Pró-Reitoria Acadêmica
Escola de Saúde e Medicina
Programa de Pós-Graduação
Stricto Sensu
em Ciências
Genômicas e Biotecnologia
ANÁLISE DO METAGENOMA VIRAL DE AMOSTRAS DE
FEZES HUMANAS DO DISTRITO FEDERAL
Brasília - DF
2015
RAYANE NOGUEIRA DOS SANTOS
ANÁLISE DO METAGENOMA VIRAL DE AMOSTRAS DE FEZES HUMANAS DO DISTRITO FEDERAL
Dissertação apresentada ao Programa de Pós –
Graduação Stricto Sensu em Ciências
Genômicas e Biotecnologia da Universidade Católica de Brasília, como requisito parcial para obtenção do Título de Mestre em Ciências Genômicas e Biotecnologia.
Orientadora: Prof.ª Dr.ª Cristine Chaves Barreto.
Coorientadora: Prof.ª Dr.ª Paula Andréia Silva.
S237a Santos, Rayane Nogueira dos.
Análise do metagenoma viral de amostras de fezes humanas do Distrito Federal. / Rayane Nogueira dos Santos – 2015.
46 f.; il.: 30 cm
Dissertação (Mestrado) – Universidade Católica de Brasília, 2015.
Orientação: Profa. Dra. Cristine Chaves Barreto Coorientação: Profa. Dra. Paula Andréia Silva
1. Biotecnologia. 2. Metagenoma viral. 3. Viroma. 4. Patógenos virais. I.
Barreto, Cristine Chaves, orient. II. Título.
Dissertação de autoria de Rayane Nogueira dos Santos, intitulada “ANÁLISE DO METAGENOMA VIRAL DE AMOSTRAS DE FEZES HUMANAS DO DISTRITO FEDERAL” apresentada como requisito parcial para obtenção de grau de mestre em Ciências Genômicas, em 27/03/2015, defendida e aprovada pela banca examinadora abaixo assinada:
_______________________________________________________ Prof.ª Dr.ª Cristine Chaves Barreto
Orientadora
(Programa de Pós-Graduação Stricto Sensu em Ciências Genômicas e Biotecnologia – UCB)
_______________________________________________________ Prof.ª Dr.ª Paula Andréia Silva
Coorientadora
(Programa de Pós-Graduação Stricto Sensu em Ciências Genômicas e Biotecnologia – UCB)
_______________________________________________________ Prof.º Dr.º Robert Edward Pogue
Membro Interno
(Programa de Pós-Graduação Stricto Sensu em Ciências Genômicas e Biotecnologia – UCB)
_______________________________________________________ Prof.º Dr.º Tatsuya Nagata
Membro Externo (Biologia Celular – UnB)
____________________________________________________ Prof.º Dr.º Fernando Lucas de Melo
Suplente
(Biologia Celular – UnB)
Dedico este trabalho aos meus pais, irmão, marido e toda família por todo amor, carinho e apoio demonstrado e aos meus
RESUMO
SANTOS, Rayane Nogueira dos. Análise do metagenoma viral de amostras de fezes humanas do distrito federal. 2015. 46 folhas. Mestrado em Ciências Genômicas e Biotecnologia – Universidade Católica de Brasília, 2015.
A gastroenterite aguda, especialmente em países em desenvolvimento, é uma importante causa de mortalidade e morbidade, que atinge pessoas de todas as classes sociais. O conhecimento do agente etiológico nestas infecções auxilia a escolha da estratégia de tratamento, como também direciona estudos epidemiológicos para medidas de controle e prevenção de doença, como o desenvolvimento de vacinas e testes diagnósticos. Abordagens metagenômicas possibilitam a detecção de sequências virais, para determinar a população viral presente nas fezes. Assim, o objetivo desse trabalho foi avaliar o viroma de fezes humanas em 4 grupos distintos, sendo composto por amostras fecais diarreicas de crianças, diarreicas de adultos, não diarreicas de adultos e de imunocomprometidos, identificando prováveis patógenos envolvidos nos quadros de gastroenterite. O método de execução da metagenômica viral consistiu na semi-purificação, extração de RNA e DNA, amplificação do RNA e DNA, construção da biblioteca, sequenciamento e análises dos dados por bioinformática. Na análise do primeiro grupo houve detecção de bacteriófagos, astrovírus e torque teno vírus; no segundo grupo foram identificados, entre outros, bacteriófagos, adenovírus e torque teno vírus; no terceiro grupo vírus da família Circoviridae e de
bacteriófagos; no quarto grupo adenovírus, torque teno vírus, gyrovirus e papilomavírus humano. Esses resultados enfatizam o que tem se identificado na literatura e fornece evidência de que a metagenômica viral tem facilitado os avanços no campo da virologia, sendo esta, uma técnica sensível para a detecção de vírus que não podem ser identificados por cultura tradicional.
ABSTRACT
Acute gastroenteritis, especially in developing countries, is a major cause of mortality and morbidity, which affects people of all social classes. The knowledge of etiological agent assists the choice of a treatment strategy, but also directs epidemiological studies for control measures and disease prevention, as the development of vaccines and diagnostic tests. Metagenomic approaches enable the detection of viral sequences to determine the virus population present in the stool. The objective of this study was to evaluate the viroma of human feces in 4 distinct groups, consisting of diarrheal stool samples from children, diarrheal from adults, no diarrhea from adults and immunocompromised, identifying likely pathogens involved in cases of gastroenteritis. The viral metagenomic execution method consists of the semi-purification, extraction of RNA and DNA, amplification of RNA and DNA, library construction, sequencing and analysis of the data by bioinformatics. In the analysis of the first group were detecting bacteriophages, astrovirus and torque teno virus; in the second group were identified, among others, bacteriophages, adenoviruses and torque teno virus; the third group of viruses Circoviridae family and bacteriophages; in the fourth group
adenovirus, torque teno virus, gyrovirus and human papillomavirus. These results emphasize what has been identified in the literature and provides evidence that viral metagenomics has facilitated advances in the field of virology, for being a sensitive technique for the detection of viruses which can not be identified by traditional culture.
LISTA DE ILUSTRAÇÕES
Figura 1. Comparação entre genoma, pan-genoma e metagenoma...13
Figura 2. Método geral de execução da metagenômica viral...15
Figura 3. Análise de qualidade das amostras realizada no Bioanalyser: A) Biblioteca de cDNA (fragmentos de cerca de 470 pares de bases); B) Biblioteca de amplificação de DNA (fragmentos de cerca de 550 pares de bases)...29
Figura 4. Relação entre as frequências e os tamanhos dos contigs nos 4 grupos. Grupo1- com
amostras diarreicas de crianças; Grupo 2- com amostras diarreicas de adultos; Grupo 3- com amostras não diarreicas de adultos; Grupo 4- com amostras de indivíduos HIV+...31
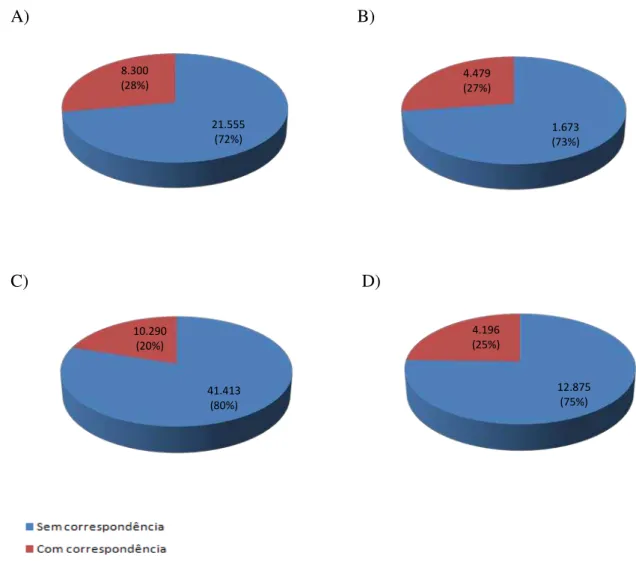
Figura 5. Relação dos contigs com/sem correspondência com o banco de dados viral. A)
Grupo com amostras diarreicas de crianças; B) Grupo com amostras diarreicas de adultos; C) Grupo com amostras não diarreicas de adultos; D) Grupo com amostras de indivíduos HIV+...32
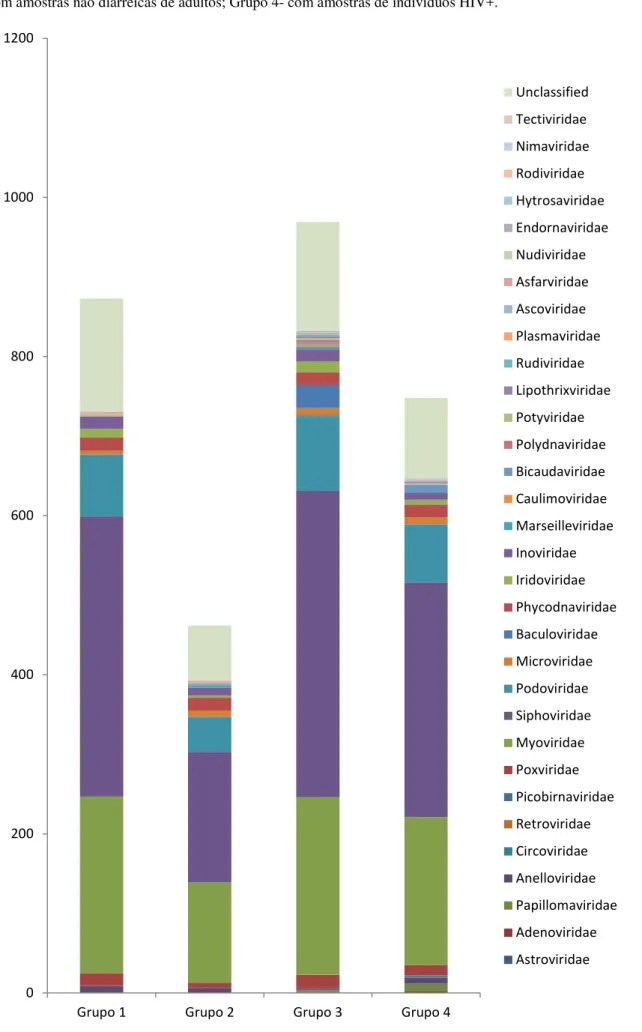
Figura 6. Distribuição dos contigs classificados em nível taxonômico de família, nos
LISTA DE TABELAS
Tabela 1. Sequenciamento obtido em número de pares de bases e reads por amostra –
experimento piloto...30
Tabela 2. Sequenciamento obtido em número de pares de bases e reads por amostra...30
Tabela 3. Avaliação dos contigs para cada amostra...31
SUMÁRIO
1. INTRODUÇÃO ... 10
2. REVISÃO BIBLIOGRÁFICA ... 11
2.1- MICROBIOTA HUMANA ... 11
2.2- GASTROENTERITES VIRAIS ... 12
2.3- METAGENÔMICA ... 13
2.4- SEQUENCIAMENTO DE ALTO DESEMPENHO ... 16
2.5- BIOINFORMÁTICA ... 17
2.6- O VIROMA ... 18
2.7- BACTERIÓFAGOS E VIROMA HUMANO ... 21
3. JUSTIFICATIVA ... 23
4. OBJETIVOS ... 24
4.1- OBJETIVO GERAL: ... 24
4.2- OBJETIVOS ESPECÍFICOS: ... 24
5. METODOLOGIA ... 25
5.1- AMOSTRAGEM ... 25
5.2- SEMI- PURIFICAÇÃO ... 26
5.3- EXTRAÇÃO E AMPLIFICAÇÃO DO MATERIAL GENÉTICO (RNA e DNA) ... 26
5.4- BIBLIOTECA E SEQUENCIAMENTO... 26
5.5- ALINHAMENTO ... 27
6. RESULTADOS ... 29
6.1- EXPERIMENTO PILOTO ... 29
6.2- COMPARAÇÃO ENTRE OS GRUPOS DO ESTUDO ... 30
7. DISCUSSÃO ... 35
8. CONCLUSÕES E PERSPECTIVAS ... 40
1. INTRODUÇÃO
A diarréia é um importante problema de saúde pública em todo o mundo, sendo que em até 40% dos casos, agentes etiológicos como bactérias, vírus e protozoários não são identificados. Estima-se que na maioria destes casos os vírus sejam os agentes causais dessas infecções. A detecção destes agentes é realizada tradicionalmente por isolamento em meio de cultura celular, mas, no entanto, muitos vírus não são cultiváveis.
As primeiras análises metagenômicas de uma comunidade viral, do viroma, de fezes humanas foi realizada por Breitbart et al (2003), e as informações adquiridas foi que a maioria
das sequências era desconhecida, e os vírus identificados eram bacteriófagos, principalmente da família Siphoviridae. Estudos posteriores, como os de Reyes et al, (2010), Minot et al,
(2011) e Kim et al, (2011), enfatizaram estes dados, pois, a maioria das sequências não
tinham correspondência com vírus presentes nos banco de dados, e que a comunidade viral era composta, principalmente, por bacteriófagos.
Métodos mais recentes como a metagenômica possibilitam a detecção de sequências genômicas virais e tem superado as principais limitações clássicas para a detecção viral. Assim, as abordagens metagenômicas têm o potencial para identificar e explorar a diversidade viral presente nas fezes, além de avaliar se estes agentes desempenham um papel causal na diarréia humana. Dessa forma, compreender o viroma, ou seja, a diversidade viral da microbiota requer uma análise profunda de sua composição, e compreender a estrutura populacional da microbiota intestinal em humanos trará implicações importantes para a saúde humana.
2. REVISÃO BIBLIOGRÁFICA
2.1- MICROBIOTA HUMANA
O ser humano durante a gestação é essencialmente estéril, mas, após o nascimento, cada superfície do corpo, incluindo a pele, boca e intestino são colonizados por uma enorme variedade de microrganismos. No corpo humano existem aproximadamente 100 trilhões de microrganismos habitando a superfície interna e externa. O conjunto de todas essas comunidades forma a microbiota humana, que varia muito nas mais diversas regiões do nosso corpo, dependendo de condições como umidade, pH, temperatura e nutrientes disponíveis. Nas regiões mais úmidas e quentes encontram-se uma maior concentração de microrganismos, enquanto que nas regiões mais secas, existe uma quantidade menor delas (MORGAN et al, 2012, RIBEIRO et al, 2014).
A maior parte dos esforços sobre a microbiota foi inicialmente centrado sobre os agentes patogênicos humanos, tais como as bactérias. O campo da bacteriologia já está em desenvolvimento há muitos anos, assim, estudos em larga escala, com o gene 16S RNA ribossomal revelou a predominância de dois filos bacterianos, no trato gastrointestinal, o Firmicutes e Bacterioides, e que constituem em mais de 90% das categorias filogenéticas conhecidas, em seguida são os filos Proteobacteria, Actinobacteria e Fusobacteria. No entanto, ainda são necessários mais estudos a respeito da diversidade de vírus no microbioma humano (Qin et al, 2010).
O trato respiratório humano, por exemplo, entra em contato com milhões de partículas em suspensão a cada dia, incluindo os vírus, e pouco se sabe sobre a microbiota das vias aéreas superiores e inferiores. Em outros locais, como no intestino, a diversidade dos vírus e seu papel na manutenção e adaptação da microbiota também continuam obscuros (WILLNER
et al, 2009; REYES et al, 2010). Assim, compreender o papel dessas populações, dita como
“viroma humano” requer uma compreensão muito mais profunda de sua composição e interação com outros seres vivos (MINOT et al, 2013). Estas comunidades microbianas são
de vital importância para a saúde, e o seu estudo leva a um melhor conhecimento da sua dinâmica complexa, que ainda pode conduzir ao desenvolvimento de novas formas de diagnóstico e até mesmo de tratamento de certas patologias. (RIBEIRO et al, 2014).
que os bacteriófagos afetam apenas as bactérias. No entanto, a comunidade científica está começando a perceber que os vírus presentes podem ser importantes para a saúde humana, ou seja, da forma com que manipulam a microbiota e por meio da interação com o sistema imune do hospedeiro (HUNTER, 2013).
2.2- GASTROENTERITES VIRAIS
A gastroenterite aguda é uma doença de ocorrência universal, que atinge pessoas de todas as classes sociais. É uma síndrome clínica caracterizada por alterações no volume e consistência das fezes, mas comumente associada com a liquidez das fezes e aumento no número de evacuações. É frequentemente acompanhada de outros sintomas como vômito, febre e cólica abdominal, podendo até apresentar muco e sangue (SILVA et al, 2004). São as
principais causas de morbidade e mortalidade em todo o mundo, numa estimativa de quase 1,5 milhões de mortes anuais de crianças até 5 anos. No Brasil, a diarréia aguda tem relação direta às condições de vida e saúde dos indivíduos, por exemplo, saneamento básico e desnutrição (BRASIL, 2010; Paz et al, 2012).
O diagnóstico para essas infecções é complexo, devido a grande variedade de agentes patogênicos, que podem apresentar os mesmos sintomas clínicos. Estima-se que, em média, 40% dos casos de diarréia são de etiologia desconhecida (NAKAMURA et al, 2009;
FINKBEINER et al, 2009). Apesar dos avanços no campo do diagnóstico, muitas síndromes
não têm sido associadas com um agente causal, embora tenham sido utilizados extensos testes de diagnóstico convencionais (CASTRIGNANO et al, 2013). Os mais frequentes agentes
etiológicos relacionados com quadros diarreicos são os vírus (rotavírus, norovírus, astrovírus e adenovírus), seguidos das bactérias e parasitas (FINKBEINER et al, 2009).
Doenças diarreicas também apresentam importância em indivíduos imunocomprometidos, uma vez que o organismo diminui sua capacidade natural de defesa contra qualquer antígeno e assim, os vírus podem estar se replicando com maior facilidade nesses indivíduos (BRASIL, 2014).
A gastroenterite aguda, a infecção respiratória aguda e a encefalite infecciosa, juntas, tem uma média de 30 % de casos onde não se tem um agente etiológico identificado, embora extensos testes de diagnóstico tenham sido utilizados, assim, muitos destes agentes podem ser vírus ainda não identificados (CASTRIGNANO et al, 2013). A dificuldade em identificar
Em meados da década de 2000, duas vacinas para rotavírus se tornaram disponíveis, uma vacina monovalente RV e outra pentavalente. Ambas as vacinas são recomendadas pela Organização Mundial de Saúde, sendo utilizadas em vários países. Estudos têm demonstrado uma redução significativa de hospitalização e mortalidade devido à gastroenterite por rotavírus. O Brasil foi um dos primeiros países a introduzir a vacinação universal contra Rotavírus A, Rotarix®, que foi fornecido gratuitamente pelo sistema de saúde pública desde março de 2006. O número de casos positivos para rotavírus diminuiu substancialmente desde então, enquanto outros patógenos agora são relatados com mais frequência (RABONI et al,
2014).
Existem cerca de 200 espécies de patógenos virais humanos reconhecidos e outras espécies continuam a ser descobertas a uma taxa de quase duas por ano. Alguns vírus têm o potencial de se transformar rapidamente, como, por exemplo, na epidemia de gripe aviária e suína. (BIBBY, 2013).
2.3- METAGENÔMICA
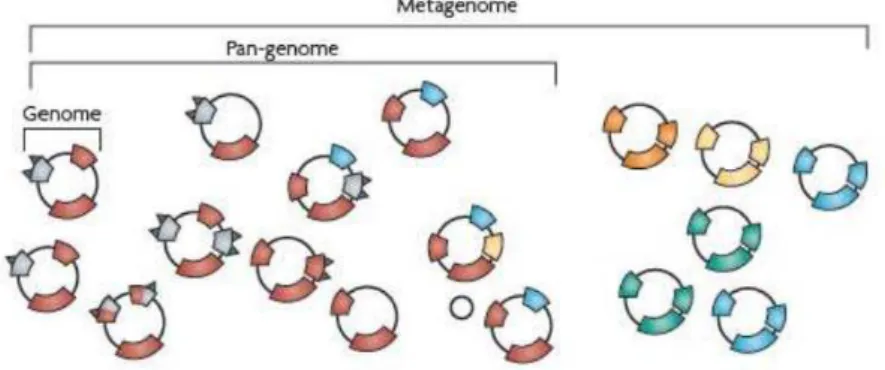
O termo genoma, criado em 1920, por Hans Winkler, designa toda a informação hereditária de um organismo que está codificada no seu DNA (ou, em alguns vírus, no RNA), isto inclui também as sequências não codificadoras. O pan - genoma compreende a análise dos conjuntos de genes de todas as estirpes de uma espécie. Já o metagenoma, por definição, é o conjunto de genomas isolados a partir de uma amostra (Figura 1) (CATANHO et al, 2010;
HANDELSMAN et al, 1998).
Figura 1: Comparação entre genoma, pan-genoma e metagenoma.
Os vírus são microrganismos que contribuem para os ciclos de vida de organismos celulares, pois, influenciam nos ciclos biogeoquímicos e impulsionam a evolução microbiana no solo, no oceano e nos seres vivos (ROHWER et al, 2009). No entanto, o estudo da
ecologia viral e a compreensão da diversidade de vírus em ambientes naturais tem sido limitado, devido às dificuldades de cultura viral aliado à falta de genes evolutivamente conservados, tal como o gene do RNA ribossomal 16S em procariotos, compartilhado por todas as bactérias e archaea (KIM et al, 2013; KLINGENBERG et al, 2013; LORENZI et al,
2011; WOMMACK et al, 2012).
Por métodos tradicionais, os agentes virais são detectados por isolamento através de cultura celular, em que as monocamadas de células apresentam os efeitos citopáticos, ou por meio de testes de neutralização de anticorpos. No entanto, muitos tipos virais não são cultiváveis, assim, a identificação viral e os métodos de diagnóstico tradicionais de pesquisa são limitados, impossibilitando uma visão completa da diversidade viral. No decorrer das últimas décadas, métodos enzimáticos e moleculares, tais como ensaio imunoenzimático (ELISA) e PCR foram utilizados para detectar e estudar os vírus não cultiváveis (BIBBY, 2013).
Considerando que a compreensão da ecologia viral das amostras é importante para a virologia clínica e diagnóstica e que existe a limitação técnica para detecção viral, métodos como a metagenômica tem sido utilizados para aumentar o conhecimento a cerca de comunidades virais em vários ambientes naturais (KIM et al, 2013; PALLEN, 2014), pois,
essas abordagens possibilitam a detecção de sequências genômicas de muitos vírus, até então desconhecidos em amostras humanas. Assim, atualmente a metagenômica viral é uma técnica poderosa e sensível para a detecção de vírus, e tem superado as principais limitações clássicas para a detecção viral, pois, como explicado anteriormente, os ácidos nucleicos virais podem ser acessados sem a necessidade do isolamento dos vírus. O interesse na metagenômica viral é também na capacidade para confirmar a presença de vírus patogênicos conhecidos, mesmo com concentrações baixas (KIM et al, 2013; FANCELLO et al, 2012).
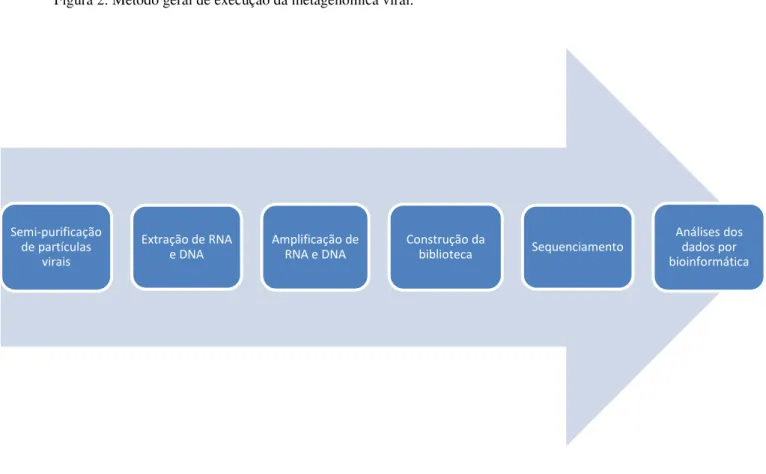
Figura 2: Método geral de execução da metagenômica viral.
A etapa inicial do metagenoma viral é a semi - purificação de partículas virais, feitos basicamente por filtrações, seguida de centrifugações (BIBBY, 2013). Assim, para que a extração de ácidos nucleicos de vírus seja mais eficiente, a utilização da filtragem feita por filtros de 0,45 e/ou 0,22 µm se torna necessária, para eliminar a contaminação por células hospedeiras e por outros microrganismos, pois, partículas virais, em geral, são menores do que os organismos eucarióticos. Uma contaminação resultaria no sequenciamento também desses genomas maiores, se transformando numa ''máscara'' nas sequências virais que por competição poderiam subestimá-las. Outro ponto relevante na execução da metagenômica é a amplificação dos ácidos nucleicos antes do sequenciamento, pois, a extração viral resulta numa pequena quantidade de ácidos nucleicos, assim, a amplificação pode facilitar a detecção dos vírus nas amostras (FANCELLO et al, 2012). No entanto, esse procedimento impede
análises a cerca da carga viral presente na amostra (KIM et al, 2013).
Semi-purificação de partículas
virais
Extração de RNA e DNA
Amplificação de RNA e DNA
Construção da
biblioteca Sequenciamento
2.4- SEQUENCIAMENTO DE ALTO DESEMPENHO
A etapa de sequenciamento pode ser realizada por diversas plataformas, sendo as novas tecnologias denominadas de sequenciamento de nova geração (NGS), comercializadas a partir de 2005. Essas novas tecnologias promovem o sequenciamento de DNA gerando milhões de pares de bases em um único ciclo. Dentre as novas plataformas de sequenciamento, duas já possuem ampla utilização em todo o mundo: a plataforma 454 FLX da Roche e a Illumina, sendo que a plataforma Illumina gera maior quantidade de dados sequenciados do que o 454 da Roche (CARVALHO et al, 2010).
A plataforma Roche 454 inicia sua execução com a construção de uma biblioteca, com sequências de fragmentos de DNA de fita única com adaptadores. Os fragmentos se ligam a esferas, a seguir ocorre amplificação nessas esferas por PCR em emulsão, a fim de aumentar a intensidade do sinal. O ideal é que durante esse processo um único fragmento seja anexado a cada grânulo, formando grupos uniformes em cada esfera. As esferas são depositadas sobre uma matriz de poços, de modo que cada poço contenha uma única esfera. Após essas etapas preparatórias, o sequenciamento começa utilizando o método de pirosequenciamento. Em cada ciclo, uma única espécie de nucleotídeo é adicionada. Nos poços onde os nucleotídeos foram incorporados ocorre a liberação de pirofosfato e depois há a presença de uma luz que é detectada utilizando um sensor, identificando qual tipo de nucleotídeo foi incorporado aos fragmentos (LEDERGERBER et al, 2010).
A plataforma Illumina é baseada na montagem de uma única biblioteca de DNA, por fragmentação aleatória das amostras de DNA. Após a adição de adaptadores universais nos fragmentos, estes, são espalhados em uma lâmina (conhecida como “flow cell”) com 8 linhas/pistas (conhecida como “lane”) imobilizadas em vidro. Após a amplificação por ponte é gerado um grande número de moldes idênticos sobre a superfície do vidro (conhecido como formação de “clusters”). Em cada ciclo, um único nucleotídeo marcado por fluorescência é incorporado a cada cadeia complementar. Após a incorporação, o marcador fluorescente é detectado e é gerada uma imagem, e os ciclos seguintes ocorrem da mesma forma (LEDERGERBER et al, 2010).
2.5- BIOINFORMÁTICA
As tecnologias estão passando por uma evolução rápida e as ferramentas da bioinformática devem ser atualizadas constantemente, para acomodar o grande volume de dados que estão sendo obtidos (PETROSINO et al, 2009). Os recentes avanços nas
tecnologias de sequenciamento produziram uma verdadeira revolução, e possibilita novas perspectivas para aplicações de diagnóstico e pesquisa, devido à alta quantidade de dados gerados. (CAPOBIANCHI et al, 2012). Ou seja, estudos metagenômicos se tornaram mais
acessíveis para a comunidade científica, o que resulta num crescimento exponencial na quantidade de dados de sequenciamento disponíveis, sendo necessária a criação de ferramentas computacionais altamente eficientes e especializadas para lidar com esses conjuntos de dados massivos (LORENZI et al, 2011)
A bioinformática analisa os dados gerados após a liberação dos resultados pelo sequenciamento, e nos fornece informações para que sejam tiradas as conclusões a cerca da população viral. Essa etapa consiste na utilização de técnicas computacionais e matemáticas relacionadas ao conhecimento químico, físico e biológico para processar suas informações a respeito de genes, proteínas, enzimas, bem como alinhamento de sequências e montagem de árvores filogenéticas. Consiste ainda, numa ciência muito dinâmica e novos programas estão continuamente sendo criados para gerir os novos dados de NGS (FANCELLO et al, 2012).
No sequenciamento há a produção de sequências curtas, denominadas de “reads”, mas
por poder tornar as buscas mais difíceis, talvez, seja necessária a montagem prévia de sequências mais longas, que são formadas a partir da sobreposição dos reads, denominadas de
“contigs” e então são alinhadas com genomas ou genes de referência, geralmente depositados
nos bancos de dados como, por exemplo, no GenBank do site do NCBI (National Center for Biotechnology Information) (BIBBY, 2013).
Em particular, os vírus geralmente não são bem representados por bancos de dados atuais, pois faltam muitas informações sobre diversos vírus, o que torna difícil obter estimativas realistas da abundância correspondente. Considerando a imensa diversidade e a variação de genomas, os vírus são notoriamente sub-representados nos bancos de dados (KLINGENBERG et al, 2013). Dessa forma, a análise dos dados dos metagenomas utilizando
a bioinformática é um dos aspectos mais desafiadores, pois, são gerados de um milhão a um bilhão de reads em plataformas de sequenciamento de alto desempenho. A maior parte das
"sequências lixo", devido à falta de bancos de dados virais adequados para sua caracterização, o que pode dificultar a determinação da diversidade viral em diversas amostras (KIM et al,
2013).
2.6- O VIROMA
A análise do viroma tem diversas aplicações, uma delas é a detecção e resposta a surtos de patógenos virais. Esta metodologia tem sido utilizada com sucesso em surtos de gripe causada pelo vírus influenza, determinando rapidamente o tipo viral circulante. Essas identificações permitem tanto a aplicação da terapêutica bem como de prevenção contra possíveis epidemias, como o desenvolvimento de imunização.
Outra aplicação possível por metagenômica viral é o diagnóstico clínico, pois, há a possibilidade de detecção de diversos patógenos. Em muitos casos, os diagnósticos clínicos de infecções virais são feitos em cultura e em testes de diagnóstico, mas para alguns patógenos estes testes podem ser insuficientes ou inconclusivos. Atualmente, a execução do metagenômica ainda é de alto custo para uso rotineiro, que requer significativos apoios técnicos e especialistas em bioinformática, mas, com a evolução das tecnologias talvez isso seja possível no futuro (PALLEN, 2014).
A primeira contribuição para a avaliação do viroma humano por metagenômica foi feita em 2003 por Breitbart et al. A comunidade de vírus de DNA, associado ao intestino
humano foi estudada utilizando - se o sequenciamento pelo método de Sanger. Nesse trabalho foi utilizada a metodologia conhecida como shotgun, (onde todo o DNA analisado é
fragmentado em milhões de pequenos pedaços) dos vírus presentes nas fezes de um adulto saudável. Entre as sequências virais identificadas, a maioria era composta por bacteriófagos e 59% das sequências geradas era desconhecida (BREITBART et al, 2003).
Observações semelhantes também foram relatadas por dois estudos sobre o viroma do intestino humano, com sequenciamento por 454 GS FLX (pirosequenciamento) em que a percentagem de sequências desconhecidas foi de 81% e 98%, respectivamente, e os bacteriófagos foram dominantes nas comunidades virais estudadas (REYES et al, 2010;
MINOT et al, 2011).
No estudo de Kim et al, (2011), as análises de sequências virais apresentaram as
aproximadamente 40 a 50%, ou, ocasionalmente, até 90 % das sequências virais das amostras são descaracterizadas (KIM et al, 2011). Assim, em diversos estudos utilizando a
metagenômica por shotgun para analisar comunidades virais, o resultado é que a maioria das
sequências virais não apresenta similaridade significativa com sequências conhecidas (WOMMACK et al, 2012), que pode ocorrer pela dificuldade de obtenção das sequências
pelos bancos de dados, que poderá ser solucionado a medida que mais estudos são realizados, mais vírus são identificados e depositados nos bancos de dados.
No estudo de Nakamura et al foi identificado norovírus e coronavírus, por
metagenômica viral, com sequenciamento por pirosequenciamento em amostras de fezes humanas (NAKAMURA et al, 2009). No estudo de Kapoor et al utilizando também a
metagenômica viral identificou-se uma nova espécie de parvovírus nas fezes humanas, cuja relação filogenética mais próxima é o bocavírus humano (HBoV) sendo denominado de HBoV2 (KAPOOR et al, 2010).
No Brasil, o grupo Adolf Lutz utilizou a metagenômica viral com uma amostra armazenada desde 2003. Antes do estudo foi observado que, alguns vírus patogênicos das plantas poderiam ser altamente abundantes nas fezes humanas, essa era a suspeita para a amostra, mas, ao invés de descobrir um vírus de planta, foram identificados dois novos genomas com características de circovírus. No entanto, esse é até o momento o único trabalho com viroma de amostras brasileiras (CASTRIGNANO et al, 2013).
Um dos maiores causadores de gastroenterites são os astrovírus humanos, estes, foram descritos pela primeira vez por Appleton e Higgins em 1975, durante um surto de diarréia aguda em uma maternidade na Inglaterra e foram denominados pela sua forma de astros de 5 a 6 pontas, vistos por microscopia eletrônica (STEWIEN et al, 1991). Os astrovírus humanos
(HAstVs) são vírus de RNA não envelopado com simetria icosaédrica. Eles são classificados em oito sorotipos (HAstVs 1-8), que são divididos em quatro subtipos (1A, 1B, 1C e 1D), com aproximadamente 6.800 nucleotídeos e apresenta uma cauda poliadenilada (Poli A) na sua extremidade 3’(VICTORIA et al, 2007; SANTOS et al, 2005).
A transmissão ocorre por contatos íntimos com pessoas infectadas, pela água e alimentos contaminados ou, provavelmente, por fômites consequentes da rota fecal-oral. O período de incubação observado em infecções por astrovírus varia de um a quatro dias. A enfermidade causada pelos vírus tende a ser leve e auto limitada e geralmente não resulta em significativo quadro de desidratação ou na necessidade de hospitalização (SANTOS et al,
Estes vírus são enteropatógenos de distribuição mundial e epidemias de diarréia associada a eles já foram relatadas em escolas e creches. A sazonalidade das infecções atribuídas a astrovírus parece variar de acordo com a região geográfica. Estudos realizados na Europa, Austrália e Argentina mostraram que há um aumento na incidência da infecção viral durante os meses mais frios do ano, enquanto em alguns países como Egito e México, a maioria das infecções pelo agente viral ocorre durante a época mais quente do ano. No Brasil, mais especificamente na cidade de Goiânia, as infecções relacionadas com este agente ocorrem, predominantemente, durante os meses de setembro a março, período no qual se observam os maiores índices pluviométricos nesta localidade (SANTOS et al, 2005).
Outro agente etiológico das gastroenterites são os adenovírus humanos. O primeiro adenovírus humano foi isolado em 1953, a partir de adenóides humanos e caracterizado de forma independente por dois grupos de pesquisadores. Infecções por HAdV afetam pacientes a nível mundial e em todos os grupos etários e são facilmente transmissíveis e além de diarréia aguda, os adenovírus podem causar outras doenças, tais como, doenças respiratórias, conjuntivite e cistite hemorrágica. Estes vírus pertencem à família Adenoviridae, são não
envelopados, mas com nucleocapsídeo que contém o genoma de DNA de cadeia dupla linear, que geralmente varia de 26 a 45 kb. A família Adenoviridae é dividida em cinco gêneros: Atadenovirus, Siadenovirus, Mastadenovírus, Aviadenovirus e Ichtadenovirus Estas divisões
são baseados nas espécies hospedeiras e a composição do DNA (ROBINSON et al, 2011).
Através das análises das sequências, foi demonstrado que os genomas de todos os adenovírus humanos têm organização genética semelhante (JONES et al, 2007). Até o ano de 2014 já
foram descritos mais de 60 tipos, agrupados em sete espécies, de A a G. O padrão de doença dos adenovírus varia de acordo com a espécie. Os adenovírus da espécie F, dos tipos 40 e 41, foram associados com gastroenterite e eles são referidos como adenovírus entéricos. Outras espécies, como A (tipos 12, 18 e 31), C (tipos 1, 2 e 5) e D (tipos 28, 29, 30, 32, 37, 43-46) também já foram associados como causadores de diarréia (MOYO et al, 2014).
Pesquisas contínuas são essenciais para o desenvolvimento e aperfeiçoamento na identificação de patógenos virais por metagenômica (BIBBY, 2013; KRISTENSEN et al,
2.7- BACTERIÓFAGOS E VIROMA HUMANO
Embora os vírus humanos estejam geralmente associados com patógenos de gastroenterites e outras doenças agudas, os bacteriófagos intestinais têm papéis significativos na diversidade genética do ecossistema intestinal por predação em seus hospedeiros bacterianos. Além disso, os bacteriófagos podem impedir a colonização por bactérias patogênicas e eliminar algumas cepas probióticas benéficas, ou introduzir novas características fenotípicas, como a resistência a antibióticos e a capacidade de produzir toxinas (PÉREZ-BROCAL et al, 2013). Assim, enquanto muitos vírus em seres humanos são
identificados com base na sua patogenicidade, agora há uma vasta comunidade viral no corpo humano que não causa doença no homem, identificados até agora no viroma humano como bacteriófagos (ABELES et al, 2014). Os bacteriófagos foram descobertos por Ernest Hanking
(1896) e Frederick Twort (1915), que descreveram sua atividade antibacteriana (WITHEY et al, 2005).
Os bacteriófagos são vírus que infectam bactérias, estimados em 1013 a 1015 a preencher o corpo humano. Eles são cerca de 50 vezes menores que as bactérias e estão presentes no solo, na água, e nos alimentos. Existem os fagos virulentos e temperados, que se diferem no seu modo de ação. O primeiro passo é a adsorção das partículas de fago com a parede da célula bacteriana, por interações específicas entre as proteínas de superfície virais e os receptores da célula hospedeira. Após a entrada na célula bacteriana, os fagos virulentos se replicam rapidamente para sintetizar proteínas dentro da célula hospedeira, finalmente, os novos fagos escapam pela ruptura da parede celular, que resulta na morte da célula. Em contraste, os fagos temperados integram o seu material genético no genoma da célula hospedeira, que é replicado juntamente com o genoma da célula hospedeira. Somente os fagos temperados participam de transferências horizontais entre as populações bacterianas (LY-CHATAIN, 2014).
A enorme diversidade torna a análise das comunidades virais humana altamente complexa, e essa diversidade na comunidade de bacteriófagos em seres humanos está ligada à diversidade de seus hospedeiros celulares e também à sua rápida evolução e transferência horizontal de genes. Os estudos estão apenas começando a ser capaz de estudar comunidades virais humanas em larga escala, principalmente pelo resultado dos avanços recentes e contínuos nas tecnologias de sequenciamento e análises de bioinformática. (ABELES et al,
3. JUSTIFICATIVA
As análises sobre a diversidade da microbiota humana estão bem avançadas no campo da bacteriologia, mas, muito tem a se conhecer a respeito da diversidade viral e assim, impulsionar estudos dos metagenomas virais.
4. OBJETIVOS
4.1- OBJETIVO GERAL:
Avaliar o viroma presente nas fezes em 4 grupos distintos, para analisar as correlações dentro e entre os viromas e os prováveis patógenos envolvidos nos quadros de gastroenterites.
4.2- OBJETIVOS ESPECÍFICOS:
- Sequenciar o genoma dos vírus presentes em amostras fecais diarréicas e não diarréicas provenientes de indivíduos brasileiros.
- Comparar o resultado do sequenciamento com sequências depositadas em banco de dados genômicos.
5. METODOLOGIA
Para avaliar a metodologia foi realizado um experimento piloto, com uma amostra de um indivíduo sadio e sem diarréia. A mesma metodologia foi utilizada com as 12 amostras incluídas no projeto.
5.1- AMOSTRAGEM
A escolha da amostragem de um estudo é uma etapa fundamental para alcançar os resultados esperados, assim, a estratégia do presente trabalho é analisar amostras diarreicas e não diarreicas provenientes de crianças e de adultos que não tiveram um agente etiológico da doença definido. Foram incluídas também amostras fecais de indivíduos com sistema imune suprimido, pois nesses casos existe a possibilidade de identificar vírus que infectam esse perfil, mas que seria de difícil detecção em indivíduos sadios.
Para determinar o número amostral foi feito um estudo na literatura de trabalhos metagenômicos, pelo site do NCI, na base de dados do PubMed. Observou-se que o número de amostras incluídas em uma corrida de sequenciamento varia 1 a 24. Assim, considerando o recurso financeiro disponível, foi determinada uma amostragem composta por 12 amostras divididas em quatro grupos detalhados a seguir:
- três amostras diarreicas “in natura” de crianças, com os seguintes critérios de inclusão: faixa
etária de 2 a 12 anos com quadro gastroentérico. Sendo cada amostra de uma criança diferente;
- três amostras diarreicas “in natura” de adultos, com os seguintes critérios de inclusão: faixa
etária acima de 18 anos com quadro gastroentérico. Sendo cada amostra de um adulto diferente;
- três amostras não-diarreicas “in natura” de adultos, assintomáticos para quadro
gastroentérico, com os seguintes critérios de inclusão: faixa etária acima de 18 anos sem sintoma gastroentérico. Sendo cada amostra de um adulto diferente;
- três amostras “in natura” de indivíduos portadores do vírus HIV, com os seguintes critérios
As amostras das crianças com diarréia e de adultos com e sem diarréia foram coletadas no Laboratório Sabin (sede – Brasília Shopping), pela parceria feita com a UCB (Universidade Católica de Brasília). Já as amostras dos pacientes com o sistema imune suprimido foram coletadas no Centro de Saúde N° 01 (Hospital Dia- W3 sul).
O presente estudo foi aprovado pelo Comitê de Ética em Pesquisa sob o número 01348312.0.0000.0029.
5.2- SEMI- PURIFICAÇÃO
Para a separação das partículas virais dos outros microrganismos e células do hospedeiro foram diluídas 500mg de fezes em 3 mL de tampão PBS (tampão fosfato salino) e as suspensões foram centrifugadas a 9.700xg por 10 minutos e os sobrenadantes colhidos. Para a filtração do sobrenadante foram utilizados os filtros de poros de 0,45 e 0,22 µm de diâmetro (Millipore). Em seguida, foi feita a ultracentrifugação com colchão de sacarose 20% a 111.132xg por 2 horas e 30 minutos.
5.3- EXTRAÇÃO E AMPLIFICAÇÃO DO MATERIAL GENÉTICO (RNA e DNA)
O pellet resultante da ultracentrifugação foi extraído utilizando o kit comercial
PureLink® Viral RNA/DNA Mini Kit (Invitrogen) conforme instruções do fabricante. Nesse momento foram separadas duas alíquotas, uma para análise de DNA e outra para RNA. A alíquota de RNA foi submetida a uma reação para obtenção do cDNA, utilizando primer
randômico e seguindo as recomendações do fabricante da enzima transcriptase reversa (Invitrogen).
Para aumentar a quantidade de material genético o DNA (direto da extração) e o cDNA foram amplificados utilizando Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare) conforme instruções do fabricante.
5.4- BIBLIOTECA E SEQUENCIAMENTO
sequenciar um total de aproximadamente 5 GB utilizando a plataforma Illumina HiSeq2000, paired end, com fragmentos de 100 pb (2x100).
Já com as 12 amostras incluídas no projeto, a construção das 4 bibliotecas foram realizadas pela empresa Macrogen, assim como, a execução do sequenciamento, para sequenciar um total de aproximadamente 40 Gb (uma lane completa) utilizando a plataforma Illumina HiSeq 2000, paired end, com fragmentos de 100 pb.
5.5- ALINHAMENTO
Para realizar o alinhamento das sequências do experimento piloto foi necessária a construção de um banco de dados viral, pela base de dados Taxonomy pelo site do NCBI (Taxonomy ID: 10239), sendo salvo todos os nucleotídeos virais no formata Fasta.Um arquivo
no formato Fasta contém sequências de nucleotídeos de DNA, em um formato simples, o que torna as sequências fáceis de analisar. Cada sequência tem uma linha de cabeçalho com informações sobre a sequência, como por exemplo, um identificador, seguido por uma ou mais linhas com as bases de DNA que foram lidas.
O envio das sequências das amostras foi em formato FastQ, pela Macrogen. O formato FastQ é um arquivo similar ao Fasta, porém possui informações sobre a qualidade do sequenciamento daquela sequência.E em seguida essas sequências foram confrontadas com o banco de dados viral criado, como descrito anteriormente.
O alinhamento foi realizado na linha de comando do sistema linux, utilizando o Bowtie, que é um alinhador de reads curtos, ou seja, de 35 a 100 pb. Primeiramente deve-se
construir os índices de referência do arquivo de referência, com o seguinte comando “bowtie-build –f sequence_fasta.fasta sequence_fasta”, sendo que o comando “-f” indica que este é o arquivo de referência, e “sequence_fasta” o nome dado à minha referência. Para iniciar o alinhamento deve-se utilizar o seguinte comando “bowtie –S –n 2 sequence_fasta Meta1_1_1.fastq Meta1_1_1.sam”, sendo que o comando “–S” indica que o arquivo terá saída no formato SAM (Sequence Alignment/Map), que é um formato de texto, e “-n” a quantidade máxima de mismatches permitos (troca de nucleotídeos numa determinada posição, quando se compara a sequência de referência com a de interesse). Tanto o alinhamento da amostra de cDNA (Meta1_1_1) quanto a de DNA (Meta1_2_1) foi realizado da maneira descrita anteriormente, sendo que, a indexação da referência só é realizada um vez.
Meta1_1_1.sam > Meta1_1_1.bam”, em que “-bS” significa que o arquivo de entrada está no formato SAM e que o arquivo de saída foi no formato BAM. Em seguida, utilizando o comando “samtools flagstat Meta1_1_1.bam” foi gerado um relatório sobre esse arquivo, resultando nos identificadores das sequências alinhadas e a quantidade de reads alinhados em
cada identificador. Esses identificadores foram copiados e colados, sem utilizar o quantitativo dos reads, em um editor de texto, como por exemplo, o word. Para recuperar e descrever o
que significa cada identicador, o Batch Entrez do site NCBI foi utilizado, assim, no campo “database” deve-se escolher “nucleotide”, já que toda a análise foi feita utilizando as bases de dados dos nucleotídeos, e no campo “file” deve-se selecionar o documento de texto que foi salvo, com isso, abrirá uma nova página no site do NCBI, que descreverá o organismo correspondente para cada identificador. Assim, o próximo passo é correlacionar a quantidade de reads para cada identificador.
Para as 4 bibliotecas sequenciadas, o software CLC genomics workbench 7 (CLC Bio,
Aarhus, Denmark) foi utilizado para realizar trimming (retirada de sequências ambíguas,
repetitivas e sem qualidade) das sequências e montagem dos contigs (conjunto de
sobreposição de segmentos de DNA que, juntos, representam uma região consenso de DNA) para otimizar as análises, feitos a partir dos reads enviados do sequenciamento, montados
com tamanhos mínimos de 500 pb.
Para as análises posteriores, o servidor web METAVIR foi utilizado, pois, auxilia a análise de um ou de vários metagenomas virais (viromas) a partir de sequências metagenômicas (reads brutos ou contigs já montados), sendo que, a composição taxonômica é
calculada a partir de uma comparação com o BLAST e o RefSeq de genomas completos com sequências de proteínas, a partir da base de dados do NCBI, utilizando BLASTp. Primeiramente foi necessário se registrar e em seguida foi feito o upload dos contigs já
6. RESULTADOS
6.1 - EXPERIMENTO PILOTO
Após a etapa de amplificação a alíquota de cDNA apresentou concentração de 65,1 ng/µL e a de DNA apresentou 26,8 ng/µL.
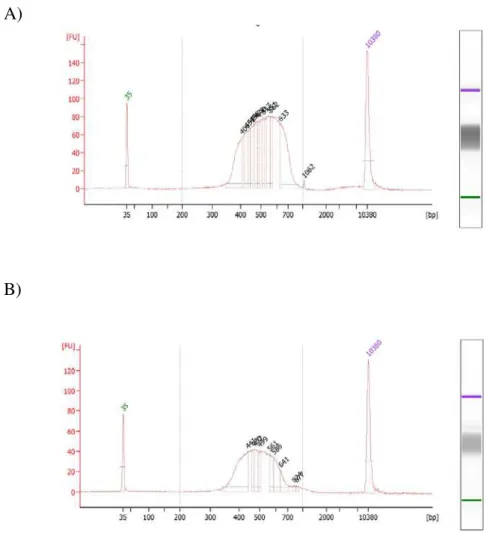
A biblioteca de cDNA foi construída com fragmentos de cerca de 470 pares de bases (Figura 3A), numa concentração de 40,9 ng/µL e a biblioteca de DNA com fragmentos de cerca de 550 pares de bases (Figura 3B), com concentração de 9,52 ng/µL. A).
Figura 3: Análise de qualidade das amostras realizada no Bioanalyser: A) Biblioteca de cDNA (fragmentos de cerca de 470 pares de bases); B) Biblioteca de amplificação de DNA (fragmentos de cerca de 550 pares de bases).
A)
A amostra de cDNA que resultou em 25.032.073 reads (tabela 1), obteve um total de
8.548 reads alinhados com as sequências obtidas pelo banco de dados viral, ou seja, 0,03% de reads foram alinhados com sequências virais. Já a amostra de DNA que resultou em
4.459.552 reads obteve 131 alinhamentos com sequências virais.
Tabela 1. Sequenciamento obtido em número de pares de bases e reads por amostra – experimento piloto.
Amostras Total de bases Reads (100pb)
cDNA 5.056.478.746 25.032.073
DNA 900.855.966 4.459.552
Com a amostra de DNA o número de alinhamentos não foi significativo para a análise. Já com a amostra de cDNA houve um número maior de alinhamentos com bacteriófagos, sendo de 3.707 reads do total de 8.548 reads; seguido dos alinhamentos com baculovírus, que
resultaram em 2891 reads. Os reads restantes foram alinhados sem significância, ou seja,
poucos reads se correlacionavam com os organismos, sendo que, a maioria destes tinham
apenas 1 read alinhado para um identificador, ou seja, para um organismo. Foi observado que
nas amostras de DNA e cDNA não foram identificados patógenos virais entéricos.
6.2- COMPARAÇÃO ENTRE OS GRUPOS DO ESTUDO
Após a análise do experimento piloto, as 12 amostras foram executadas como já descrito e os 4 grupos foram sequenciados, resultando em pouco mais de 40 Gb (Tabela 2):
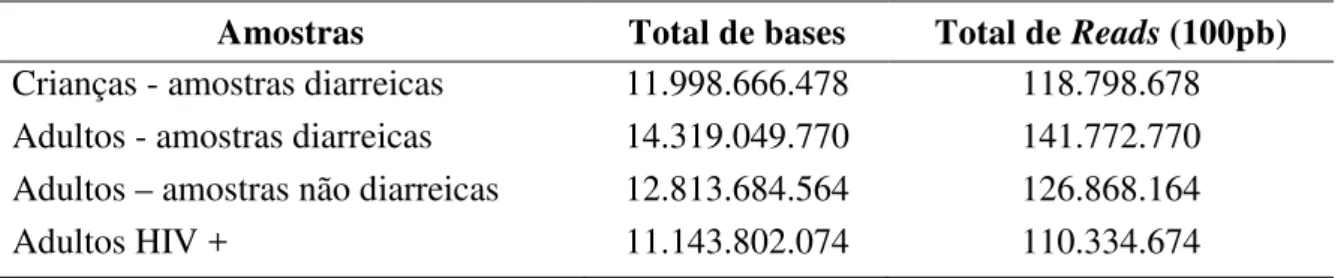
Tabela 2. Sequenciamento obtido em número de pares de bases e reads por amostra.
Amostras Total de bases Total de Reads (100pb)
Crianças - amostras diarreicas 11.998.666.478 118.798.678
Adultos - amostras diarreicas 14.319.049.770 141.772.770
Adultos – amostras não diarreicas 12.813.684.564 126.868.164
A quantidade de contigs obtidos foi diferente para cada tipo de amostra, sendo que a
biblioteca 1, amostras diarreicas de crianças, apresentou o maior número de contigs, sendo
51.703, já a biblioteca 2, amostras diarreicas de adultos, apresentou o menor número de
contigs, sendo 6.152 (Tabela 3).
Tabela 3. Avaliação dos contigs para cada amostra.
Amostras Nº de Contigs sequência (nt) Mínimo de sequência (nt) Máximo de
Crianças – amostras diarreicas 29.855 465 428.276
Adultos – amostras diarreicas 6.152 483 112.694
Adultos – amostras não diarreicas 51.703 500 250.730
Adultos HIV + 17.071 500 168.151
Contigs de tamanhos de 400 e 1.000 nucleotídeos foram os mais frequentes nos 4
grupos, ou seja, mais de 50% de todos os contigs obtidos estão nessa faixa de tamanho.
Enquanto que contigs com tamanhos maiores de 9.500 nucleotídeos representam de 3,01 a
6,3% da frequência dos contigs obtidos (Figura 4).
Figura 4 – Relação entre as frequências e os tamanhos dos contigs nos 4 grupos. Grupo 1- com amostras
diarreicas de crianças; Grupo 2- com amostras diarreicas de adultos; Grupo 3- com amostras não diarreicas de adultos; Grupo 4- com amostras de indivíduos HIV+.
0 10 20 30 40 50 60 40 0 999 10 00 14 99 15 00 19 99 20 00 24 99 25 00 29 99 30 00 34 99 35 00 39 99 40 00 44 99 45 00 49 99 50 00 54 99 55 00 59 99 60 00 64 99 65 00 69 99 70 00 74 99 75 00 79 99 80 8 499 85 00 89 99 90 00 94 99 95 00 42 8. 276 F re q u ê n ci a d o s cont ig s
Tamanho dos contigs
Grupo 1
Grupo 2
Grupo 3
A relação dos contigs com e sem correspondência com o banco de dados viral mostra
que menos de 30% dos contigs em cada grupo de estudo apresentou correspondência com
sequências do banco de dados (Figura 5).
Figura 5- Relação dos contigs com/sem correspondência com o banco de dados viral. A) Grupo com amostras
diarreicas de crianças; B) Grupo com amostras diarreicas de adultos; C) Grupo com amostras não diarreicas de adultos; D) Grupo com amostras de indivíduos HIV+.
A) B)
C) D)
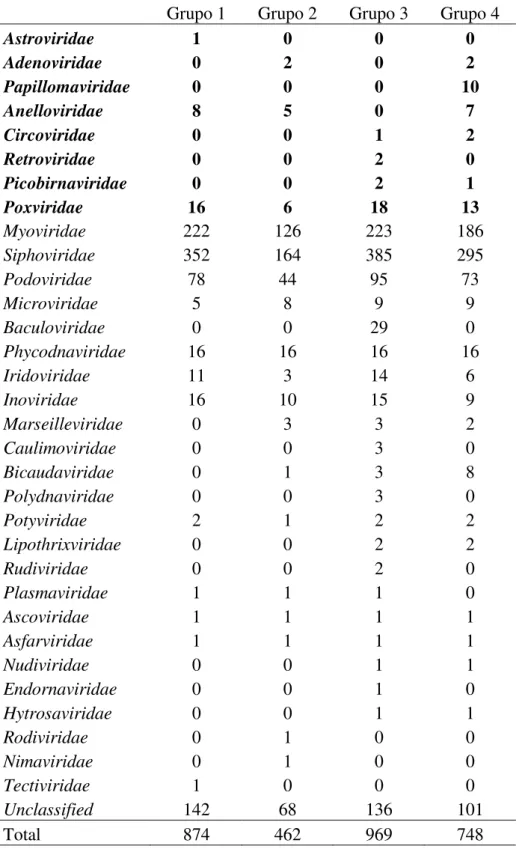
As espécies virais estão agrupadas nas suas famílias, para cada grupo do estudo (Figura 6; Tabela 4), e em destaque estão os vírus humanos. O grupo com amostras diarreicas de adultos (grupo 2) foi o que obteve o menor número de espécies, que foi de 462 espécies, já o grupo com amostras não diarreicas de adultos (grupo 3) apresentou o maior número de espécies, sendo de 969. O número de famílias identificadas foi de 15 para o grupo 1, 18 para o grupo 2, 25 para o grupo 3 e 22 para o grupo 4.
21.555 (72%) 8.300
(28%)
1.673 (73%) 4.479
(27%)
41.413 (80%) 10.290
(20%)
12.875 (75%) 4.196
Figura 6 – Distribuição dos contigs classificados em nível taxonômico de família, nos diferentes grupos
estudados: Grupo1- com amostras diarreicas de crianças; Grupo 2- com amostras diarreicas de adultos; Grupo 3- com amostras não diarreicas de adultos; Grupo 4- com amostras de indivíduos HIV+.
0 200 400 600 800 1000 1200
Grupo 1 Grupo 2 Grupo 3 Grupo 4
Tabela 4. Número de espécies para cada família viral nos quatro grupos. Grupo1- com amostras diarreicas de crianças; Grupo 2- com amostras diarreicas de adultos; Grupo 3- com amostras não diarreicas de adultos; Grupo 4- com amostras de indivíduos HIV+.
Grupo 1 Grupo 2 Grupo 3 Grupo 4
Astroviridae 1 0 0 0
Adenoviridae 0 2 0 2
Papillomaviridae 0 0 0 10
Anelloviridae 8 5 0 7
Circoviridae 0 0 1 2
Retroviridae 0 0 2 0
Picobirnaviridae 0 0 2 1
Poxviridae 16 6 18 13
Myoviridae 222 126 223 186 Siphoviridae 352 164 385 295 Podoviridae 78 44 95 73 Microviridae 5 8 9 9 Baculoviridae 0 0 29 0 Phycodnaviridae 16 16 16 16 Iridoviridae 11 3 14 6 Inoviridae 16 10 15 9 Marseilleviridae 0 3 3 2 Caulimoviridae 0 0 3 0 Bicaudaviridae 0 1 3 8 Polydnaviridae 0 0 3 0 Potyviridae 2 1 2 2 Lipothrixviridae 0 0 2 2 Rudiviridae 0 0 2 0 Plasmaviridae 1 1 1 0 Ascoviridae 1 1 1 1 Asfarviridae 1 1 1 1 Nudiviridae 0 0 1 1 Endornaviridae 0 0 1 0 Hytrosaviridae 0 0 1 1 Rodiviridae 0 1 0 0 Nimaviridae 0 1 0 0 Tectiviridae 1 0 0 0 Unclassified 142 68 136 101
7. DISCUSSÃO
O objetivo desse estudo foi avaliar o viroma de amostras fecais humanas de 12 indivíduos brasileiros divididos em quatro grupos (Grupo 1: 3 amostras fecais diarreicas de crianças; Grupo 2: 3 amostras fecais diarreicas de adultos; Grupo 3: 3 amostras fecais não diarreicas de adulto; Grupo 4: 3 amostras fecais de imunocomprometidos), e assim, identificar vírus presentes na microbiota intestinal, tanto vírus participantes da microbiota normal, quanto potenciais vírus causadores de doenças diarreicas.
Mais de 20 diferentes tipos de vírus têm sido identificados como agentes etiológicos das gastroenterites agudas, mas os principais vírus associados com a diarréia em crianças podem ser divididos em quatro famílias diferentes: Reoviridae, Caliciviridae, Astroviridae e Adenoviridae (RABONI et al, 2014).
No experimento piloto, a ausência de vírus humanos pode ser explicado pela raridade de sequências de vírus eucarióticos em amostras de viromas de intestino de indivíduos saudáveis, assim como no estudo de Minot et al (2013), em que foi analisado o viroma de um
indivíduo saudável, e nenhum vírus humano foi identificado durante o estudo (MINOT et al,
2013). Já a identificação de baculovírus nas amostras é resultado de contaminação, ocorrida no momento da ultracentrifugação, visto que, no laboratório de Biologia Celular da UnB muitos experimentos utilizam os baculovírus, sendo assim, é importante evitar e eliminar focos de contaminações, para que não mascare outras sequências e que diminua o viés do estudo. Assim, a realização desse experimento piloto foi uma etapa importante para padronização dos experimentos e detecção de possíveis ajustes na metodologia.
A diversidade viral foi avaliada nos 4 grupos distintos. Com a grande quantidade de
reads gerados no sequenciamento, ou seja, pouco mais de cem mil reads em cada biblioteca,
foi necessária a montagem de contigs no software CLC genomics workbench 7, como citado
no artigo de Smits et al (2014), com uma das ferramentas de montagem disponíveis.
A quantidade gerada de contigs foi bem diversa em cada grupo, variando de 6.152 contigs (no grupo 2 - amostras diarreicas de adultos) a 51.703 contigs (no grupo 3 – amostras
não diarreicas de adultos). A frequência dos contigs montados em cada faixa de tamanho foi
semelhante em todos os grupos, o que indica uma boa proporcionalidade entre os grupos, e que mais de 50% dos contigs, em todos os grupos, apresentaram tamanhos entre 400 e 999
nucleotídeos. A proporcionalidade também foi mantida na relação dos contigs com e sem
sequências sem correspondência variaram de 72% (grupo 1) a 80% (grupo 3). Como descrito por Breitbart et al (2003), a maioria das sequências geradas foram desconhecidas (59%) e
entre as sequências virais identificadas, a maioria era composta por bacteriófagos (BREITBART et al, 2003, SMITS et al, 2014).
Os vírus possuem alta diversidade genética, o que limita a probabilidade de se identificar vírus não conhecidos. Uma maneira de se analisar é com a utilização do BLASTx ou BLASTp, ao invés de BLASTn, pois, as mutações sinônimas são ignoradas na etapa de tradução, tornando assim, este método mais sensível para a recuperação de vírus conhecidos, e foi desta forma que as sequências foram analisadas(FANCELLO et al, 2012).
Na análise do primeiro grupo em questão (crianças com amostras diarreicas) não houve detecção de rotavírus, mas sim, bacteriófagos, astrovírus e torque teno. O torque teno vírus (TTV), assim como adenovírus, enterovírus, rotavírus e astrovírus são agentes excretados nas fezes. Este vírus está atualmente classificado na família Anelloviridae,
podendo infectar muitas espécies de vertebrados, incluindo humanos. Até o momento, no entanto, não há consenso sobre o papel da infecção pelo TTV (VECCHIA et al, 2012). Os
TTVs são caracterizados por uma elevada prevalência na população em geral e o possível envolvimento desses vírus em patologias humanas tem sido debatido desde sua descoberta. Certamente, é improvável que seja patogênico por ser um vírus quase onipresente. Estudos têm mostrado que as partículas variam de 30 a 50 nm, mas há poucos estudos que investigaram a estrutura e função. O componente mais bem estudado dos TTVs é o seu genoma, que consiste numa molécula de DNA circular, cadeia simples, polaridade negativa, com uma região rica em GC de 117 nucleotídeos (89% - 90,6%), sendo que, o tamanho do genoma varia de 3,6 a 3,9 kb (SPANDOLE et al, 2015).
A identificação de astrovírus no grupo com amostras de crianças era esperada, pois, em um dos primeiros estudos que relataram a ocorrência de casos de diarréia aguda foi associada a astrovírus, realizado por Stewien et al, (1991), no qual observaram 3% de
positividade em amostras fecais provenientes de 67 crianças hospitalizadas, residentes na cidade de São Paulo. Um estudo realizado no Rio de Janeiro, em 2004, mostrou uma prevalência de 14% de astrovírus nas amostras coletadas de crianças hospitalizadas com gastroenterite aguda (VICTORIA et al. 2007). No estudo de Alam et al (2015) foi descrito
astrovírus, sendo estes, os grandes responsáveis por causar gastroenterites em diversas crianças (ALAM et al, 2015).
No segundo grupo, composto de adultos com amostras diarreicas, foi identificado bacteriófagos, adenovírus e torque teno vírus. Os torque teno vírus, como já descrito, possuem prevalência elevada em toda população, podendo ser identificado em qualquer indivíduo. Os adenovírus humano (HAdV) provocam diarréia aguda esporadicamente, bem como surtos.
No terceiro grupo, com amostras não diarreicas de adultos, houve a detecção da família Circoviridae, mas, não foi possível identificar a espécie presente. Como esperado,
houve a identificação de bacteriófagos, já que os indivíduos se apresentavam sadios e assintomáticos.
Para investigar a origem e a evolução do viroma no intestino humano, o grupo de Minot et al (2013) analisou a comunidade viral de um único indivíduo adulto e sadio por 2
anos e 6 meses, por métodos de sequenciamento, assim, as amostras de fezes (n = 24) foram coletadas de um homem saudável em 16 pontos no tempo, distribuídos por 884 dias. Foram identificadas as seguintes famílias: Microviridae, Podoviridae, Myoviridae e Siphoviridae,
ressaltando a enorme variação de bacteriófagos. Microviridae predominou, mas essa
predominância pode ser uma consequência da amplificação favorecida por Φ29 polimerase
em pequenos genomas circulares, e aproximadamente 80% dos tipos de bacteriófagos persistiram durante todo o período do estudo, indicando estabilidade global em longo prazo e nenhum dos contigs foi correlacionado a vírus que infectam as células eucariotas (MINOT et al, 2013) Assim como no trabalho em questão, grupo com amostras não diarreicas de adultos,
não houve identificação de vírus humanos, pois, a raridade de sequências de vírus eucarióticos é típica em amostras de intestino de indivíduos saudáveis, indicando o tamanho enorme das populações de bacteriófagos do intestino.
A presença de gyrovírus foi identificada no estudo em questão como também por Minot et al (2013), onde foi caracterizado na família Circoviridae e gênero Circovirus, com
tamanhos pequenos de genoma (~2.3 kb) e que recentemente foi correlacionado a infectar os seres humanos (MINOT et al, 2013). E desde 2011 outras espécies virais tem sido encontrada
em soros e tecidos de galinhas, fezes humanas, e pele humana (GIA et al, 2013). No estudo de
Chu et al (2013), foi detectado gyrovirus (GyV4) em pele de galinha e nas fezes humanas,
mas a epidemiologia e o papel patogênico do vírus em humanos e em frangos requer uma investigação mais aprofundada, e estudos futuros devem investigar se esses gyrovírus estão se replicando em seres humanos (CHU et al, 2013).
Os papilomavírus (PVs) são uma grande família de vírus que infectam os epitélios da mucosa e da pele dos vertebrados, incluindo os seres humanos (HPV). A doença varia desde lesões benignas, como verrugas comuns, até carcinomas malignos do colo do útero, vulva, vagina, pênis e ânus. Em todo o mundo, as infecções por HPV têm sido associados a vários tipos de câncer, incluindo pulmão, mama, ovário, próstata, bexiga, uretra e câncer de cólon retal, embora os dados entre os vírus e o câncer ainda sejam controversos. O estudo de Di Bonito et al (2015) foi o primeiro a investigar a presença de HPV em amostras fecais de
pacientes hospitalizados, com sinais clínicos de diarréia, de etiologia desconhecida, utilizando reação em cadeia da polimerase (PCR), e das 103 amostras, 13 (12,6%) foram positivas para HPV, demonstrando a possibilidade de identificar HPV nas fezes. (DI BONITO et al, 2015).
Tanto no experimento piloto, quanto na comparação entre os quatro grupos se observa a grande presença de fagos, o que enfatiza a abundância dessas populações presentes no intestino, comprovando que, a maioria das sequências identificadas é de bacteriófagos.
Nos quatro grupos do estudo, as mesmas famílias identificadas por Minot et al (2013),
também foram detectadas, sendo estas as famílias com mais diversidade de espécies de bacteriófagos, mas, a família predominante em todos os quatro grupos foi a Siphoviridae,
diferentemente do que foi encontrado por Minot el al (2013), em que a família predominante
foi a Microviridae (MINOT et al, 2013).
No estudo de revisão de Abeles et al (2014) foi observado que o foco dos estudos até
agora em viromas do intestino humano, tem classificado vírus de DNA de cadeia dupla na ordem Caudovirales (incluindo as famílias Podoviridae, Siphoviridae, e Myoviridae) ou vírus
de DNA de cadeia simples, nas famílias Microviridae e Inoviridae (ABELES et al, 2014).
O grupo com amostras diarreicas de adultos (grupo 2) foi o que obteve o menor número de espécies, que foi de 462, já o grupo com amostras não diarreicas de adultos (grupo 3) apresentou o maior número de espécies, sendo de 969, assim, esses números podem ter influência direta da quantidade de contigs, ou seja, o grupo 2 apresentou o menor número, e o
grupo 3 o maior número de contigs. Assim, o grupo com maior riqueza de espécies foi o de
adultos sadios, e essa relação pode ser explicada por estarem saudáveis, já os outros três grupos obtiveram uma menor riqueza, sendo que estes estavam envolvidos com alguma patologia, que pode afetar diretamente os microrganismos presentes na microbiota intestinal. No geral, as famílias virais nos quatro grupos apresentaram alto grau de semelhança entre as comunidades de fagos (Figura 6 e Tabela 4), que pode ser causado pela sazonalidade da região do Distrito Federal (DF), mas, mais estudos detalhados são necessários para confirmar essa informação.
8. CONCLUSÕES E PERSPECTIVAS
O presente estudo fornece uma evidência adicional a respeito do viroma intestinal humano, e enfatiza a ideia de que o surgimento da metagenômica viral tem facilitado os avanços em virologia, e tem permitido a compreensão da ecologia viral de uma variedade de tipos de amostras. Atualmente, a metagenômica viral é uma técnica sensível para a detecção de vírus que não podem ser identificados por cultura tradicional, mas, podem produzir erros técnicos, com a amplificação, por exemplo, e fazer sub-estimativas, pois, os bancos de dados públicos virais são muito limitados, o que pode dificultar a determinação da diversidade viral. Muitos dos vírus em humanos são bacteriófagos, como identificado em todos os grupos desse trabalho e também por Breitbart et al, (2003), Reyes et al, (2010) e Minot et al
(2011). Os dados apresentados no estudo afirmam tanto a presença de astrovírus em amostras de crianças, quanto de adenovírus em amostras de adultos com gastroenterite e imunocomprometidos. O grupo de pacientes imunocomprometidos obteve o maior número de espécies para vírus humanos, inferindo que, a situação em que o organismo se encontra pode estar intimamente relacionada com o aumento da probabilidade de infecção, consequentemente da identificação de patógenos.
Muitas sequências foram geradas, consequentemente, muitos dados. Com isso, no futuro é possível analisar as sequências que não apresentaram correspondências com vírus, para que se tenha uma visão completa de toda a microbiota presente em cada grupo, além de estudos de filogenia para os vírus humanos identificados. Há ainda a possibilidade de se identificar vírus novos, pois, até o momento, a análise foi qualitativa, ou seja, análise da presença ou ausência dos vírus. A maioria dos estudos tem sido descritivo, assim, ainda há muito a aprender sobre a interação entre espécies na microbiota e também entre o hospedeiro e os microrganismos que habitam.
9. REFERÊNCIAS
ABELES, S. R.; PRIDE, D. T. Molecular bases and role of viruses in the human microbiome.
Journal of Molecular Biology. v. 426, n. 23, p. 3892-3906, 2014.
ALAM, M. M.; KHURSHID, A.; SHAUKAT, S.; RANA, M. S.; SHARIF, S.; ANGEZ, M.; NISAR, N.; AAMIR, U. B.; NAEEM, M.; ZAIDI, S. S. Viral Etiologies of Acute
Dehydrating Gastroenteritis in Pakistani Children: Confounding Role of Parechoviruses.
Viruses. v. 7, n. 1p. 378–393, 2015.
BIBBY, K. Metagenomic identification of viral pathogens. Trends in Biotechnology. v. 31, n. 5, p. 275-279, 2013.
BRASIL. MINISTÉRIO DA SAÚDE (2014). Sintomas e fases da AIDS. Disponível em: <http://www.aids.gov.br/pagina/sintomas-e-fases-da-aids>. Acesso em: 04/11/2014.
BRASIL. MINISTÉRIO DA SAÚDE (2010). Capacitação em Monitorização das Doenças Diarreicas Agudas-MDDA. Disponível em:
<http://bvsms.saude.gov.br/bvs/publicacoes/capacitacao_monitoramento_diarreicas_monitor. pdf>. Acesso em: 04/11/14.
BREITBART, M.; HEWSON I.; FELTS B.; MAHAFFY J.M.; NULTON J.; SALAMON P.; ROHWER F. Metagenomic Analyses of an Uncultured Viral Community from Human Feces.
Journal of Bacteriology. v. 185, n. 20, p. 6220–6223, 2003.
CAPOBIANCHI M. R.; GIOMBINI E.; ROZERA G. Next-generation sequencing technology in clinical virology. Clinical Microbiology and Infection. v. 19, n. 1, p.15–22, 2012.
CARVALHO, M. C. C. G.; SILVA, D. C. G. Sequenciamento de DNA de nova geração e suas aplicações na genômica de plantas. Ciência Rural. v. 40, n. 3, p.735-744, 2010.
CASTRIGNANO S. B.; NAGASSE-SUGAHARA T. K.; KISIELIUS J. J.; UEDA-ITO M.; BRANDÃO P. E.; CURTI S. P. Two novel circo-like viruses detected in human feces: complete genome sequencing and electron microscopy analysis. Virus Research. v. 78, n. 2, p. 364– 373, 2013.
CHU, D. K.; POON, L. L.; CHIU, S. S.; CHAN, K. H.; NG, E. M.; BAUER, I.; CHEUNG, T. K.; NG, I. H.; GUAN, Y.; WANG, D.; PEIRIS, J.S. Characterization of a novel gyrovirus in human stool and chicken meat. Journal of Clinical Virology. v. 55, n. 3, p. 209–213, 2012. DI BONITO, P.; DELLA L.; PETRICCA, S.; IACONELLI, M.; SANGUINETTI, M.; GRAFFEO, R.; ACCARDI, L.; LA ROSA, G. A LARGE SPECTRUM OF ALPHA AND BETA PAPILLOMAVIRUSES ARE DETECTED IN HUMAN STOOL SAMPLES.
Journal of General Virology. v. 96, n. 3, p. 607-613, 2015.
FANCELLO, L.; RAOULT, D.; DESNUES, C. Computational tools for viral metagenomics and their application in clinical research.Virology. v. 434, n. 2, p. 162–174, 2012.
FINKBEINER S.R.; ALLRED A. F.; TARR P. I.; KLEIN E. J.; KIRKWOOD C. D.; WANG D. Metagenomic Analysis of Human Diarrhea: Viral Detection and Discovery. PLOS
Pathogens. v. 4, n. 2, p.1000011, 2008.
GIA, P. T.; PHUNG V. N.; SDIRI-LOULIZI, K.; AOUNI, M.; POTHIER, P.; AMBERT-BALAY, K.; DENG, X.; DELWART, E. Divergent gyroviruses in the feces of Tunisian children. Virology. v. 446, n. 1-2, p. 346-348, 2013.
HANDELSMAN J.; RONDON M. R.; BRADY S. F.; CLARDY J.; GOODMAN R. M. Molecular biology access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry & Biology. v. 5, n. 10, p. R245-R249, 1998.
HUNTER, P. The secret garden's gardeners. Research increasingly appreciates the crucial role of gut viruses for human health and disease. EMBO reports. v. 14, n. 8, p. 683-685, 2013. JONES, M. S.; HARRACH, B.; GANAC, R. D. New Adenovirus Species Found in a Patient Presenting with Gastroenteritis . Journal of Virology. v. 81, n. 11, p. 5978-5984, 2007. KAPOOR, A.; SLIKAS, E.; SIMMONDS P.; CHIEOCHANSIN, T.; NAEEM A.;
SHAUKAT, S.; ALAM, M. M.; SHARIF, S.; ANGEZ, M.; ZAIDI, S.; DELWART, E. A new bocavirus species in human stool. TheJournal of Infectious Diseases. v. 199, n. 2, p. 196– 200, 2009.