Rev. paul. pediatr. vol.35 número2
Texto
Imagem
Documentos relacionados
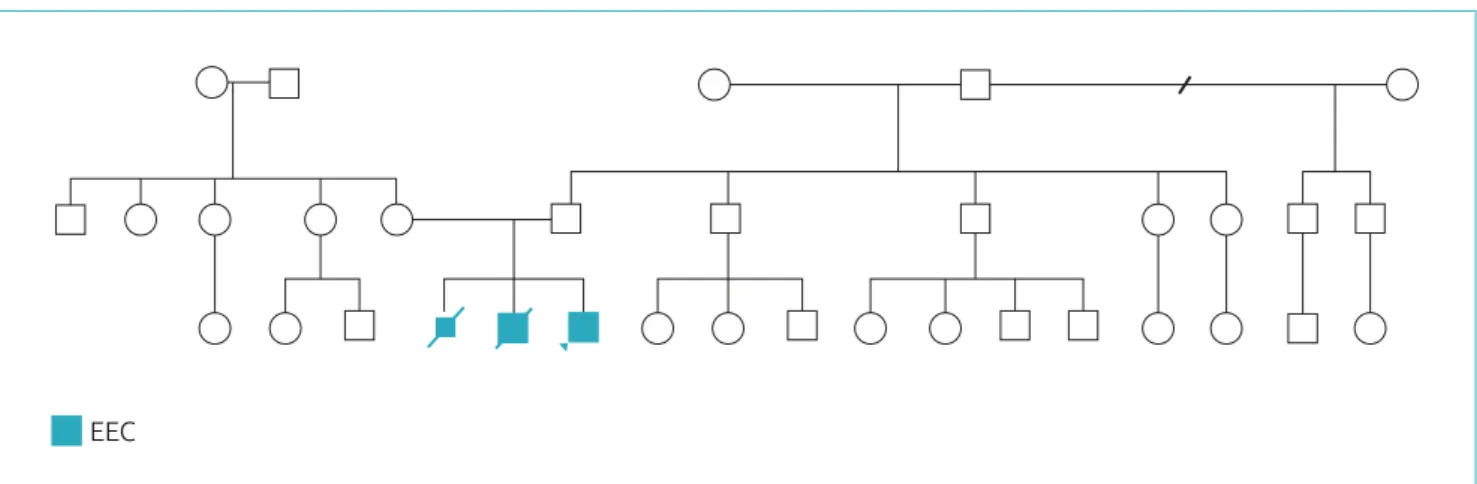
13 na Clínica Cleveland, foram encontrados os seguintes preditores de recorrência de FA após cirurgia de CM associados à correção valvar mitral: maior duração da
Pain in the affected region was the initial symptom in 52% of patients, and 45.5% of the patients had low back pain, and those with dorsal discitis had back pain as the main
Our sample of patients with cleft lip and palate and hearing loss fitted with hearing aids may be characterized mainly as patients with cleft lip and palate with a history of
Consi- dering the type of cleft, 2 patients had incomplete direct cleft lip associated with the incomplete palate cleft (Fig. 1); 2 had incomplete left cleft lip plus incomplete
It was also noted, by analyzing mul- tinomial logistic regression that the risk of occurrence of cleft lip in relation the cleft palate was 2.19 times in males compared to
We describe a rare case of a patient with 18p deletion syndrome presenting focal dystonia, Hashimoto thyroidi- tis, vitiligo and cerebral white matter
Luciana e Mariana - Se pensarmos a dramaturgia fora do logocentrismo como um conceito expandido que inclui a articulação de sentidos e efeitos num espetá- culo, é possível pensar
Os motores ciclo Otto com aspiração natural são os que mais sofrem com essa diferença de pressão, já os motores equipados com turbo não sofrem tanto com
