eV) utilizado na excitac¸˜ao.
2.4
Detalhes do experimento
Como fonte de luz para excitac¸˜ao da amostra utilizamos um laser de argˆonio da
Coher-ent (λ = 514,5 nm). Para focalizac¸˜ao empregamos um microsc´opio Olympus modelo BX-40
equipado com uma lente Nikon (20X, f = 20 mm). Para acumular os espectros utilizamos um
espectrˆometro Yvon Jobin modelo T64000 equipado com um detector tipo CCD refrigerado por
nitrogˆenio l´ıquido. A aquisic¸˜ao dos dados foi feita atrav´es de microcomputador com o software
LabSpec.
Nosso experimento de espectroscopia Raman em altas press˜oes consiste em variar a press˜ao
aplicada ao material, aguardar um tempo de relaxac¸˜ao necess´ario para a press˜ao
estabilizar-se, medir o espectro do rubi para obter o valor da press˜ao aplicada, acumular os espectros
didas Raman da amostra executadas em intervalos de press˜ao crescentes, entre 0,2 GPa a 0,5
GPa. Ao atingir o valor m´aximo de press˜ao, fazemos uma etapa de descompress˜ao, seguindo
metodologia similar `a etapa de compress˜ao, por´em, com intervalos de press˜ao decrescentes.
2.5
Tratamento dos dados experimentais
Obtidos os espectros Raman da amostra, precisamos fazer a deconvoluc¸˜ao dos modos
vi-bracionais utilizando uma func¸˜ao distribuic¸˜ao conhecida. Abaixo mostramos as distribuic¸˜oes
Lorentziana e Gaussiana, respectivamente:
y= a0 [1+ (x−a1
a2 )
2] (2.24)
y=a0exp[−ln(2)(
x−a1
a2
)2] (2.25)
Os parˆametros a0, a1 e a2 s˜ao respectivamente a intensidade, o centro e a meia largura
`a meia altura (HWHM - Half Width at Half Maximum). A distribuic¸˜ao Lorentziana ´e
ade-quada para o ajuste dos espectros. Quando o erro estat´ıstico da acumulac¸˜ao dos espectros ´e
significativo, torna-se conveniente ajustar os espectros Raman utilizando uma convoluc¸˜ao das
distribuic¸˜oes Lorentziana e Gaussiana, chamada func¸˜ao Voigt. Neste trabalho, utilizamos o
pa-cote Fityk para ajustar os espectros do molibdato de ´ıtrio com func¸˜oes Voigt, lembrando que o
Fityk emprega a seguinte forma para a Voigt:
y=
a0Rdt e
−t2
a23+[x−a1
a2 −t]
R dt et2
a23+t2
(2.26)
O ajuste dos espectros fornece os parˆametros de cada modo vibracional: intensidade, centro
e largura total `a meia altura (FWHM - Full Width at Half Maximum). Com os parˆametros
fornecidos pelos ajustes, podemos construir gr´aficos da evoluc¸˜ao da frequˆencia e da largura de
linha dos modos Raman em func¸˜ao da variac¸˜ao da press˜ao, cuja an´alise permite inferir sobre as
18
3
Resultados e Discuss˜oes
Apresentamos nesse cap´ıtulo os resultados obtidos para o molibdato de ´ıtrio (Y2(MoO4)3)
em altas press˜oes hidrost´aticas. Utilizamos uma an´alise de Teoria de Grupos e c´alculos da
dinˆamica de rede como suporte para a caracterizac¸˜ao inicial do material. Observamos uma
transic¸˜ao da fase ortorrˆombica para outra fase cristalina em 0,3 GPa. A reduc¸˜ao na quantidade
de modos observados no espectro Raman entre 1,4 GPa e 1,8 GPa foi interpretada como sendo
uma transic¸˜ao para uma fase desordenada de alta press˜ao. Discutimos o car´ater irrevers´ıvel
desta transic¸˜ao de fase em 2,4 GPa, visto que os modos caracter´ısticos do material observados
`a press˜ao ambiente n˜ao foram recuperados ap´os descompress˜ao.
3.1
An´alise de Teoria de Grupos
O molibdato de ´ıtrio utilizado em nosso estudo apresenta-se na fase cristalina ortorrˆombica
em condic¸˜oes de press˜ao e temperatura ambiente. Esse material pertence ao grupo espacial
Pbcn, D142h(mmm), comZ=4. A c´elula unit´aria ´e formada por 8 ´atomos de ´ıtrio ocupando s´ıtios de simetria C1equivalentes, 8 tetraedros MoO4ocupando s´ıtios de simetria C1equivalentes e 4
tetraedros MoO4ocupando s´ıtios de simetria C2n˜ao equivalentes, totalizando 68 ´atomos assim
distribu´ıdos:
D142h(P21/b2/c21/n) =dC1(8) +cC2Y(4) + (b+a)C1(4) (3.1)
Isoladamente, o tetraedro MoO4pertence ao grupo de ponto Tde pelo m´etodo da correlac¸˜ao
de s´ıtios, podemos obter as simetrias dos modos de vibrac¸˜ao quando esse ´ıon for incorporado na
rede cristalina do material cuja simetria ´e D142h. Resumimos nas tabelas 3.1 e 3.2 os resultados da an´alise de Teoria de Grupos para designar a simetria dos modos relativos aos tetraedros MoO4
localizados nos s´ıtios de simetrias C1e C2, respectivamente. A nomeclaturaν1se refere ao
esti-ramento sim´etrico,ν2 `a flex˜ao sim´etrica,ν3ao estiramento assim´etrico,ν4 `a flex˜ao assim´etrica,
A1(ν1) A Ag 1ν11S 2ν23R 3ν33ν43T
A2(S) Au 1ν11S 2ν23R 3ν33ν43T
E(ν2) B1g 1ν11S 2ν23R 3ν33ν43T
T1(R) B1u 1ν11S 2ν23R 3ν33ν43T
T2(ν3,ν4,T) B2g 1ν11S 2ν23R 3ν33ν43T
B2u 1ν11S 2ν23R 3ν33ν43T
B3g 1ν11S 2ν23R 3ν33ν43T
B3u 1ν11S 2ν23R 3ν33ν43T
Tabela 3.2: Tabela de correlac¸˜ao dos modos para 4MoO4ocupando s´ıtio C2na simetria D2h.
Td C2 D2h Modos
A1(ν1) A Ag 1ν11S 2ν21R 1ν31ν41T
A2(S) Au 1ν11S 2ν21R 1ν31ν41T
E(ν2) B1g 2R 2ν32ν42T
B1u 2R 2ν32ν42T
T1(R) B B2g 2R 2ν32ν42T
T2(ν3,ν4,T) B2u 2R 2ν32ν42T
B3g 1ν11S 2ν21R 1ν31ν41T
B3u 1ν11S 2ν21R 1ν31ν41T
Atrav´es das tabelas 3.1 e 3.2, obtemos o total de 180 modos vibracionais dos tetraedros
MoO4:
ΓMoO4 =22Ag+22Au+23B1g+23B1u+23B2g+23B2u+22B3g+22B3u (3.2)
Os modos vibracionais dos ´atomos de ´ıtrio ocupando s´ıtios de simetria C1s˜ao dados por:
ΓY =3Ag+3Au+3B1g+3B1u+3B2g+3B2u+3B3g+3B3u (3.3)
Somando os modos dos ´atomos de ´ıtrio e dos tetraedros MoO4, obtemos o total de modos
vibracionais do Y2(MoO4)3, assim distribu´ıdos nas representac¸˜oes irredut´ıveis do grupo de
ponto D2h:
ΓTotal =25Ag+25Au+26B1g+26B1u+26B2g+26B2u+25B3g+25B3u (3.4)
Temos ao todo 204 graus de liberdade no centro da zona de Brillo¨uin Γ. Pelas regras de
selec¸˜ao do grupo D142h (mmm), apenas os modos de simetria Ag, B1g, B2g e B3g s˜ao ativos na
espectroscopia Raman, pois s˜ao os ´unicos cujas func¸˜oes de base s˜ao quadr´aticas. Os modos com
20
simetria Aus˜ao ac´usticos. Ent˜ao, temos um total de 102 modos previstos pela Teoria de Grupos
que podem ser observados pela t´ecnica de espectroscopia Raman no molibdato de ´ıtrio:
ΓRaman=25Ag+26B1g+26B2g+25B3g (3.5)
3.2
C´alculos de dinˆamica de rede
Efetuamos os c´alculos de dinˆamica de rede utilizando o software GULP desenvoldido por
J.J. Gale [43]. Esse programa trata o sistema em estudo de acordo com a teoria do ´ıon r´ıgido
dis-cutida no cap´ıtulo anterior. Os valores listados na Tabela 3.3 referentes aos parˆametrosbi j,ρi j e
ci jdo potencial de Buckingham s˜ao ajustados de forma a se ter melhor correspondˆencia entre os
c´alculos e os experimentos. Adotamos as cargas efetivaszi j dos ´ıons (indicados pelos ´ındices)
com valores: ZY =3, ZMo =6, ZO=−2.84819. Como fator de correc¸˜ao, adicionamos um
potencial harmˆonico com constante de molak0=74.92mDynA˚−1 representando as interac¸˜oes
entre os ´atomos de oxigˆenio.
Tabela 3.3: Valores dos parˆametros utilizados no potencial de Buckingham. Ligac¸˜ao b(eV) ρ( ˚A) c(eV ˚A6)
Mo - O 1285,2 0,375 0
Y - O 16,19x106 0,14 0
O - O 22764 0,149 27,00
As frequˆencias dos fˆonons foram calculadas utilizando o m´etodo da matriz FG de Wilson
e o software VIBRATZ desenvolvido por Dowty [44]. As constantes de forc¸a iniciais foram
obtidas atrav´es da relac¸˜ao:
fi j =−
1
r
∂Ui j(r)
∂r (3.6)
onde os ´ındices i e j indicam os ´ıons interagentes e r ´e a distˆancia que os separa. Podemos
observar os resultados dos c´alculos das constantes de forc¸a versus a distˆancia das ligac¸˜oes na
Figura 3.1.
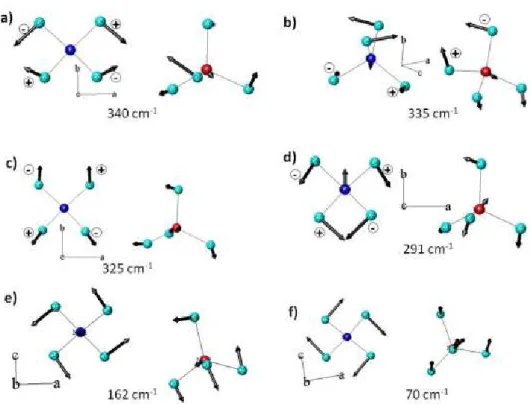
Nossos c´alculos mostram que os modos Raman do tetraedro MoO4na regi˜ao 800-970 cm−1
correspondem `as vibrac¸˜oes de estiramento e os modos na regi˜ao 200-380 cm−1correspondem aos modos de flex˜ao. Os resultados tamb´em mostram que os modos abaixo de 200 cm−1 s˜ao fortemente acoplados e envolvem librac¸˜oes e translac¸˜oes das unidades MoO4e translac¸˜oes dos
´atomos deY. As frequˆencias calculadas para os modos vibracionais e suas respectivas esp´ecies
s˜ao mostradas na tabela 3.4. As vibrac¸˜oes dos tetraedros MoO4est˜ao ilustradas nas Figuras 3.2
Tabela 3.4: Frequˆencias e identificac¸˜ao dos modos vibracionais dos tetraedros MoO4calculados
usando dinˆamica de rede.
Modos Descric¸˜ao dos modos Frequˆencias calculadas cm−1
T translac¸˜ao Y - MoO4 70
162
L librac¸˜ao 291
ν2,ν4 flex˜ao sim´etrica e assim´etrica 325
335 340
ν3 estiramento assim´etrico 828
842
ν1 estiramento sim´etrico 896
937 952 994
22
Figura 3.2: Autovetores e autovalores para os modos de baixa energia dos tetraedros MoO4para
a representac¸˜ao irredut´ıvel Ag.
Figura 3.3: Autovetores e autovalores para os modos de alta energia dos tetraedros MoO4para
O molibdato de ´ıtrio (Y2(MoO4)3) ´e altamente higrosc´opico [36], exigindo alguns cuidados
quando o objetivo ´e medir o espectro Raman da fase anidra. A amostra Y2(MoO4)3·3H2O foi
colocada em um criostato e aquecida `a temperatura de 120◦C em condic¸˜oes de v´acuo de 10−3 torr. Deixamos o sistema resfrirar at´e a temperatura ambiente em condic¸˜oes de v´acuo por 12
horas antes da medida do espectro Raman da fase anidra, mostrado na Figura 3.4. Para efeito
de comparac¸˜ao, mostramos na mesma Figura o espectro Raman obtido para a fase hidratada.
A partir da an´alise de deconvoluc¸˜ao dos espectros usando func¸˜oes do tipo Voigt, obtivemos os
deslocamentos Raman e as larguras de linha.
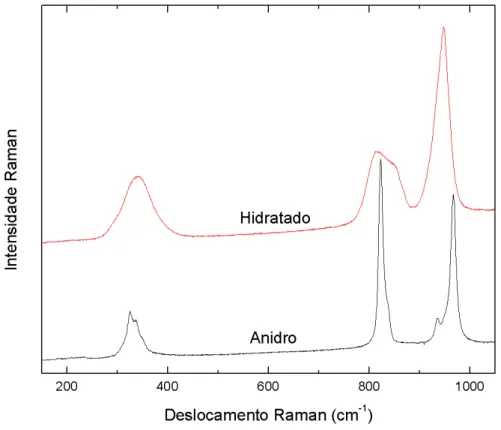
Figura 3.4: Espectros Raman do molibdato de ´ıtrio nas fases anidra (Y2(MoO4)3) e hidratada
(Y2(MoO4)3·3H2O) medidos em condic¸˜oes de temperatura e press˜ao ambiente.
Em nosso experimento, a fase hidratada mostra trˆes bandas largas e altamente degeneradas,
centradas em 340, 831 e 952 cm−1. Lianget al. relatam essas bandas em torno de 340, 828 (836 em outro trabalho) e 945 cm−1, classificando-as respectivamente como flex˜ao sim´etrica e assim´etrica (ν2eν4), estiramento assim´etrico (ν3) e estiramento sim´etrico (ν1) [32, 36].
24
Notamos que os modos de baixa frequˆencia (atribu´ıdos `a rede cristalina) apresentam intensidade
relativa baixa. Comparando estes resultados com a literatura, constatamos que nossas amostras
de Y2(MoO4)3est˜ao na fase ortorrˆombica e com boa qualidade cristalina [32, 36].
No espectro da Figura 3.4, podemos distinguir apenas 13 modos. Essa grande diferenc¸a
entre a quantidade de modos previstos pela Teoria de Grupos e a quantidade de modos observada
experimentalmente ´e devido ao fato de que realizamos medidas em amostras policristalinas e
que o espectro deve ter muitas degenerescˆencias acidentais. Comparando nossos resultados
obtidos pela aplicac¸˜ao da Teoria de Grupos com a literatura [33], podemos identificar os modos
observados experimentalmente. Nomeamos e listamos esses modos na tabela 3.5 para facilitar
nossa an´alise dos experimentos em condic¸˜oes de altas press˜oes.
Tabela 3.5: Identificac¸˜ao do modos do Y2(MoO4)3observados experimentalmente.
Modo Descric¸˜ao Frequˆencia (cm−1) Nomeclatura Esp´ecie
ν2,ν4 flex˜ao sim´etrica e assim´etrica 324 A Ag+B2g
337 B B3g
352 C Ag+B2g
ν3 estiramento assim´etrico 825 D B1g
836 E B3g
ν1 estiramento sim´etrico 937 F B3g
955 G
969 H
3.4
Y
2(MoO
4)
3sob altas press˜oes hidrost´aticas
A amostra policristalina de Y2(MoO4)3foi aquecida num cadinho `a temperatura de 120◦C,
sendo em seguida envolvida com o meio compressor (´oleo mineral comercial - Nujol) e
imedi-atamente colocada dentro da c´elula de press˜ao a extremos de diamante. Ap´os o resfriamento,
constatamos que o ´oleo “blindou” a amostra contra a umidade, conservando-a anidra, como
podemos verificar no primeiro espectro medido dentro da c´elula de press˜ao (Figura 3.5) ao
compar´a-lo com o espectro medido dentro do criostato (Figura 3.4), nos quais identificamos
imediatamente os mesmos modos. Isto nos permite continuar o experimento sem preju´ızo para
nossas considerac¸˜oes feitas anteriormente sobre a caracterizac¸˜ao da amostra.
Em cada medida, tomamos duas acumulac¸˜oes de 30 segundos. A baixa intensidade dos
modos do espectro da amostra dentro da c´elula de press˜ao, por causa do volume reduzido da
amostra, tornou necess´ario o ajuste da potˆencia de sa´ıda do laser para 200 mW (dobro do valor
Figura 3.5: Espectro Raman do Y2(MoO4)3 logo ap´os ser transferido para o interior da c´elula
de press˜ao.
(tempo de relaxac¸˜ao do sistema) ap´os variarmos a press˜ao. Ressaltamos que durante cada
in-tervalo de acumulac¸˜ao n˜ao notamos variac¸˜ao significativa na energia da linha do rubi, ou seja,
a press˜ao aplicada manteve-se constante para cada medida. Ao todo, obtivemos 26 espectros
durante o regime de compress˜ao (de 0 a 9,6 GPa) e mais 16 espectros durante o regime de
descompress˜ao (at´e o m´ınimo de 0,2 GPa).
O ajuste do espectro da Figura 3.5 mostra que as frequˆencias e larguras dos picos observados
s˜ao quase idˆenticos `aqueles medidos para o Y2(MoO4)3fora da c´elula, cujo espectro mostramos
na Figura 3.4. A diferenc¸a notada entre as frequˆencias dos modos observados no experimento
`a press˜ao ambiente e no primeiro espectro do experimento sob press˜ao (ver as Figuras 3.4 e
3.5) n˜ao ´e significativa e pode ser devida a uma diferenc¸a muito pequena entre a press˜ao
ambi-ente e a press˜ao inicial medida dentro da c´elula, tomada como zero no experimento. Os modos
observados com frequˆencia inferior `a 250 cm−1possuem baixa intensidade relativa, sendo des-considerados em nossa primeira an´alise ao observarmos que esta caracter´ıstica acentua-se com o
aumento da press˜ao, tornando qualquer an´alise quantitativa imprecisa. Apesar desses modos de
26
os modos da rede), veremos que o comportamento dos modos de flex˜ao (‘bending’) e
estira-mento (‘stretching’) em func¸˜ao da press˜ao revela caracter´ısticas suficientes para descrevermos
o comportamento do molibdato de ´ıtrio sob altas press˜oes.
Figura 3.6: Espectros Raman do Y2(MoO4)3medidos durante a compress˜ao entre 0 e 2,4 GPa.
Na Figura 3.6 mostramos os espectros Raman caracter´ısticos da evoluc¸˜ao do Y2(MoO4)3
com o aumento da press˜ao. Partindo da press˜ao m´ınima (0 GPa), notamos de imediato a perda
de intensidade em todos os modos com o aumento da press˜ao. Observamos tamb´em que os
modos de flex˜ao A e B gradualmente perdem intensidade relativa em comparac¸˜ao com o modo
C (flex˜ao) entre 0 e 1,1 GPa, tendendo progressivamente a colapsar em uma ´unica banda. Em
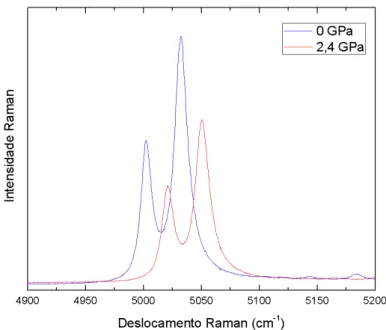
torno de 0,3 GPa, verificamos que a largura do modo C tem um grande aumento (4x
aprox-imadamente). Como pode ser conferido na Figura 3.9, o modo H (estiramento sim´etrico),
centrado em 969 cm−1, se divide (‘splitting’) e observamos um novo modo centrado em 978 cm−1 (chamaremos de modo I), indicando uma transic¸˜ao da fase ortorrˆombica para outra fase cristalina menos sim´etrica (maior quantidade de modos vibracionais implica simetria mais baixa
do sistema). Em torno de 0,6 GPa podemos notar que a separac¸˜ao dos modos H e I torna-se
bastante evidente. Tamb´em notamos que o modo G (estiramento sim´etrico) ganha intensidade
abruptamente em relac¸˜ao ao modo F (estiramento sim´etrico), implicando na presenc¸a de uma
em altas press˜oes aplicadas ao molibdato de escˆandio (Sc2(MoO4)3) mostra uma transic¸˜ao da
fase ortorrˆombica (em condic¸˜ao ambiente) para outra fase cristalina em 0,29 GPa [21]. Uma
an´alise comparativa dos resultados em altas press˜oes com experimentos Raman em baixas
tem-peraturas para este material sugere tratar-se da fase monocl´ınica, de simetria menor que a
or-torrˆombica [21]. Devido a semelhanc¸a desses dois materiais em termos de estrutura cristalina
(em condic¸˜oes de press˜ao e temperatura ambiente), ´e prov´avel que a fase do molibdato de ´ıtrio
estabilizada para press˜oes maiores que 0,3 GPa possa ser associada tamb´em a uma fase com
estrutura monocl´ınica.
A partir de 1,4 GPa, vemos na Figura 3.6 que os modos de flex˜ao A e B desaparecem,
a largura do modo C duplica (ver Figura 3.9 e este passa a ocupar o espac¸o onde antes
en-contr´avamos os trˆes modos de dobramento. O modo H (estiramento sim´etrico) tamb´em n˜ao ´e
mais observado para este valor de press˜ao. Os modos restantes (C, D, E, F, G e I) tornam-se
menos resolvidos no espectro, apresentando-se como bandas largas. Para esta press˜ao,
verifi-camos tamb´em o alargamento dos modos de estiramento sim´etrico F e G, como pode ser
con-statado nos detalhes da Figura 3.11). A partir dessas observac¸˜oes, sugerimos o in´ıcio de outra
transic¸˜ao de fase, mas desta vez, para uma fase de alta press˜ao possivelmente n˜ao cristalina
devido `a acentuada largura das bandas que traduzem a desordem estrutural induzida no material
pela press˜ao hidrost´atica [21, 23, 31]. Uma observac¸˜ao meramente qualitativa da evoluc¸˜ao da
forma dos espectros com a press˜ao ilustrada na Figura 3.6 mostra uma tendˆencia de alargamento
e supress˜ao de todos os modos. Entre 1,4 GPa e 1,8 GPa, verificamos atrav´es da Figura 3.10 um
comportamento crescente da largura dos modos D e E (estiramento assim´etrico) com o aumento
da press˜ao.
Em torno de 2,4 GPa (ver Figura 3.6), os modos de estiramento sim´etrico F e I tamb´em
deixam de ser distingu´ıveis. A partir deste valor de press˜ao, os espectros apresentam apenas
quatro modos (C, D, E e G) extremamente largos e de baix´ıssima intensidade. Os espectros
medidos em press˜oes maiores acentuam a tendˆencia j´a observada entre 1,4 GPa e 2,4 GPa,
havendo gradual perda na intensidade e aumento da largura de linha. Os picos bem resolvidos
dos trˆes conjuntos observados em baixa press˜ao colapsam para formar em alta press˜ao trˆes
bandas largas. O alargamento pronunciado dos modos restantes nos permite concluir que houve
perda da simetria translacional e da correlac¸˜ao de longo alcance do cristal, em outras palavras,
alcanc¸amos uma fase desordenada de alta press˜ao em 2,4 GPa e possivelmente temos o in´ıcio
do processo de amorfizac¸˜ao induzida pela press˜ao. O espectro (ilustrado na Figura 3.8) medido
28
tungstato de ´ıtrio (Y2(WO4)3) em 10,8 GPa, j´a caracterizado na sua fase amorfa [23]. Isto nos
permite sugerir que o molibdato de ´ıtrio pode tornar-se amorfo em press˜oes superiores ao limite
estudado neste trabalho.
Figura 3.7: Espectros Raman do Y2(MoO4)3medidos durante a descompress˜ao entre 5,3 GPa
e 0,3 GPa).
A reversibilidade das transformac¸˜oes estruturais observadas fornece informac¸˜oes
impor-tantes sobre as mudanc¸as cristalino-desordenado. Realizamos medidas de espalhamento Raman
durante o processo de descompress˜ao, conforme ilustrado na Figura 3.7. Podemos observar que
a descompress˜ao n˜ao provocou variac¸˜oes significativas na forma dos espectros entre 9,1 GPa
e 5,7 GPa. A Figura 3.7 mostra as oito ´ultimas medidas no regime de descompress˜ao, onde
podemos notar uma leve diminuic¸˜ao nas larguras de linha, indicando uma reduc¸˜ao no grau de
desordem `a medida que a press˜ao diminui. O comportamento da largura de linha (ver Figura
3.12) e da posic¸˜ao dos modos (ver Figura 3.13) indicam a manutenc¸˜ao da fase desordenada,
indicando que a transformac¸˜ao cristalino-desordenado ´e irrevers´ıvel.
Percebemos que o ´ultimo espectro medido no regime de descompresss˜ao em 0,2 GPa ´e
muito semelhante ao espectro medido no regime de compress˜ao em 2,4 GPa, valor para o qual
estabelecemos o aparecimento da fase desordenada de alta press˜ao. A Figura 3.8 apresenta um
comparativo espec´ıfico entre estes espectros. Essas observac¸˜oes mostram claramente que a fase
Figura 3.8: Comparac¸˜ao dos espectros tomados em 2,4 GPa (regime de compress˜ao), 9,6 GPa (m´axima press˜ao atingida em nosso experimento) e 0,2 GPa (regime de descompress˜ao).
0,3 GPa. Ao fim do experimento, obtivemos a mesma fase desordenada alcanc¸ada em 2,4 GPa,
mostrando que apenas a descompress˜ao n˜ao ´e capaz de devolver a fase original do molibdato
de ´ıtrio.
Procedemos com uma an´alise mais detalhada sobre a dependˆencia da frequˆencia dos modos
com a variac¸˜ao da press˜ao. Montamos ent˜ao o gr´aficoω (cm−1) versus P (GPa) mostrado na Figura 3.13, cujos pontos representam as frequˆencias onde os modos est˜ao centrados para os
respectivos valores de press˜ao nos quais foram observados. As linhas s´olidas representam os
ajustes para esses pontos utilizando a equac¸˜ao:
ω(P) =ω0+αP. (3.7)
Determinamos os coeficientes α =∂ ω/∂P das retas ajustadas na Figura 3.13. Os
valo-res das frequˆenciasω0 obtidas das retas est˜ao listados na tabela 3.6. A ocorrˆencia de valores
negativos paraα ´e comum em compostos que apresentam expans˜ao t´ermica negativa [14]. Os
30
Figura 3.9: Variac¸˜ao da largura dos modos de flex˜ao com a press˜ao.
Figura 3.10: Variac¸˜ao da largura dos modos de estiramento assim´etrico com a press˜ao.
que os valores encontrados para os modos de mesma esp´ecie na fase cristalina. Este
compor-tamento ´e invertido para os modos C e E. Podemos relacionar isto com a reduc¸˜ao do volume
do material, lembrando que a press˜ao aplicada forc¸a uma reduc¸˜ao das distˆancias interatˆomicas
Figura 3.11: Variac¸˜ao da largura dos modos de estiramento sim´etrico com a press˜ao.
Figura 3.12: Variac¸˜ao da largura dos modos remanescentes durante a descompress˜ao.
Na literatura [21] ´e relatado para o Sc2(MoO4)3 que se a press˜ao aplicada for
suficiente-mente alta para empurrar os tetraedros MoO24− uns contra os outros, a forte repuls˜ao entre eles em conjunto com a reduc¸˜ao do volume impedem uma reconFigurac¸˜ao do material, levando `a
perda da correlac¸˜ao de longo alcance e consequente desordenamento da estrutura cristalina.
32
Figura 3.13: Variac¸˜ao da posic¸˜ao dos modos do Y2(MoO4)3durante a compress˜ao.
Tabela 3.6: Valores deα (cm−1GPa−1) eω0(cm−1) determinados para: Fase 1 (entre 0,3 e 1,1
GPa) e Fase 2 (entre 2,4 GPa e 9,6 GPa).
Fase 1 Fase 2
Modo ω0 α ω0 α
A 323,8 -0,6
B 336,0 -0,9
C 349,6 14,9 351,6 7,1
D 824,6 4,4 728,5 5,2
E 834,7 8,6 849,8 3,1
F 937,6 -2,7
G 958,5 -1,5 940,4 0,6
H 968,3 -2,6
I 976,9 2,7
considerac¸˜oes te´oricas.
Para finalizar nosso estudo, ainda precisamos descobrir qual fase cristalina foi obtida em
0,3 GPa para o Y2(MoO4)3. Como mais ind´ıcios, podemos lembrar que diversos molibdatos e
tungstatos (inclu´ındo nossa amostra) apresentam uma sequˆencia de transic¸˜oes de fase sob altas
press˜oes, passando de uma fase cristalina para outra fase cristalina de simetria mais baixa e
pos-teriormente para uma fase desordenada [21] e estudos em molibdatos de terras raras sugerem
que estes compostos transitam da fase ortorrˆombica para monocl´ınica antes de amorfizar-se
experi-3.5
Y
2(MoO
4)
3·
xH
2O sob altas press˜oes hidrost´aticas
Embora apenas a fase anidra do molibdato de ´ıtrio mostre o fenˆomeno de expans˜ao t´ermica
negativa e portanto o torna um material mais atrativo, realizamos tamb´em experimentos de
espectroscopia Raman em altas press˜oes para a fase hidratada. Nosso objetivo ´e comparar os
resultados com aqueles obtidos para a fase anidra na tentativa de revelar o papel das mol´eculas
de H2O na estabilidade do molibdato de ´ıtrio.
Colocamos uma amostra de molibdato de ´ıtrio policristalina e hidratada dentro da c´elula
de press˜ao a extremos de diamante, utilizando novamente o ´oleo comercial Nujol como meio
transmissor de press˜ao. Em cada medida, foram realizadas duas acumulac¸˜oes de 180 segundos.
Obtivemos 16 espectros durante o regime de compress˜ao (de 0 a 6,2 GPa) e mais 12 espectros
durante o regime de descompress˜ao (press˜ao ambiente).
Figura 3.14: Espectro Raman do Y2(MoO4)3·xH2O (x<3) ap´os ser transferido para a c´elula
de press˜ao.
O primeiro espectro medido no interior da c´elula de press˜ao exibe as trˆes bandas
anterior-mente identificadas durante a caracterizac¸˜ao da amostra: 340 cm−1 (ν2), 831 cm−1(ν3) e 952
34
931 cm−1, como pode ser observado no espectro ilustrado na Figura 3.14. Lianget al.atribuem esses trˆes modos adicionais `a fase hidratada com f´ormula Y2(MoO4)3·xH2O (x<3) [36], que
identificamos como sendo a fase investigada neste experimento de press˜ao. A evoluc¸˜ao dos
espectros com o aumento da press˜ao est´a ilustrada na Figura 3.15.
Figura 3.15: Espectros do Y2(MoO4)3·xH2O (x<3) em func¸˜ao da press˜ao hidrost´atica.
A partir de 0,8 GPa, os modos com frequˆencias de partida iguais a 814 cm−1, 831 cm−1 e 850 cm−1 tendem a colapsar em uma ´unica banda com frequˆencia em torno de 834 cm−1, formando claramente uma banda larga para valores de press˜ao em torno de 1,2 GPa. A Figura
3.16 mostra que os modos de frequˆencias iniciais 814 cm−1e 850 cm−1n˜ao s˜ao mais observa-dos em 0,8 GPa e 1,2 GPa, respectivamente. A partir de 3,3 GPa, os moobserva-dos com frequˆencias de
partida iguais a 931 cm−1e 952 cm−1 evoluem para uma banda com frequˆencia de 945 cm−1, exibindo comportamento idˆentico ao observado para a fase anidra em torno dos mesmos
valo-res de pvalo-ress˜ao, indicando poss´ıvel in´ıcio de um processo de amorfizac¸˜ao induzida pela pvalo-ress˜ao
hidrost´atica nessa fase hidratada. Tamb´em ressaltamos que o espectro medido em 6,2 GPa
in-dica uma reduc¸˜ao significativa na correlac¸˜ao de longo alcance do material, apresentando forma
t´ıpica de um espectro de fase amorfa, como ´e observado na literatura para o Y2(WoO4)3 [23].
Ap´os a descompress˜ao do sistema, a amostra retornou para o estado desordenado observado em
2,4 GPa durante a compress˜ao, como pode ser conferido na Figura 3.15.
![Figura 2.3: Detalhe da bigorna de diamante, gaxeta e amostra. Adaptac¸˜ao do original [41].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15258108.539076/27.892.296.645.694.983/figura-detalhe-bigorna-diamante-gaxeta-amostra-adaptac-original.webp)
![Figura 2.5: C´elula de press˜ao tipo NBS. Adaptac¸˜ao do original [41].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15258108.539076/28.892.219.717.318.521/figura-c-elula-press-tipo-nbs-adaptac-original.webp)