UNIVERSIDADE DE LISBOA
F
ACULDADE DE
C
IÊNCIAS
D
EPARTAMENTO DE
Q
UÍMICA E
B
IOQUÍMICA
Clonagem, Expressão e Purificação da
Sinfilina-1 e da Parkina
Maria Helena Coelho de Sousa Franco
Mestrado em Bioquímica
Especialização em Bioquímica Médica
UNIVERSIDADE DE LISBOA
F
ACULDADE DE
C
IÊNCIAS
D
EPARTAMENTO DE
Q
UÍMICA E
B
IOQUÍMICA
Clonagem, Expressão e Purificação da
Sinfilina-1 e da Parkina
Dissertação orientada por:
Prof. Doutor Alexandre Luís Quintas
Prof. Doutora Ana Ponces Freire
Maria Helena Coelho de Sousa Franco
Mestrado em Bioquímica
Especialização em Bioquímica Médica
i
Resumo
A Doença de Parkinson (DP) é a doença neurodegenerativa mais comum a seguir à doença de Alzheimer e está associada à degeneração dos neurónios dopaminérgicos da pars compacta da
substantia nigra (SNpc). Nesta doença, o misfolding, o unfolding e a agregação proteica, em conjunto
com uma resposta disfuncional da célula ao stress induzido pelas espécies parcialmente desnaturadas, pode estar na origem de danos celulares que conduzem à degeneração dos neurónios dopaminérgicos.
A DP é essencialmente idiopática, à excepção de um número reduzido de indivíduos que são portadores de um gene mutante (<10 % dos casos de Parkinson) ou que foram expostos a agentes indutores de parkinsonismo, como o MPTP.
A etiologia da DP resulta da interacção dinâmica de factores genéticos e ambientais, em que a acumulação de proteínas parcialmente desnaturadas, a presença de agregados e de proteínas ubiquitinadas nos corpos de Lewy, sugere um motivo comum a interrelacionar estes factores.
Dentro da DP hereditária, existe um grupo restrito cuja sintomatologia precoce e a ausência de Corpos de Lewy é denominada como Parkinsonismo Juvenil Autossómico Recessivo. Neste contexto, os estudos genéticos revelaram a presença de variadas mutações associadas ao gene da Parkina, uma proteína com 465 resíduos de aminoácidos com uma massa molecular aparente de 52 kDa. A sua função está relacionada com as vias de degradação de proteínas, sendo considerada um ligase entre a ubiquitina e a proteína alvo possuindo também um domínio N-terminal idêntico à ubiquitina, cuja função permanece desconhecida.
Actualmente, os estudos in vitro desenvolvidos com a Parkina humana têm sido realizados exclusivamente com domínios desta, uma vez que a purificação da Parkina de forma integral tem-se revelado uma tarefa árdua. Esta dificuldade tem se observado devido à elevada complexicidade do processo de folding, uma vez que a proteína apresenta variados domínios e complexa com iões zinco.
A Sinfilina-1, uma proteína de interacção da α-sinucleína, é um dos substratos ubiquitinados pela Parkina. Desde que foi descoberta, em 1999, têm sido desenvolvidos estudos que demonstram a existência de uma relação entre a Sinfilina-1 e a doença de Parkinson. No entanto, apesar de ainda não ser conhecido o seu papel na patologia está cientificamente aceite que a Sinfilina-1 é capaz de promover a formação de inclusões semelhantes a corpos de Lewy.
O objectivo fundamental deste trabalho consistiu na expressão e purificação de duas proteínas de relevância para a doença de Parkinson: a Parkina e a Sinfilina-1. A expressão e a purificação destas proteínas potenciam estudos estruturais, estudos de folding e de estabilidade, bem como as alterações da interacção entre a Parkina, Sinfilina-1 e α-sinucleína.
No presente estudo desenvolveu-se uma metodologia de purificação da Parkina e da Sinfilina-1, de forma integrada, com o objectivo de elucidar o envolvimento da Parkina na patogénese da Doença de Parkinson.
Palavras-chave: Corpos de Lewy, Parkina, PARK2, Parkinsonismo Juvenil Autossómico Recessivo, purificação de proteínas, Sinfilina-1, Subclonagem.
ii
Abstract
Parkinson's Disease (PD) is the most common neurodegenerative disease after Alzheimer's disease and is associated with degeneration of dopamine neurons in the substantia nigra pars
compacta (SNpc). In this disease, misfolding, unfolding and protein aggregation, together with a
dysfunctional response to cell stress induced by the partially denatured species, could cause cell damage that, in turn, leads to degeneration of dopaminergic neurons.
PD is primarily idiopathic, with the exception of a few individuals who are carriers of a mutated gene (<10% of cases of Parkinson's) or who were exposed to agents that induce Parkinsonism, such as MPTP.
The etiology of PD results from the dynamic interplay of genetic and environmental factors in the accumulation of partially denatured, aggregated and ubiquitinated proteins in Lewy bodies, suggesting a common motif connecting these factors.
Within hereditary PD, a small group, known as autosomal recessive juvenile Parkinsonism, is characterized by early symptoms and the absence of Lewy bodies. In this context, genetic studies revealed the presence of various gene mutations associated with Parkin gene, a protein with 465 amino acid residues with an apparent molecular mass of 52 kDa. Parkin plays a role in protein degradation, being considered an ubiquitin ligase and possesses an N-terminal domain similar to ubiquitin, whose function remains unknown.
Currently, in vitro studies developed with human Parkin have been performed exclusively with protein domains, since the purification of complete Parkin has proved to be an arduous task. This difficulty has been observed due to the high complexity of folding, since the protein has different domains and complexes with zinc ions.
Synphilin-1, an interaction protein of α-synuclein, is also a Parkin substrate. Since its discovery in 1999, studies have been developed that demonstrate the existence of a relationship between Synphilin-1 and Parkinson's disease. Although its contribution to the pathology is still unknown, it is scientifically accepted that Synphilin-1 can promote the formation of inclusions similar to Lewy bodies.
The basic aim of this work consisted in the expression and purification of two proteins of relevance to Parkinson's disease: Synphilin-1 and Parkin. The expression and purification of these proteins potentiate structural, folding and stability studies, as well as modifications in the interaction between Parkin, Synphilin-1 and α-synuclein.
In this study a methodology was developed for purification os Parkin and Synphilin-1, as well as protein with the aim of elucidating the involvement of Parkin in the pathogenesis of Parkinson´s disease in future studies.
Keywords: Lewy bodies, Parkin, PARK2, Parkinsonism Juvenile Autosomal Recessive, protein purification, Sinfilina-1, subcloning.
iii
Agradecimentos
Ao Professor Doutor Alexandre Quintas, orientador do projecto, pela total liberdade e todo o apoio ao logo do desenvolvimento do projecto. Obrigado pela amizade e compreensão. À Professora Doutora Ana Ponces, orientadora interna, que sempre se mostrou disponível para apoiar e motivar. Pela inspiração ao longo da Licenciatura e Mestrado na Faculdade de Ciências da Universidade de Lisboa. À FFCUL, pelo financiamento no âmbito do projecto PTDC/QUI/73430/2006. À Ana Lajes, colega de tantas horas de trabalho, pelo suporte e companheirismo. Ao Carlos Família e à Joana Couceiro, pela amizade. Ao Henrique Neves pelas brincadeiras. Ficarám sempre na minha memória os passeios fantásticos, pensados e marcados à última da hora. Com vocês, quaisquer 5 minutos de pausa podem ser uma verdadeira aventura… À Susana Bandarra, pela amizade e orientações no campo da Biologia Molecular. À Teresa Calejo, à Inês Cavaleiro e ao Paulo Mascarenhas pela boa disposição e amizade. À Professora Doutora Madalena Oom pela supervisão nos procedimentos de Biologia Molecular. À minha família, pelo apoio incondicional que os fez atravessar Lisboa para me virem buscar a “altas” horas da noite. Dedico este trabalho ao meu pai, sempre presente na saudade.
Der Mensch, der den Berg versetzte,
war derjenige, der anfing, kleine Steine aus dem Weg zu räumen. Chin.Sprichwort
Wer immer sich zum Schüler macht, wird immer einen Meister finden Friedrich von Hagedorn
iv
Abreviaturas
CL – Corpos de Lewy CSF – Common fragil site DA – Doença de Alzheimer DMJ - Doença de Machado-Joseph DP – Doença de Parkinson
DP-AD – Doença de Parkinson Autossómica Dominante DP-AR – Doença de Parkinson Autossómica Recessiva DUB – Deubiquitinating Enzyme
EGF – Factor de crescimento epidermal
EGFR – Receptor do factor de crescimento epidermal
Eps15 – Epidermal growth factor receptor pathway substrate 15
ERAD – Mecanismo de degradação associado ao retículo endoplasmático ERQC – Mecanismo de controlo de qualidade do retículo endoplasmático HECT – Domínio Homologous to E6-Associated Protein (E6AP) C-Terminus HSP70 - Heat shock protein 70
IBR – Domínio In-between-RING K - Lisina
LN - Neuritos de Lewy
LRRK2 - Gene leucine-rich repeat kinase 2 NAC - Componente beta não-amilóide NO - Óxido nítrico
PACRG - Gene co-regulador da Parkina
Pael-R - Parkin associated endothelin-like receptor PI – Ponto isoeléctrico
PI3K - Fosfoinositol 3-cinase PINK1 - PTEN-induced kinase 1
PJ-AR – Parkinsonismo Juvenil Autossómico Recessivo RBR – Superdomínio RING-IBR-RING
RE – Retículo endoplasmático
RING – Domínio Really interesting new gene ROS - Espécies reactivas de oxigénio SN – Substância nigra
SNC – Sistema Nervoso Central SNCAIP – SNCA Interacting Protein SNpc – Pars compacta da substância nigra SytXI – Sinaptotamina XI
U – Unidades enzimáticas (moles de substrato convertido por unidade de tempo). 1U corresponde 1 μmol min-1
UBC – Ubiquitin conjugation UBL – Domínio do tipo ubiquitina
UIM – Motif de interacção com a ubiquitina WT – Wild-type
v
Í
NDICE
:
Resumo ... ii
Abstract ... ii
Agradecimentos ... iii
Abreviaturas ... iv
1.
Introdução ... 1
2.
Aspectos Gerais da Doença de Parkinson ... 3
2.1. Incidência ... 6
2.2. Etiologia ... 6
2.3. Mecanismos de Morte Celular ... 7
2.3.1. Disfunção mitocondrial ... 7
2.3.2. Stress oxidativo ... 8
2.3.3. Excitotoxicidade ... 8
2.4. Parkinson idiopática e hereditária ... 8
3.
Parkinson Juvenil Autossómico Recessivo (PJ-AR) ... 10
4.
O locus PARK2: Estrutura e função ... 13
5.
Factores genéticos envolvidos na patogénese hereditária ... 16
5.1. PARK1 e 4-SNCA (α-sinucleína) ... 16
5.2. PARK2 (Parkina) ... 16 5.3. PARK5 (UCH-L1) ... 16 5.4. PARK6 (PINK1) ... 17 5.5. PARK7 (DJ1) ... 17 5.6. PARK8 (LRRK2) ... 17
6.
Estruturas e funções ... 19
6.1. Estrutura da Parkina: Domínios ... 19
6.1.1. Domínio UBL ... 19
6.1.2. Super-domínio RING-IBR-RING (RBR) ... 20
6.1.3. In between RINGs ... 20
6.1.4. Domínio RING0 ... 21
6.2. Função da Parkina ... 22
6.2.1. Via de degradação pelo proteassoma ... 23
6.3. Estrutura da Sinfilina-1: Domínios ... 27
6.3.1. Domínio ankyrin-like motifs ... 27
6.4. Função da Sinfilina-1 ... 28
vi
7.1. A Ubiquitina como substrato ... 30
7.2. Sinfilina-1 ... 31
7.2.1. Complexo Parkina, α-sinucleína e Sinfilina-1 ... 31
7.3. Alfa-sinucleína... 31
7.4. HSP70 ... 33
7.5. Parkina associated endothelin-like receptor ... 34
7.6. CDCrel-1 e Sept5 ... 35
7.7. LRRK2 ... 36
7.7.1. Complexo Parkina, PINK1 e DJ1 ... 36
8.
Importância funcional das mutações Parkina ... 38
9.
Bioquímica da Parkina ... 39
9.1. Parkina na via do Fosfatidilinositol / Akt ... 39
9.2. Parkina na via IKK/NFkb ... 41
9.7. S-Nitrosilação ... 44
9.8. Formação de Agressomas ... 44
10.
A Parkina noutras Patologias ... 45
10.1. Susceptibilidade à lepra ... 45
10.2. Supressor tumoral ... 45
Metodologia, resultados e discussão ... 46
1.
Parkina ... 47
1.1. Subclonagem ... 47
1.1.1. Procedimentos e Considerações ... 48
1.2. Expressão e Purificação ... 51
1.2.1. Lise celular e Purificação dos corpos de inclusão ... 52
1.2.2. Refolding proteico ... 53
2.
Sinfilina-1 ... 55
2.1. Expressão e Purificação ... 56
2.1.1. Lise celular e Purificação dos corpos de inclusão ... 57
2.1.2. Refolding proteico ... 58 2.1.3. Purificação da Sinfilina-1 ... 59
3.
Considerações finais ... 61
4.
Anexos ... 63
4.1. Anexo 1 ... 63 4.2. Anexo 2 ... 64vii
Í
NDICE DE FIGURAS
:
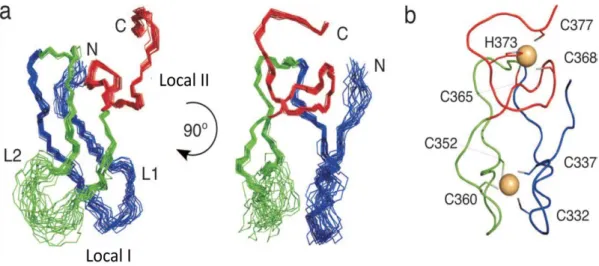
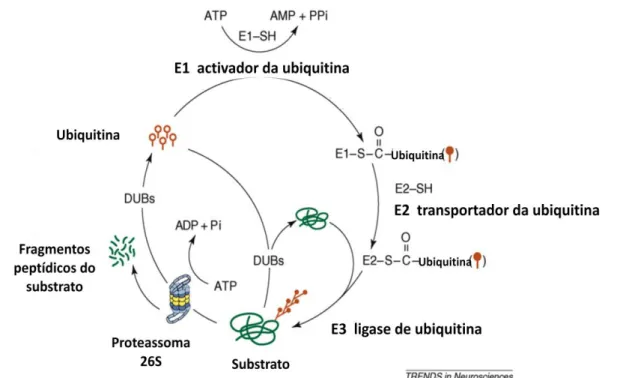
Figura 1 – Os Corpos de Lewy do tronco cerebral e do proencéfalo basal apresentam um diâmetro superior a 15µm e são constituídos por um centro hialinico esférico e um halo distinto. Os CL corticais, por sua vez, possuem dimensões mais reduzidas, não apresentam um core disto sendo facilmente observados com coloração anti-ubiquitina. A imagem A mostra um CL no citoplasma de um neurónio dopaminérgico pigmentado na substancia nigra (hematoxylin-eosin and Luxol fast blue, X100). A imagem B mostra um exame ultra-estrutural de um Corpo de Lewy onde é visível a acumulação de filamentos e material granular com um centro denso e filamentos periféricos soltos (X21,560). Adaptado de Lang e A M Lozano (Lang & A M Lozano 1998). ____________________________________________ 4 Figura 2 - Loci envolvidos na genética da DP, localização cromossómica, gene, hereditariedade e função provável adaptado de Gil (Gil 2009). ______________________________________________________ 15 Figura 3- Estrutura modular da Parkina. UBL: Domínio semelhante à ubiquitina; R0: Domínio RING 0; R1: Domínio RING 1; R2: Domínio RING 2; IBR: Domínio In-Between-RING. Adaptado de Mata, von Coelln e Hristova (Mata et al. 2004; von Coelln et al. 2004; Hristova et al. 2009) __________________________ 19 Figura 4 – Sequencia especifica característica de domínios RING em que X representa qualquer aminoácido. Retirado de Borden, Morett e Bork (Borden 2000; Morett & Bork 1999). ______________ 20 Figura 5 - Estrutura do domínio IBR (307-384) na zona 320-377 (NMR). A zona N-terminal 307-319 e C-terminal 378-384 não surgem representadas devido à falta de estrutura. (a) Sobreposição do backbone de 20 estruturas usando CYANA. Local I de ligação do zinco compreende as extensas regiões em loop L1 (azul) e L2 (verde), e o local II de ligação do zinco encontra-se a vermelho. (b) Representação do IBR da Parkina, demonstrando a posição dos resíduos coordenados com os zincos. Adaptado de Beasley, Hristova e Shaw (Beasley, Hristova & Shaw 2007a). __________________________________________ 21 Figura 6 - Ilustração da degradação associada ao retículo endoplasmático. (a) Proteínas misfolded intramembranares do citoplasma e lúmen do RE são reconhecidas por chaperones citoplasmáticos e do lúmen do RE e por factores associados, como membros da família das Hsp70, calnexina e calreticulina, e isomerases de ligações persulfureto. (b) Marcação proteica. Substratos do ERAD são retrotranslocados, após marcação, para a maquinaria de retrotranslocação (o translocão) e/ou E3 ligases. (c) Iniciação da retrotranslocação. A retrotranslocação do substrato para o citoplasma é iniciada pelo complexo Cdc48 (cell-division cycle-48) ou outros componentes, como chaperones moleculares ou proteassoma. A energia proveniente da hidrólise do ATP pelo Cdc48, que é um AAA+ ATPase, é usada na retrotranslocação. (d) Ubiquitinação e finalização da retrotranslocação. Após a passagem pelo translocão, a proteína é poliubiquitinada por uma E3 ligase de ubiquitina. A reacção finaliza a translocação com o auxílio de complexos citoplasmáticos que ligam ubiquitina a proteínas. (e) Marcação e degradação proteassomal. Após a poliubiquitinação, o substrato disponível no citoplasma é reconhecido pelos receptores do topo 19S pertencentes ao proteassoma 26S. Os enzimas envolvidos na desubiquitinação (não apresentados) removem a marcação e a N-glicase de péptidos (não apresentado) que é potencialmente necessária para uma degradação eficiente. O substrato é introduzido no core catalítico do proteassoma onde é degradado em péptidos. A ubiquitina proveniente do processo de degradação pode ser reciclada. Adaptado de Vembar e Brodsky (Vembar & Brodsky 2008). _________ 24 Figura 7 - Via da ubiquitina-proteassoma. O enzima E1 adenila o C-terminal do resíduo Glicina da ubiquitina com hidrolise de ATP. O adenilato de ubiquitina intermediário liga-se ao grupo tiol da cisteína do centro activo do enzima E1 formando um tioéster de ubiquitina e AMP livre. Seguidamente, o tioéster de ubiquitina é transferido para o resíduo de cisteína do centro activo do E2, um enzima de conjugação da ubiquitina. Posteriormente o enzima E2 transfere a cadeia de ubiquitinas directamente para o substrato ou, em alternativa, para o E3 que se encarrega de a ligar ao substrato (dependendo do E3). Várias moléculas de ubiquitina são ligadas sequencialmente pelo E3 ligase, formando uma cadeia de poliubiquitinas. Os substratos poliubiquitinados são degradados pelo complexo proteolítico denominado de proteassoma 26S, num processo dependente de ATP. A ubiquitinação é um processo reversível em que as DUBs degradam a cadeia de poliubiquitinas prevenindo a degradação do substrato necessário, reciclando a ubiquitina. Ubiquitina – círculos vazios com caudas; ubiquitina activada – círculos preenchidos. De referir que a conjugação da ubiquitina pode ocorrer, independentemente da via da ubiquitina-proteassoma, de uma forma não canónica (por activação da UBL, activação do híbrido
E1-viii
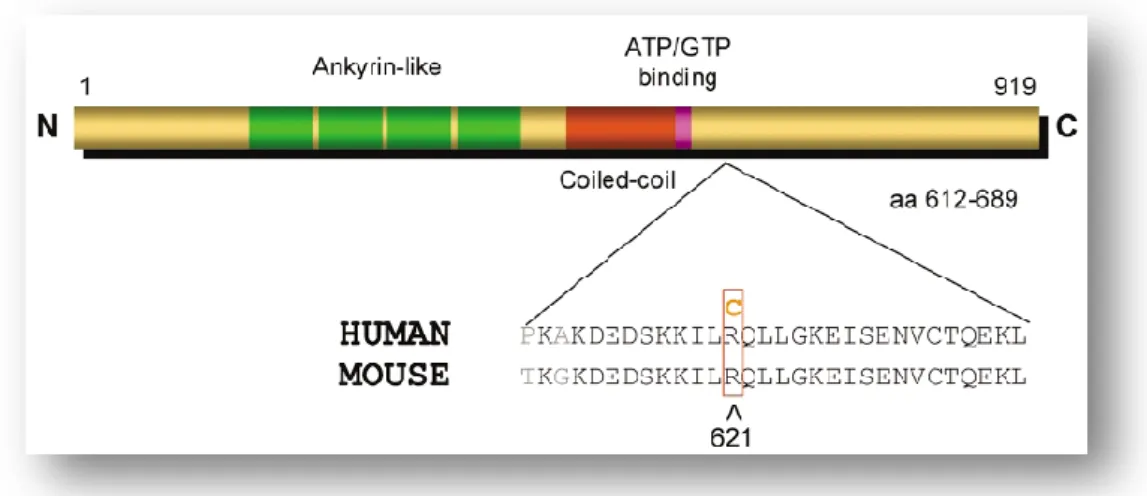
E2 ou E2-E3 ou por elongação das cadeias curtas de ubiquitina pela E4) Adaptado de Hegde e Upadhya (Hegde & Upadhya 2007). ______________________________________________________________ 26 Figura 8 - Vista esquemática da proteína Sinfilina-1, com comparação de sequências entre a variante humana e a de murganho. Retirado de Marx (Marx 2003). ____________________________________ 27 Figura 9 - (a) Modelo proposto sobre a interacção da Parkina com o Esp15. O EGF estimula a ligação entre a Parkina e o Eps15 numa interacção dependente de UBL e UIM (esquerda). A ligação ao Eps15 estimula a actividade E3 ligase da Parkina e promove a ubiquitinação do Eps15 (centro). A ligação intramolecular da ubiquitina no Eps15 pelo UIMs interfere com a ligação de Ubl por parte da Parkina e destrói a interacção entre a Parkina e o Eps15 (direita). (b-c) O modelo explica como é que a ubiquitinação mediada pela Parkina consegue regular o trafficking e sinalização do EGFR. (b) Na presença de Parkina, a ubiquitinação do Eps15 previne a sua ligação ao complexo EGFR, o que atrasa a endocitose do receptor e prolonga a sinalização via neuronal de sobrevivência PI3K-Akt. (c) Na ausência de Parkina: Defeitos na ubiquitinação mediada pela ubiquitina, como é o caso de DP associada à Parkina, aumenta a disponibilidade de Esp15 capaz de se ligar ao complexo EGFR, o que por sua vez acelera a endocitose e menospreza a sinalização via PI3K-Akt. Adaptado de Fallon (Fallon et al. 2006). ________________________________________ 40 Figura 10 - Modelo do papel funcional da Parkina na via de sinalização IKK/NFkB. Durante o stress e em condições fisiológicas a Parkina promove regulação por ubiquitinação do TRAF2 e IKKγ, levando à activação do NF-κB. Consequentemente, a transcrição de genes prosurvival é upregulated. A actividade da Parkina pode ser modelada pelo stress. Sob stress moderado, a Parkina é upregulated. Para uma resposta imediata, é possível que a actividade da Parkina possa ser regulada por modificações pos-traducionais. Em contraste, no caso de um severo stress proteotoxico, induzido por exemplo por dopamina oxidada, ocorre misfolding e inactivação da Parkina. Mutações no gene da Parkina, relacionado com parkinsonismo juvenil autossómico recessivo, também interferem com a capacidade da Parkina de estimular a via de sinalização IKK/NFkB. Adaptado de Henn e Bouman (Henn et al. 2007).__ 42 Figura 11 - A: Mapa do vector circular pCI-neo (Promega). B: Mapa do vector circular pGEX-4T-1 (Amersham) _________________________________________________________________________ 47 Figura 12 - Primer Forward (A) e Primer Complementary Reverse (B) usados para amplificar a região codificante do gene PARK2 no processo de subclonagem. (A) apresenta um Tm a 61,6ºC e (B) a 65, 6ºC. Os enzimas Bam HI e EcoR I não possuem local de restrição na sequência da PARK2. O Bam HI permite uma introdução em frame da PARK2 no pGEX-4T-1. O design dos primers teve em conta a adição de um conteúdo “GC” no início da sequência de forma a facilitar a ligação deste à cadeia de DNA molde e a posterior actuação da polimerase. _______________________________________________________ 48 Figura 13 - Multiple cloning site do vector de destino pGEX-4T-1 (Amersham) _____________________ 48 Figura 14 - Condições do PCR para amplificação da região codificante do gene PARK2. ______________ 49 Figura 15 – Controlo da eficácia da subclonagem por restrição do DNA plasmídico resultante com os enzimas Bam HI e EcoR I, utilizados no passo da restrição. Poços 2 a 10: restrição do DNA plasmídico de 9 colónias independentes resultante da subclonagem, obtiveram-se dois fragmentos de tamanhos correspondentes ao vector digerido (≈4900pb) e Parkina (1395pb); Poços 11 a 19: verificação da integridade dos plasmídeos obtidos a partir das 9 colónias respectivas. _________________________ 50 Figura 16 - Primers externos pGEX_For (A) pGEX_Rev (B) usado para sequenciação do plasmídeo clonado com a região codificante do gene PARK2 no processo de subclonagem __________________________ 50 Figura 17 - Gel SDS-PAGE 12% contendo aliquotas provenientes da fracção solúvel da indução de expressão. M: marcador; T0: 0h; T1: 1h; T2:2h; T3:3h; T4:4h; T5:6h; T6:8h. As setas a vermelho indicam a banda a aproximadamente 80kDa. _______________________________________________________ 51 Figura 18 - Gel SDS-PAGE 12% contendo aliquotas provenientes da fracção insolúvel da indução de expressão após a sonicação. M: marcador; T0: 0h; T1: 1h; T2:2h; T3:3h; T4:4h; T5:6h; T6:8h __________ 52 Figura 19 - Sequência de diálises utilizadas no refolding da proteína de fusão GST-Parkina. Condições de diálise I, II e III. _______________________________________________________________________ 53 Figura 20 - Gel SDS-Page a 12% com amostras cujo pellet foi previamente ressuspenso após as diálises com diferentes tempos de indução. M: marcador; T0: 0h; T1: 1h; T2:2h; T3:3h; T4:4h; T5:6h. _____________ 54
ix
Figura 21 – Mapa do vector pET30A (Novagen) _____________________________________________ 55 Figura 22 - Multiple cloning site do vector pTE30A (Novagen) __________________________________ 55 Figura 23 - Gel de agarose com digestão simples e dupla de duas amostras de plasmídeo pET30A. 1) Marcador; 2) pET30A digerido com Xho I (amostra 1); 2) pET30A digerido com XhoI e BamHI (amostra 1); 3) pET30A digerido com Xho I (amostra 2); 4) pET30A digerido com XhoI e BamHI (amostra 2). ______ 56 Figura 24 - Gel SDS-PAGE 12% contendo aliquotas provenientes da fracção solúvel da indução de expressão. M: marcador; T0: 0h; T1: 2h; T2: 4h; T3: 6h; T4: 8h __________________________________ 57 Figura 25 - Sequência de diálises utilizadas no refolding da Sinfilina-1. Condições de diálise I, II e III. ___ 58 Figura 26 - Gel SDS-Page a 12% com amostras de Sinfilina-1 após diálises I, II e III. ___________________ 59 Figura 27 – Fragmento dos resultados da chapa fotográfica proveniente do Western Blot com detecção da Sinfilina-1 com Anticorpo Anti-Sinfilina-1 aos diferentes tempos. ____________________________ 59 Figura 28 - Cromatograma proveniente da coluna de caudas de Histidina após injecção da amostra de Sinfilina-1. ___________________________________________________________________________ 60 Figura 29 - Gel SDS-Page a 12% com fracções recolhidas na cromatografia de afinidade. M: marcador; A1: fracção recolhida após injecção da amostra (0-5mL); A2: fracção recolhida ao equilibrar da coluna com a amostra (5-10 mL); A3: fracção recolhida antes da troca do tampão para o Elution Buffer (100-105mL); A4: fracção recolhida após a adição do Elution Buffer (105-110mL); A5: fracção recolhida após a adição do Elution Buffer (110-115mL) A6: fracção após a adição do Elution Buffer (115-120mL); A7: fracção recolhida após a adição do Elution Buffer (120-125mL); A8: fracção recolhida após a adição do Elution Buffer (125-130mL) A9: fracção recolhida após a adição do Elution Buffer (135-140mL). ______________________ 60
1
1. Introdução
Rudolf Virchow descreveu pela primeira vez, em 1854, uma substância eosinófila depositada em diversos tecidos com aparência hialina quando observada em microscopia óptica e com algumas características de coloração histoquímica semelhantes ao amido, que designou por substância amilóide (semelhante ao amido). Embora Friedrich e Kekule tenha demonstrado que estes compostos tinham natureza proteica, a designação “amilóide” perdurou e ainda hoje é usada para denominar depósitos de fibras resultantes da agregação de proteínas características de determinada doença (Costa 1994).
Em 1971, Glenner e colaboradores caracterizaram pela primeira vez uma proteína responsável pela formação de depósitos amilóides (G. G. Glenner et al. 1971). Desde então, observou-se que muitas proteínas solúveis podem produzir depósitos amilóides. Actualmente, existem cerca de várias amiloidoses conhecidas entre as quais a doença de Alzheimer (DA), a doença de Parkinson (DP), a Encefalopatia Espongiforme Bovina e a Polineuropatia Amiloidótica Familiar.
As amiloidoses dividem-se em localizadas, quando os depósitos ocorrem num único órgão, como é o caso do cérebro na DP, ou sistémicas quando a deposição afecta múltiplos órgãos. Apesar da heterogeneidade das amiloidoses, todas apresentam características comuns como depósitos amilóides com elevada resistência à proteólise e insolúveis, estruturalmente apresentam predominantemente folhas β ({ excepção das redes de neurofibrilhas), possuem propriedades cromóforas e ópticas comuns, como coloração verde após coloração com vermelho do Congo e a fluorescência característica da ligação à tioflavina T, para além da capacidade de associação a outras proteínas (Ghiso et al. 1994).
Uma característica importante dos depósitos amilóides é a estrutura secundária predominante de folha-β, com as cadeias β das proteínas orientadas perpendicularmente ao longo do eixo das fibrilhas (GLENNER et al. 1968) cuja análise por espectroscopia de infra-vermelhos revelou que a orientação das proteínas polimerizadas é antiparalela (Termine et al. 1972). Genericamente as fibras amilóides resultam da formação de agregados insolúveis a partir de proteínas nativas ou suas variantes que produzem intermediários com elevado potencial de agregação no processo de folding ou de unfolding (Chiti et al. 1999; Chiti et al. 2000; Jaenicke 1995; Taubes 1996; Uversky 1998; WETZEL 1996); de proteínas nativas ou variantes que sofrem alterações conformacionais após o processo de folding (Booth et al. 1997; Carrell & Gooptu 1998; Perutz 1997; Telling et al. 1996); a partir da superprodução de proteínas que devido à elevada quantidade tendem a agregar (Benson & Wallace 1989; Guela et al. 1998; Gupta et al. 1998); ou proteólise de uma proteína precursora (C Haass 1993; Kosik 1999).
2
As várias proteínas amiloidogénicas (precursoras dos depósitos amilóides nas várias amiloidoses) não apresentam qualquer homologia estrutural ou sequencial relevante, mas originam fibrilhas amilóides in vivo com conformação similar. Este facto sugere a existência de um mecanismo comum na formação dos depósitos amilóides a partir das diferentes proteínas precursoras (Massimo Stefani 2008). Os mecanismos moleculares de agregação proteica têm sido objecto de atenção crescente devido ao seu papel central em diversas patologias degenerativas. A abordagem mais directa a este problema, apesar dos diversos factores celulares presentes e específicos de cada patologia, consiste em estabelecer relações entre a estrutura, estabilidade e dinâmica das proteínas mutantes, modificadas pós-traducionalmente, ou intermediários potencialmente amiloidogénicos com a proteína normal.
3
2. Aspectos Gerais da Doença de Parkinson
Originalmente descrita por James Parkinson, em 1817, no ensaio intitulado “An Essay of the Shaking Palsy” (Parkinson 2002) a DP é a doença degenerativa com maior incidência depois da doença de Alzheimer, afectando cerca de 4 milhões de pessoas em todo o mundo (Bushnell & M. L. Martin 1999). Esta pode ser caracterizada por três sintomas fundamentais: bradicinesia (lentidão anormal dos movimentos voluntários), tremor de repouso e rigidez muscular (Fahn 2003), sintomatologia que se manifesta como consequência da perda progressiva e selectiva dos neurónios dopaminérgicos no sistema nervoso central.
Na DP vários grupos de neurónios são afectados. No entanto, esta patologia é caracterizada pela perda selectiva e em grande escala de neurónios dopaminérgicos pigmentados com neuromelanina, atingindo valores de 80% num universo de cerca 450.000 neurónios dopaminérgicos presentes na SNpc humana (Lang & A M Lozano 1998), e em menor escala, perda dos neurónios não pigmentados da área ventral tegmental vizinha, impulsionados pela diminuição dos níveis de dopamina (E Hirsch et al. 1988; German et al. 1989). O facto de apenas neurónios específicos serem vulneráveis à DP é uma questão ainda em aberto e crítica no contexto da desordem. Uma explicação poderá residir na capacidade de certos neurónios assimilarem os compostos tóxicos endógenos e extrínsecos através e mecanismos transportadores selectivos, como é o caso do transportador de dopamina. Outras hipóteses incluem o aumento do stress metabólico, elevados níveis fisiológicos de proteína oxidada, degeneração selectiva de toxinas ou falha no sistema de destoxificação ou orientação desta (possivelmente devido à presença de neuromelanina) bem como a necessidade de requisitos específicos para o suporte neurotrófico (Lang & A M Lozano 1998).
A morte neuronal observada tem tipicamente duas origens: 1) no decréscimo da dopamina disponível no corpo estriado, ou 2) no numero reduzido de receptores de dopamina no mesmo local, sendo que o primeiro caso constitui cerca de 80% dos casos existentes e é genericamente denominado de doença de Parkinson (Fahn 2003; Sulzer 2007).
A actividade normal da função motora depende da síntese e libertação de dopamina pelos neurónios dopaminérgicos na substância nigra que incidem sobre o striatum (Chase et al. 1998). Fisiologicamente a morte gradual dos neurónios dopaminérgicos da SNpc ocorre com a idade, acompanhado pela redução dos níveis de dopamina no striatum. No entanto, a degeneração aumentada dos neurónios dopaminérgicos localizados na substância nigra pars compacta (SNpc) e a subsequente perda dos nervos dopaminérgicos terminais no striatum são responsáveis pela maior parte dos distúrbios de movimento (Chase et al. 1998; Nagatsu et al. 2000). Desta forma, o controlo sintomático da DP com levodopa e outras drogas dopaminérgicas dominam a actual terapia, sendo eficazes no controlo das discinesias, apenas nas fases iniciais da doença. No entanto, a longo prazo, estas terapias perdem eficácia com reincidência dos distúrbios de movimento e psicoses. Com o
4
propósito de melhorar a terapia a longo prazo, foram desenvolvidos vários fármacos não-dopaminérgicas, que se verificou serem eficazes na redução das discenisias quando aplicadas em modelos animais da DP, são actualmente discutidas como potenciais drogas terapêuticas (Bibbiani et al. 2003).
Outra característica patológica da DP são os neuritos distróficos, designados “neuritos de Lewy” (LNs) e as inclusões intraneurais eosinófilas citoplasmática, conhecidas por corpos de Lewy (CLs) (L. S. Forno 1996; FEARNLEY & LEES 1991) (Figura 1). Os CL encontram-se localizados a nível das células nervosas na substancia nigra e de outras regiões do sistema nervoso e estão presentes nos vários estágios da doença.
Apesar de, à semelhança com os agregados de proteínas, os CL serem tóxicos para as células neuronais uma vez que sequestram proteínas relevantes para a sobrevivência celular, pensa-se que poderão ter um efeito neuroprotector pois capturam agregados proteicos prejudiciais à célula (F. J. S. Lee & F. Liu 2008) impedindo a sua toxicidade. Desta forma o diagnóstico patológico da DP requer a presença de corpos de Lewy, apesar de continuar a não ser evidente a participação destes na patologia, e a perda de neurónios dopaminérgicos na substancia nigra (Thomas & Beal 2007; Paul S. Fishman & G. a Oyler 2002).
Figura 1 – Os Corpos de Lewy do tronco cerebral e do proencéfalo basal apresentam um diâmetro superior a 15µm e são constituídos por um centro hialinico esférico e um halo distinto. Os CL corticais, por sua vez, possuem dimensões mais reduzidas, não apresentam um core disto sendo facilmente observados com coloração anti-ubiquitina. A imagem A mostra um CL no citoplasma de um neurónio dopaminérgico pigmentado na substancia nigra (hematoxylin-eosin and Luxol fast blue, X100). A imagem B mostra um exame ultra-estrutural de um Corpo de Lewy onde é visível a acumulação de filamentos e material granular com um centro denso e filamentos periféricos soltos (X21,560). Adaptado de Lang e A M Lozano (Lang & A M Lozano 1998).
5
Os mecanismos moleculares subjacentes à formação de DP ainda não são totalmente conhecidos. Existem factores genéticos associados ao desenvolvimento da patologia, uma vez que indivíduos com mutações pontuais em genes relevantes para a DP, desenvolvem parkinsonismo juvenil (referencia). No entanto, estes factores parecem apenas ter influência na aceleração do processo patológico, visto que indivíduos que possuem mutações desenvolvem a doença num período significativamente mais curto. Por outro lado, estudos epidemiológicos têm demonstrado que vários factores aumentam o risco de desenvolver DP (C. M. Tanner & Langston 1990). Numerosas toxinas exógenas têm sido associadas com o desenvolvimento de parkinsonismo, no entanto nenhuma delas foi encontrada no cérebro de doentes com DP. Desta forma, esta patologia é definida como multifactorial, resultando da acção de factores ambientais sobre indivíduos geneticamente predispostos e idosos (Riess & R Krüger 1999). Mas, a relação entre factores genéticos e ambientais não se encontra estabelecida, para além de que a maioria dos modelos de DP apresenta uma abordagem redutora do problema, focando apenas determinado gene ou toxina. Desta forma, um dos grandes desafios da biologia pós-genómica é relacionar os mecanismos moleculares modificados pelos alelos associados à doença e os factores ambientais susceptíveis de provocar DP (Dauer et al. 2002).
Apesar da progressão lenta da DP, a ausência de tratamento tende a que a sintomatologia característica da DP evolua para um estado acinético-rígido. Em alguns casos, o quadro clínico evolui para demência podendo na sequência de complicações relacionadas com a imobilidade levar à morte do doente (Lang & A M Lozano 1998).
Presentemente, a diminuição de dopamina no corpo estriado, característica desta patologia, poder quantificada por técnicas de imagiologia in vivo (Eckert et al. 2007), contudo não existe qualquer tipo de marcador biológico que confirme inequivocamente a DP, pelo que recorre-se vulgarmente a exames neuropatológicos (L. S. Forno 1996; FEARNLEY & LEES 1991; F. J. S. Lee & F. Liu 2008). Por outro lado, algumas das características clínicas presentes na DP podem ser facilmente encontradas noutras doenças, sendo obrigatório observar um conjunto de sintomas típicos para que seja possível confirmar o diagnóstico. Por outro lado, patologias como o parkinsonismo secundário e o parkinsonismo atípico podem dificultar o diagnóstico de DP pois apresentam quadros clínicos semelhantes, exigindo por isso despiste (Lang & A M Lozano 1998).
O parkinsonismo secundário pode ter origem medicamentosa, sendo potencialmente reversível, ou pode estar relacionado com doenças como encefalites, meningites, tumores e complicações vasculares, enquanto o parkinsonismo atípico é incapacitante e o processo de degeneração não se limita à substantia nigra, afectando outras regiões do cérebro e evoluindo para desordens como a Demência com Corpos de Lewy e a Atrofia Multi-Sistémica (Lang & A M Lozano 1998).
6
A investigação das formas de parkinsonismo hereditário levou a revelações sobre os mecanismos moleculares da morte dos neurónios dopaminérgicos subjacente à DP idiopática. Após uma prenunciada perda neuronal, característica de estágios intermédios da doença, assiste-se à neurodegeneração generalizada do sistema nervoso central (SNC) (H. Braak et al. 2003). Independentemente das diferentes manifestações da DP, todas convergem para a formação de CL e posterior morte dos neurónios dopaminérgicos da SNpc (Sulzer 2007). Estudos genéticos e moleculares da DP hereditária permitirão a uma melhor compreensão da relação entre os corpos de Lewy e perda de células neuronais dopaminérgicas (Paul S. Fishman & G. a Oyler 2002).
É ainda de realçar que as características clínicas dos doentes com DP incluem tanto as incapacidades motoras, anteriormente referidas, como sintomas autonómicos, cognitivos e psiquiátricos (Thomas & Beal 2007). Especificamente, dentro dos sintomas autónomos surge ainda a seborreia frontal, a sialorreia, a hiperhidrose e a retenção urinária.
2.1. Incidência
A DP é uma patologia neurodegenerativa esporádica e idiopática descrita como complexa e multifactorial que presentemente não tem cura, apesar de ser possível atenuar os seus sintomas, nomeadamente através da terapia com levodopa (Paul S. Fishman & G. a Oyler 2002).
A incidência da DP encontra-se sobretudo correlacionada com a idade e sexo. Verifica-se um aumento da prevalência em idades compreendidas entre 44 e 85 anos (Fahn & Sulzer 2004) em que a afectação varia de 1 a 2% da população com idade superior a 65 anos (de Rijk et al. 1997) aumentando para 4%-5% por volta dos 85 anos (L. S. Forno 1996; Kachergus et al. 2005). Relativamente à incidência nos diferentes sexos, vários estudos apontam para uma prevalência superior nos homens na razão de uma vez e meia face às mulheres, sendo esta tendência geralmente explicada pelo efeito neuroprotector do estrogénio e dos factores genéticos de risco associados ao cromossoma X, entre outros (Wooten 2004).
2.2. Etiologia
Os resultados provenientes da etiologia da DP ainda estão em discussão. Presentemente, vários estudos sugerem que a degeneração neuronal associada à patologia encontra-se estritamente relacionada com o stress oxidativo, disfunções mitocondriais, uma anormal degradação de proteínas, a acção de excitotoxinas, o deficiente suporte neurotrófico e com mecanismos de imunidade associados à inflamação (Muqit et al. 2004; Lang & A M Lozano 1998; D. J. Moore, Andrew B West, et al. 2005; Abou-Sleiman et al. 2006; M. T. Lin & Beal 2006; Wersinger & Sidhu 2006). A ênfase na etiologia idiopática tenderá a diminuir com o tempo, enquanto os mecanismos genéticos e ambientais da morte neuronal na DP são definidos.
7
2.3. Mecanismos de Morte Celular
A DP é caracterizada por uma degeneração progressiva de neurónios dopaminérgicos da SNpc. No entanto, o mecanismo de morte celular não está bem compreendido. Diferentes formas de morte celular devem contribuir para a perda de células dopaminérgicas: apoptose, necrose ou degeneração autofágica, tendo sido demonstrado que neurónios da SNpc de doentes com DP apresentam características de apoptose e degeneração autofágica (Anglade et al. 1997; William G Tatton et al. 2003). No entanto, são necessários mais estudos para elucidar as cascatas intracelulares que conduzem à morte celular nesta doença neurodegenerativa crónica e progressiva.
Recentemente, tem sido dada especial atenção a vários mecanismos conceptualmente distintos, com participação activa na indução da morte celular, a disfunção mitocondrial, o stress oxidativo e a excitotoxicidade. É possível que todos eles interajam e se amplifiquem (Dunnett & Bjorklund 1999), conduzindo a uma disfunção neuronal e atrofia que culmina na morte celular.
2.3.1. Disfunção mitocondrial
Diversos estudos equacionam a hipótese de a disfunção mitocondrial estar na origem ou ser um factor importante no desenvolvimento de DP. Um defeito no complexo I, com diminuição da actividade do mesmo, poderá estar na origem da diminuição na síntese de ATP. Na verdade, mutações no complexo I estão directamente relacionadas com a agregação da α-sinucleína, uma proteína envolvida na DP, que contribui para a diminuição do número de neurónios dopaminérgicos (Ted M Dawson & Valina L Dawson 2003).
A toxicidade do MPP+, o metabolito neurotóxico derivado do 1-metil-1-4-fenil-1,2,3,6-tetrahidropiridina (MPTP), inibe a formação de NADH, o que resulta na inibição do complexo I do mitocôndrio, levando a uma insuficiência energética da célula (Nicklas et al. 1987). Além disso, a toxina rotenona também actua ao nível do mitocôndrio, inibindo selectivamente o complexo I (P Jenner 2001; J T Greenamyre et al. 1999) e a cadeia transportadora de electrões (E. R. D. L. T. Gil 2009). No entanto, parece que a disfunção mitocondrial não está exclusivamente relacionada com os neurónios dopaminérgicos da SNpc, visto ser também observada a nível do striatum e outros tecidos (W D Parker et al. 1989; Cardellach et al. 1993; W Davis Parker & Swerdlow 1998).
A Dieldrina e Maneb são dois pesticidas que inibem de forma selectiva o complexo III e a cadeia transportadora de electrões. Estudos comprovam que a concentração de Dieldrina em cérebros de doentes com DP é maior que os respectivos controlos e que a exposição crónica ao Maneb provoca o síndrome parkinsónico crónico (E. R. D. L. T. Gil 2009).
8
2.3.2. Stress oxidativo
Diversas observações têm suportado a hipótese do stress oxidativo, através da peroxidação lipídica, contribuir para a DP (T. Yoshikawa 1993). O cérebro depende fundamentalmente da energia produzida nos mitocôndrios, associado à produção de espécies reactivas de oxigénio (ROS). Existem várias fontes potenciais indutoras de stress oxidativo, incluindo a disfunção mitocondrial, níveis de ferro livre aumentados e mecanismos de defesa contra radicais livres danificados (Foley & Riederer 2000). Sob circunstâncias específicas, o stress oxidativo, poderá desenvolver-se ao nível da
substantia nigra, provocando danos nas células e conduzindo à morte celular (Olanow & W G Tatton
1999). O herbicida Paraquat é um dos exemplos que incrementam o risco de DP actuando através da produção de ROS.
2.3.3. Excitotoxicidade
Os danos causados pelo excesso de glutamato, resultantes de uma alteração da permeabilidade das células ao cálcio, através dos receptores N-metil-D-aspartato (NMDAr), são considerados como fazendo parte da neurodegeneração. Pensa-se que a excitotoxicidade induzida pelo glutamato representa um importante papel na isquémia (Rothman & Olney 1986; Ikonomidou & Turski 1996) e epilepsia (Olney et al. 1986) e deverá ser um importante factor na DP. A activação massiva de receptores de glutamato poderá resultar em aumentos excessivos da pool de Ca2+, que em última instância provoca morte neuronal (Mody & MacDonald 1995). A morte celular induzida pelo excesso de glutamato ocorre por dois mecanismos fundamentais: formação excessiva de óxido nítrico (NO)(Peter Jenner 2003) e disfunção neuronal (Schinder et al. 1996). A suportar a importância deste mecanismo de excitotoxicidade na DP existem dados que demonstram que antagonistas do NMDA protegem os neurónios dopaminérgicos da morte celular, induzida pelo tratamento de ratos e primatas (Turski et al. 1991; J T Greenamyre et al. 1994) com MPTP.
2.4. Parkinson idiopática e hereditária
As vias metabólicas subjacentes ao quadro patológico da DP não são bem conhecidas, no entanto, sabe-se que podem resultar da combinação de factores ambientais e genéticos. Os casos idiopáticos e hereditários de DP apresentam características semelhantes, mas os casos hereditários surgem em idades precoces (<40 anos) e encontram-se associados a características clínicas atípicas, como é o caso do Parkinsonismo Juvenil Autossómico Recessivo (PJ-AR) (Yamamura et al. 1993). Estudos epidemiológicos revelam que menos de 10% dos doentes com DP apresentam uma estrita etiologia hereditária (DP mendelianas), apresentando DP autossómica recessiva (DP-AR) ou dominante (DP-AD), no entanto, na maioria dos doentes os casos são idiopáticos (Thomas & Beal 2007).
9
Apesar da raridade dos casos de DP hereditária, a descoberta de cada vez mais genes envolvidos nesta patologia tem vindo a confirmar a importância e o papel dos factores genéticos no desenvolvimento da doença, fornecendo pistas essenciais na compreensão da patogénese molecular da DP idiopática (Thomas Gasser 1998; Thomas & Beal 2007). Apesar da relação mencionada ser pouco clara, o facto de partilharem das mesmas características patofisiológicas sugere o envolvimento de mecanismos patogénicos comuns (John Hardy 2003).
10
3. Parkinson Juvenil Autossómico Recessivo (PJ-AR)
O PJ-AR é uma doença neurodegenerativa, inserida na DP hereditária, que surge antes dos 40 anos manifestando-se de forma lenta e progressiva (H Takahashi et al. 1994). Esta doença é sintomaticamente diferente da DP idiopática, no entanto, apresenta características clínicas semelhantes. Os doentes com PJ-AR apresentam um leve parkinsonismo com resposta ao tratamento com levodopa, sendo os principais sintomas a instabilidade da postura, distonia do pé, hiperreflexia, óbvias flutuações diurnas dos sintomas parkinsónicos e uma melhoria dos sintomas após um sono ocasional que pode durar algumas horas (Ishikawa & Tsuji 1996).
A condição autossómica recessiva, sendo encontrada em doentes cujos progenitores partilham consanguinidade e é observada numa única geração. A média de idades de início da doença ronda os 25 anos (24.6 ±3.2 [mean ± SEM, range; 8 a 38, n=9]) nos homens e 23 anos (22.5 ±2.7 [mean ± SEM, range; 8 a 43, n=17]) nas mulheres (M. Saito et al. 1998) sendo que a manifestação clínica ocorre por volta dos 40 anos (E. R. D. L. T. Gil 2009).
Esta condição encontra-se descrita predominantemente na população Japonesa, podendo ter diferentes denominações, das quais se destaca o parkinsonismo autossómico recessivo de inicio precoce com flutuações diurnas, forma hereditária de parkinsonismo e parkinsonismo juvenil (Yamamura et al. 1973; Takubo et al. 1996).
Alterações no gene PARK2 (MIM 602544) foram identificadas como preponderantes para o desenvolvimento do PJ-AR (MIM 600116), estando descritas mais de cem alterações onde as mais graves levam ao consequente misfold e perda de função da proteína codificada (Shimura et al. 2000).
O estudo do gene indutor do PJ-AR, permitirá elucidar os mecanismos moleculares subjacentes à degeneração selectiva dos neurónios dopaminérgicos da SNpc no PJ-AR e impulsionará a compreensão dos mecanismos de degeneração neuronal na DP (M. Saito et al. 1998).
A nível histológico, os doentes apresentam uma perda neuronal óbvia, com uma marcada despigmentação da SNpc, resultante da diminuição dos neurónios que contêm neuromelanina nesta zona. Para além desta perda neuronal, é observada a deposição extraneuronal de melanina e gliose na zona central e ventrolateral da SNpc (van de Warrenburg et al. 2001). A zona cerebral do cerúleo, que possui a maior densidade de neurónios dopaminérgicos, apresenta também um acentuado decréscimo neuronal e uma diminuição do conteúdo em neuromelanina.
Apesar de estas características também estarem presentes em doentes com DP, no caso dos doentes com PJ-AR (e contrariamente à DP) não é observada a presença de CLs no conteúdo celular dos neurónios da substância nigra (SN) e cerúleo (SAITO 2000; H Takahashi et al. 1994) . A ausência de CLs nesta doença sugere que a função da Parkina seja crucial na formação dos corpos de inclusão (Sasaki et al. 2004). É no entanto importante referir a existência de uma excepção descrita na literatura (M Farrer et al. 2001).
11
A relação entre a PJ-AR e a DP idiopática foi inicialmente questionada porque os doentes de PJ-AR são atípicos quando comparados com os doentes de DP idiopática, nomeadamente na sintomatologia e na idade do surgimento dos sintomas clínicos (21,9 vs 58 anos de idade, respectivamente) (H. Mori et al. 1998; H Takahashi et al. 1994). A distonia e as flutuações diurnas sintomáticas são frequentemente observadas em doentes com DP precoce, mesmo sem historial hereditário (H. Mori et al. 1998). Algumas das características atípicas da PJ-AR podem estar relacionadas com a idade do surgimento da patologia em vez de uma PJ-AR particular.
Mutações no gene da Parkina foram estabelecidas como base na genética da PJ-AR assim como com os casos mais típicos de DP, e a PJ-AR cumpre uma definição comummente utilizada da DP hereditária (Paul S. Fishman & G. a Oyler 2002). Estudos iniciais em cérebros post-mortem com PJ-AR contribuem para a polémica sobre a relação entre a PJ-AR e a DP idiopática. A análise de um número restrito de cérebros com PJ-AR mostra duas características comuns: 1) a perda de neurónios dopaminérgicos na substantia nigra e 2) o tronco cerebral gliose não apresentar corpos de Lewy (H. Mori et al. 1998; H Takahashi et al. 1994). Autópsias de doentes de PJ-AR com mutações na Parkina também revelam ausência de corpos de Lewy, com uma excepção (Portman et al. 2001; Hayashi et al. 2000; M Farrer et al. 2001). Embora a ausência de corpos de Lewy na PJ-AR exclua esse distúrbio patológico de cumprir os critérios estabelecidos para o diagnóstico da DP, a PJ-AR e o gene da Parkina subjacentes são susceptíveis de ter um relacionamento próximo com DP idiopática (Paul S. Fishman & G. a Oyler 2002).
A ausência de Corpos de Lewy na PJ-AR impulsionou o interesse na relação da PJ-AR heredit|ria e a DP causada por mutações na sinucleína. Quando as famílias com mutações de α-sinucleína foram descritas, a relevância das formas hereditárias de parkinsonismo esporádico com resposta à levodopa tornou-se evidente (Polymeropoulos et al. 1997). A descoberta de que a α-sinucleína era um dos principais componentes de Corpos de Lewy estabeleceu uma característica comum entre parkinsonismo hereditário responsivo à levodopa e à DP idiopática (Spillantini et al. 1997). Esta constatação reforçou a ideia de que formas hereditárias de parkinsonismo responsivo à levodopa deveriam ser definidas como DP hereditária, e ilustram a importância de identificar genes para a DP hereditária, mesmo entre grupos restritos de doentes que apresentam características atípicas.
Alguns estudos revelam ainda que ocasionalmente são observadas inclusões basofílicas, diferentes dos CLs, com uma forma esférica que apresentam na sua constituição α-sinucleína e ubiquitina (Sasaki et al. 2004), sendo de evidenciar que, a variedade de aberrações que se verificam na Parkina poderá provocar diferenças patológicas tanto em grau como na distribuição da neurodegeneração, incluindo a presença ou ausência de CLs. Esta hipótese vem reforçar uma possível ligação entre parkinsonismo induzido pela Parkina e a DP idiopática (Sasaki et al. 2004).
12
Estudos de imagiologia de doentes de PJ-AR mostram uma perda da função no transporte de dopamina semelhante à DP típica, apoiando a relevância da PJ-AR para a DP típica, determinando a perda da função dos neurónios dopaminérgicos (Broussolle et al. 2000; Hilker et al. 2001).
13
4. O locus PARK2: Estrutura e função
Desde a identificação do locus PARK2, o conhecimento genotípico e fenotípico das mutações genéticas da Parkina tem expandido (Abbas et al. 1999; C B Lücking et al. 2001). O gene Park2 encontra-se entre os maiores genes humanos, com um comprimento de 1.380.245pb, possui 12 exões e várias formas de splicing alternativo (E. R. D. L. T. Gil 2009). Encontra-se localizado no cromossoma 6q25.2-q27 (Matsumine et al. 1997) e foi identificado por clonagem posicional em 1998 (T Kitada et al. 1998). O seu elevado tamanho deve-se aos extensos intrões que resultam numa open
reading frame de apenas 1395pb que codifica para uma proteína de 465 aminoácidos com um peso
molecular aparente de 52kDa (T Kitada et al. 1998). Ao estudar este gene verificou-se codificar uma proteína de origem anteriormente desconhecida denominada de Parkina (T Kitada et al. 1998), uma proteína evolutivamente conservada que está presente, com elevado grau de homologia, em
Caenorhabditis elegans, Drosophila melanogaster, Mus musculus, Rattus rattus e em outras espécies
(Culetto & Sattelle 2000; Tohru Kitada et al. 2000; Horowitz et al. 1999; Y.-J. Bae et al. 2003).
O promotor do gene da Parkina é um promotor bidireccional, regulando a transcrição da Parkina e do gene upstream antissense (Andrew B. West et al. 2003) que possui 5 exões e um comprimento total de 0.6Mb. O gene upstream antissense constituí o gene co-regulador da Parkina (PACRG), no entanto, a sua relevância fisiológica ou patofisiológica é desconhecida.
As mutações no gene da Parkina foram as primeiras a serem associadas ao PJ-AR hereditário (T Kitada et al. 1998). Ao contr|rio da α-sinucleína, uma proteína envolvida na DP, as mutações na Parkina parecem ser relativamente comuns na DP hereditária e são responsáveis por muitos casos de incidência precoce da DP (C B Lücking et al. 2000; Paul S. Fishman & G. a Oyler 2002) uma vez que cerca de 50% dos doentes com DP precoce apresenta mutações na Parkina (C B Lücking et al. 2000).
As primeiras mutações no locus PARK2 que levaram à identificação do gene correspondem a grandes delecções homozigóticas, de um e cinco exões respectivamente (Tohru Kitada et al. 2000). Desde então, uma variedade de mutações foi identificada, entre elas, delecções de um ou vários exões, duplicações ou triplicações de exões, mutações em frame-shift, mutações pontuais que podem ser subdivididas em mutações missense (substituição de um resíduo de aminoácido por outro), non-sense (resultando num codão stop) e splice-site (intrónicas) (A. West et al. 2002; C B Lücking et al. 2000; Abbas et al. 1999; E. R. D. L. T. Gil 2009).
Delecções homozigóticas na Parkina, semelhantes às do Japão, foram encontradas em linhagens da Europa, juntamente com a identificação de linhagens contendo delecções adicionais (exão 8-9, na Argélia) e mutações pontuais homozigóticas (Paisán-Ruíz et al. 2004; Zimprich et al. 2004; Thomas & Beal 2007). Na Europa foram identificadas 35 famílias com DP de início precoce, possuindo um padrão autossómico recessivo com mutações na Parkina. A partir dessas linhagens,
14
outras nove famílias portadoras de novas mutações, sobretudo mutações pontuais, foram descobertas com idade de início tardio (38 ± 12 anos) manifestando um fenótipo mais parecido com a DP idiopática (Abbas et al. 1999).
Juntamente com os doentes que apresentavam mutações heterozigóticas na Parkina e a combinação de mutações pontuais ou delecções com uma mutação pontual, foi identificada uma família com uma herança pseuso-dominante onde a mutação no segundo alelo não foi identificada, mesmo após o screening de todos os exões da Parkina, sugerindo que mutações no promotor ou nos intrões da Parkina também poderão originar a doença (C Klein et al. 2000). Fica, no entanto, por determinar se esses doentes expressam Parkina funcional (Paul S. Fishman & G. a Oyler 2002).
Quando o interesse se centrou sobre os casos com início precoce, sem histórico hereditário conhecido, as mutações homozigóticas da Parkina também foram detectadas. No Japão, 2% dos doentes de DP aparentemente idiopática, com incidência precoce (6,3% abaixo de 50 anos de idade), são portadores de mutações na Parkina. Apesar da vasta gama de sintomas que foram observados nestes doentes, eles tendem a ser jovens e a ter um quadro clínico atípico descrito para PJ-AR, apresentando distonia, apresentação simétrica e excelente resposta inicial a levodopa, mas com aparecimento precoce de discinesia.
As mutações na Parkina associadas à PJ-AR ocorrem tanto na forma homozigótica como heterozigótica apresentando os alelos com diferentes mutações. Por outro lado, sendo o PJ-AR uma condição autossómica recessiva, é espectável que ambos os alelos se encontrem afectados por uma mutação que interfira com a expressão ou função da proteína. No entanto, foram reportados alguns casos onde apenas um dos alelos surge mutado, mesmo após um intenso screening (M Farrer et al. 2001; A. West et al. 2002), sendo estes casos explicados por um modelo de haploinsuficiência (M Farrer et al. 2001) ou pelo facto da Parkina poder ser inibida pós-traducionalmente. Um exemplo da inibição pós-traducional observa-se com a S-nitrosilação in vivo da Parkina, que leva à inibição da actividade E2 ligase removendo a sua acção protectora (K. K. K. Chung et al. 2004). Assim sendo, uma mutação heterozigótica associada ao stress nitrosativo pode promover a manifestação da haploinsuficiência, reforçando a observação das mutações heterozigóticas associadas à doença. Um ganho de função tóxica ou um efeito negativo dominante transportando algumas mutações
missense é também uma hipótese plausível. No entanto, descobertas de que proteínas truncadas não
são expressas a níveis detectáveis em cérebros de doentes com delecções dos exões 3 ou 4 (Shimura et al. 1999; Shimura et al. 2001) reforça o conceito da ocorrência de mutações que levam à perda de função como sendo o mecanismo patológico predominante no PJ-AR.
Globalmente, as formas monogénicas de DP são raras entre os genes identificados até à data, sendo que as mutações na Parkina parecem ser a causa mais comum de PJ-AR. Num estudo europeu, com doentes com idade de surgimento da doença até aos 45 anos de idade ou indivíduos com um irmão afectado pela mesma condição, foram identificadas mutações na Parkina em cerca de 50% dos casos hereditários e em 18% de casos idiopáticos de DP (C B Lücking et al. 2000). Em doentes sem historial hereditário, foram observadas mutações na Parkina em 44% dos doentes que tinham
15
idade de surgimento inferior aos 30 anos e apenas em 3% dos que tinham idade de surgimento superior aos 30 anos de idade (Martin Kann et al. 2002). Outros relatórios suportam esta ordem de magnitude, sendo as variações descritas atribuídas aos diferentes critérios usados.
Em geral os irmãos gémeos heterozigóticos de indivíduos com PJ-AR não são afectados, no entanto, alguns desses irmãos têm resultados parcialmente discrepantes na tomografia de fluorodopa por emissão de positrões (PET), sugerindo que indivíduos com um gene Parkina anómalo podem ter mudanças pré-clínicas na DP (Broussolle et al. 2000). Note-se que alguns doentes têm apenas uma mutação na Parkina identificada sendo provável que os doentes portadores de uma mutação no locus da Parkina não tenham sido identificados por razões técnicas ou porque a mesma se encontra numa região não-exónica, como o promotor de Parkina. No entanto, é possível que alguns desses doentes tenham de facto a doença associada a apenas um alelo de Parkina mutante (C Klein et al. 2000; J. Satoh & Y Kuroda 1999; Paul S. Fishman & G. a Oyler 2002).
Ao longo de vários anos, treze loci foram identificados e relacionados com os casos DP hereditária, apresentando um traço mendeliano autossómico dominante ou recessivo.
Existem, no entanto, seis causas genéticas claramente definidas (figura 2) associadas aos genes da α-sinucleína (PARK1) (Polymeropoulos et al. 1997), Parkina (PARK2) (T Kitada et al. 1998), UCHL-1 (PARK5)(Leroy et al. 1998), PINK1 (PARK6)(Valente et al. 2004), DJ-1 (PARK7) (Vincenzo Bonifati et al. 2003) e LRRK2/dardarina (PARK8) (Zimprich et al. 2004; Paisán-Ruíz et al. 2004; Thomas & Beal 2007).
Locus Cromossoma Gene Hereditariedade Função provável
PARK1/4-SNCA 4q21.3 α-sinucleína AD Proteína pre-sináptica/ Chaperone
PARK2 6q25.2-27 Parkina AR Ligase E3 ubiquitina
PARK5 4p14 UCH-L1 AD Hidrolase c-terminal ubiquitina
PARK6 1p35-36 PINK-1 AR Cinase
PARK7 1p36 DJ-1 AR Chaperone
PARK8 12p11.2q-13.1 LRRK2 AD Cinase
Figura 2 - Loci envolvidos na genética da DP, localização cromossómica, gene, hereditariedade e função provável adaptado de Gil (Gil 2009).
O fenótipo associado à Parkina não é consistente o suficiente para permitir a identificação dos doentes caso a caso. Uma grande variedade de genótipos resultantes de mutações da Parkina parece causar fenótipos semelhantes na população europeia, com 19 diferentes rearranjos exónicos (e uma delecção intrónica) e 16 mutações pontuais exónicas diferentes identificadas nessas famílias, apesar dos sintomas semelhantes (C B Lücking et al. 2000; M Periquet et al. 2001). O rastreio dos vários tipos de mutações associadas a DP, em diversos grupos hereditários, sugere uma origem comum para as mutações pontuais. Já os rearranjos exónicos mais prováveis ocorrerão de forma independente nas diferentes famílias (M Periquet et al. 2001).
16
5. Factores genéticos envolvidos na patogénese hereditária
5.1. PARK1 e 4-SNCA (α-sinucleína)
Os genes PARK1 e 4-SNCA codificam para a α-sinucleína, sendo o principal constituinte dos CLs devido ao elevado potencial para a agregação, formando agregados insolúveis e tóxicos para a célula, induzindo stress proteolítico, neurodegeneração (Thomas & Beal 2007) e pensa-se que poderá estar envolvida na patogénese da DP e da Demência com CLs. Espécies mutadas incrementam a propensão para agregar em diferentes graus, sendo que, diferentes tipos de mutações nos genes produzem manifestações diferentes da doença (Polymeropoulos et al. 1997).
Os genes que codificam para a α-sinucleína foram a primeira causa genética identificada associada à DP hereditária, estando esta relacionada com um fenótipo autossómico dominante. Contudo, foi a identificação de todos estes genes e da relação entre as suas mutações e a forma hereditária de DP que veio fornecer uma ajuda considerável na compreensão da patogénese da DP (Portman et al. 2001).
A identificação da α-sinucleína como o principal componente dos Corpos de Lewy, tanto na forma da DP hereditária como na idiopática, guiou a investigação para o estudo do papel da α-sinucleína na patogénese da DP. Uma tendência semelhante de generalizar as conclusões para a função da Parkina na PJ-AR à DP idiopática também está a ocorrer (Paul S. Fishman & G. a Oyler 2002).
5.2. PARK2 (Parkina)
Mutações no gene da Parkina (PARK2) surgem com elevada frequência, sendo observadas em cerca de 50% dos casos de DP hereditária (Betarbet et al. 2005). Quando o gene se encontra mutado manifesta-se um fenótipo autossómico recessivo juvenil da DP. Uma vez que o tema do presente trabalho recai especificamente sobre este gene e proteína, ao longo deste capítulo, mais informação será apresentada relativamente a este assunto.
5.3. PARK5 (UCH-L1)
O UCH-L1 encontra-se associado a várias doenças neurodegenerativas, entre elas a DP hereditária e a DA (Betarbet et al. 2005). Este gene codifica para a proteína homónima que pertence ao grupo de enzimas desubiquitinadores responsáveis pela hidrólise de cadeias de poliubiquitina em monómeros de ubiquitina (Pickart 2000) encontrando-se exclusivamente em neurónios. Quando apresenta mutações a sua actividade sofre uma redução em cerca de 50% levando à diminuição da ubiquitinação e consequente deposição de proteínas anormais nos neurónios (Pickart 2000).