Ciclo Celular e novas terapias contra o cancro
(o ano do Nobel)
Introdução
A capacidade de uma célula se dividir em duas é uma propriedade fundamental dos organismos vivos, e por isso o mecanismo pelo qual as células se dividem desde sempre fascinou os biólogos. A formação de uma nova célula tem sempre origem numa célula já existente, através de um processo denominado divisão celular, sendo o ciclo celular definido como o intervalo entre duas divisões celulares sucessivas.
É interessante verificar que a duração do ciclo celular varia enormemente entre diferentes organismos, entre células em diferentes fases do desenvolvimento desses organismos, e entre diferentes tipos de células no mesmo organismo (enquanto que alguns tipos de células se dividem num espaço de horas, outros necessitam de meses). Ainda dentro de um mesmo organismo multicelular adulto as células podem dividir-se em três classes, relativamente à sua capacidade de divisão: As que possuem um elevado grau de especialização e que perderam a capacidade de divisão, como por exemplo as do sistema nervoso, ou glóbulos vermelhos; as que normalmente não se dividem, mas que podem ser induzidas a fazê-lo quando estimuladas (eg células hepáticas e linfócitos); finalmente, as células que possuem uma actividade mitótica elevada (eg células das gónadas ou epiteliais). No entanto, e apesar destas diferenças, observa-se que quando crescidas em cultura, muitas células de diferentes animais
atravessam um ciclo completo em cerca de 20 horas, o que indica que, num organismo multicelular, a taxa de proliferação de cada tipo celular é regulada por factores extrínsecos à célula, não dependendo apenas de características celulares intrínsecas. Como se estima que, num corpo humano adulto, em cada segundo se encontrem em divisão mais de 25 milhões de células, tem-se noção que a coordenação da divisão celular é um aspecto essencial ao organismo.
Para além do interesse científico inerente ao estudo dos mecanismos envolvidos, o estudo do ciclo celular tem implicações práticas enormes no campo da saúde humana, nomeadamente no que respeita ao combate ao cancro, uma vez que esta doença resulta, essencialmente, do facto de a célula perder o controlo da sua própria divisão. As células tumorais dividem-se persistente-mente em situações em que tal não deveria acontecer, uma vez que perderam os controlos moleculares que as fazem pertencer a uma das três classes acima descritas.
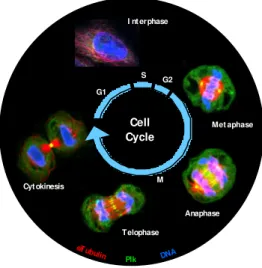
Ciclo celular
O ciclo celular divide-se tradicional-mente em interfase e mitose. A fase de divisão, denominada mitose ou fase M, é separada da divisão seguinte pela interfase. A observação de que a duplicação (replicação) dos cromossomas ocorre durante um período específico da interfase originou a sua divisão em três partes, tendo o período de síntese do DNA
sido denominado fase S, a fase que o antecede G1 e o período entre a fase S e a mitose seguinte, G2.
A mitose é uma fase relativamente curta do ciclo celular sendo a sua duração bastante constante entre diferentes tipos de células (frequentemente cerca de uma hora). O mesmo acontece relativamente às fases S e G2, verificando-se que, em termos de duração, a maior variabilidade ocorre na fase G1. Esta fase corresponde ao período em que a célula cresce e executa as suas funções e verifica-se que, com raras excepções, as células que pararam de se dividir, quer temporária quer definitivamente, seja no organismo seja em cultura, fazem-no num ponto imediatamente anterior à iniciação da síntese de DNA. As células nestas condições dizem-se em G0, distinguindo assim as células que abandonaram o ciclo celular daquelas que se encontram numa fase G1 típica. A re-entrada de células em G0 no ciclo celular, passando a G1, dá-se por resposta a estímulos externos (eg hormonas). A passagem da fase G1 para a fase S depende de um sinal interno que é gerado pela célula, e determina o início da replicação do DNA. Este sinal marca um ponto no ciclo celular, denominado start, que constitui um ponto sem retorno uma vez que a partir do momento em que é desencadeado, o ciclo celular não pode “voltar atrás” - a célula invariavelmente completará a dupli-cação do DNA e entrará em mitose.
Sabemos hoje que os acontecimentos que se relacionam com a duplicação e divisão do material genético, constituem acontecimentos-chave do ciclo celular. Com efeito, o facto de os cromossomas, contrariamente a quase todos os componentes celulares existirem nas células numa única cópia e serem portadores da informação genética faz com que a sua duplicação e separação exactas Joana Perdigão2 e Álvaro Tavares1,2
1 Secção de Biotecnologia, Departamento de Engenharia Química, Instituto Superior Técnico,
Av. Rovisco Pais, 1049-001 Lisboa
pelas duas células-filhas quando uma célula se divide constituam um requisito essencial à viabilidade celular.
O início da compreensão
do celular
A clonagem dos genes que controlam o ciclo celular constituiu um passo-chave para a compreensão da divisão celular. A aplicação da genética, associada à biologia molecular, foi particularmente eficaz no estudo do ciclo celular em leveduras. Nestes organismos, os estudos genéticos iniciais permitiram a identificação de um grande grupo de genes envolvidos no controlo da divisão celular (genes cdc; Hartwell et al. 1970). Mutantes nestes genes foram isolados, caracterizados fisiologica-mente, clonados por complementa-ção, e os genes clonados utilizados como ponto de partida para análises bioquímicas subsequentes. A análise de mutantes em Saccharomyces cerevisiae e em Schizosaccharo-myces pombe permitiu verificar que havia um gene – cdc2 em S. pombe -com uma função central no ciclo celular (Nurse et al. 1976).
Simultaneamente, no início dos anos 70 foi feita uma descoberta importante por embriologistas a trabalhar com oócitos de Xenopus: a transferência de citoplasma de um oócito maduro para um outro imaturo era capaz de provocar a entrada deste último em mitose (Masui e Market, 1971). Ao factor citoplasmático responsável por esta maturação, que mais tarde se demonstrou estar presente em todos os tipos de células mitóticas, chamou-se MPF (Maturation ou M-phase Promoting Factor). Desenvolveu-se assim uma segunda abordagem experimental, in vitro, utilizando-se extractos celulares derivados de oócitos ou ovos de anfíbios e de invertebrados marinhos, para criar sistemas capazes de executar os passos do ciclo celular. A possibilidade de depletar e purificar certas moléculas destes extractos permitiu uma análise
bioquímica de cada um destes passos, da qual resultou a purificação do MPF (Lohka et al. 1988).
Estas duas abordagens experimen-tais, a genética e a bioquímica, complementam-se e o facto de o ciclo celular e o seu controlo serem altamente conservados significou que os estudos puderam ser feitos e comparados numa diversidade de organismos biológicos, cada um com as suas vantagens específicas. Por exemplo, a purificação do factor MPF de anfíbios revelou que consistia num complexo proteíco de duas subunidades, sendo uma delas uma cinase proteíca com elevada homologia com a cdc2 de levedura (Labbe et al. 1988). A clonagem do gene cdc2 humano por complemen-tação do mutante cdc2 em levedura de fissão (Lee e Nurse, 1987) demonstrou que a cinase cdc2 é um regulador universal do ciclo celular.
A segunda subunidade do MPF é uma ciclina, um tipo de proteína entretanto isolada de embriões de ouriço-do-mar, e que é sintetizada e degradada de um modo cíclico, em sincronia com o ciclo celular (Evans et al. 1983). A ciclina identificada como componente do MPF era uma ciclina mitótica (com pico de abundância durante a mitose), denominada ciclina B (Lohka et al. 1988). A convergência destas linhas
independentes de investigação permitiu que, nos finais dos anos 80, o factor MPF fosse identificado como um regulador universal do início da fase M.
CDKs e ciclinas
Sabe-se hoje que a proteína cdc2 pertence a uma família conservada de cinases, conhecidas como Cdks (cyclin-dependent kinases). As Cdks são inactivas como monómeros, necessitando de se ligar a um parceiro ciclina para se tornarem activas. Em todas as células eucariotas a progressão do ciclo celular é controlada pela activação e inactivação sucessivas de diferentes complexos ciclina-CDK (para revisão ver por exemplo Pines, 1999).
Enquanto que em eucariotas inferiores, como as leveduras, há apenas um ou dois genes tipo cdc2, em mamíferos há pelo menos nove CDKs (Cdk1-9) (Johnson e Walker, 1999). Em S. cerevisiae, a activação e especificidade da única Cdk existente (a CDC28) é definida por nove ciclinas diferentes (Levine et al. 1999). Três ciclinas G1 regulam os acontecimentos que ocorrem durante G1 ou até ao ponto em que as células entram na fase S e seis outras ciclinas são requeridas para a iniciação da replicação do DNA durante a fase S e Plk
α αTubuli
n DNA
Met aphase
Anaphase
Telophase G2
Cell Cycle
G1 S
Cyt okinesis
I nt erphase
M
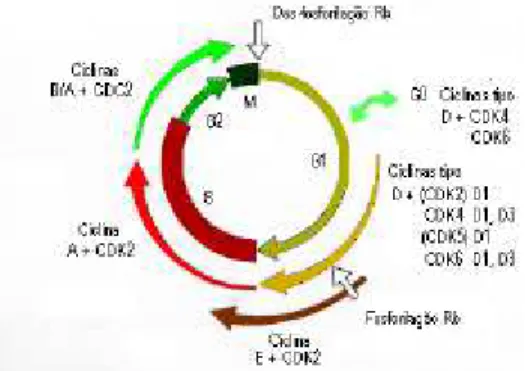
a mitose subsequente (Nasmyth, 1996). Em mamíferos a situação é muito mais complexa pois para além das 9 CDKs já isoladas identificaram-se até ao momento dezasseis ciclinas (denominadas A, B1, B2, C, D1, D2, D3, E, F, G1, G2, H, I, K, T1 e T2) . Todas estas ciclinas têm em comum uma região de homologia, a caixa ciclina, que é na realidade um domínio de ligação às Cdks (Hunt, 1991). As ciclinas são muitas vezes referidas como mitóticas ou da fase S, consoante o seu pico de expressão ocorre durante a fase M ou fase S, respectivamente.
Demonstrou-se posteriormente que os complexos ciclina-CDK que controlam o início da fase S e da mitose são distintos, e que aumentando a actividade destas CDKs se conseguia acelerar essas as fases. A actividade cinásica das CDKs é regulada pela abundância intracelular das subunidades ciclina, por mudanças no seu estado de fosforilação (que é controlado por actividades antagónicas de outras cinases e fosfatases), e pela associação aos inibidores CKI.
A título de exemplo, a transição de G2 para M (transição G2/M) requer a activação de um complexo Cdk/ciclina. A ciclina B é a principal ciclina mitótica em eucariotas superiores e em conjunto com a cdc2 (agora chamada Cdk1) forma o MPF. Contudo, a activação completa do MPF, necessária para o início da mitose, depende de outros níveis de controlo, para além da junção das duas subunidades. Em primeiro lugar, é necessário que ocorra a fosforilação
da cdc2 num resíduo de treonina conservado, o T161, pela cinase activadora CAK (Cdk-activating kinase). Por outro lado, a Cdk1/ciclina B pode ser mantida num estado inactivo por fosforilação dos resíduos T14 e Y15 da CDK1, ambos no local de ligação da cinase ao ATP. Estas fosforilações inibidoras são executadas por duas cinases diferentes chamadas Wee1 e Myt1. A activação é executada por membros da família de fosfatases cdc25 que removem essas fosforilações inibidoras do local de ligação da Cdk ao ATP. Após esta desfosforilação, o complexo Cdk/ciclina B está finalmente completamente activo e a mitose inicia-se.
A proteólise e a transcrição constituem dois outros processos reguladores importantes para a progressão do ciclo. A proteólise controlada tem um papel directo na regulação das CDKs controlando os níveis de ciclinas, e contribuindo também para outros passos do ciclo celular tais como alterações na
coesão das cromátidas-irmãs, que ocorrem quando as cromátidas se separam, na transição metafase/ anafase. As ciclinas são degradadas de um modo selectivo sendo “marcadas” para destruição pela conjugação com moléculas de ubiquitina, e assim reconhecidas e degradadas pelo complexo proteolítico proteossoma. Por exemplo, durante a mitose diferentes ciclinas são ubiquitinadas por acção de um complexo proteico denomi-nado APC. Este complexo, por associação a diferentes proteínas reguladoras, tem especificidade diferente para as várias ciclinas em diferentes alturas da mitose. Deste modo as ciclinas mitóticas não são todas degradadas simultaneamente mas sim em alturas muito específicas. Assim, o aumento e diminuição sucessivas na concentração de cada ciclina são obtidos pela coordenação entre a sua transcrição e a sua degradação pelo proteossoma. Este duplo controlo assegura que a concentração de cada ciclina atinge o nível necessário para activar o seu parceiro catalítico (uma Cdk), na fase correcta do ciclo. A destruição programada das ciclinas é, por outro lado, um passo importante que permite à célula passar à próxima etapa do ciclo. Por esta razão, células mutantes que não conseguem degradar as ciclinas mitóticas ficam paradas em mitose e não reentram em G1.
Figura 2 - Acumulação ao longo do ciclo celular dos diferentes complexos ciclina/Cdk
Pontos de controlo
De modo a executar as diferentes etapas do ciclo numa ordem correcta as células possuem controlos que monitorizam o estado dos acontecimentos ao longo do ciclo celular, tais como a replicação dos cromossomas ou a formação do fuso mitótico, e que determinam se o ciclo deve ou não continuar. Se o sistema detectar problemas despoleta uma resposta que parará temporariamente a progressão do ciclo. A célula poderá então proceder às reparações necessárias em vez de continuar para a nova etapa do ciclo, algo que poderia conduzir à sua morte ou à transformação numa célula cancerosa. Se, no entanto, o sistema de vigilância falha na detecção de anomalias, o ciclo prosseguirá para as etapas seguintes mesmo em presença de erros. É de notar que estes controlos são extraordinariamente sensíveis: A presença de uma única quebra numa das moléculas de DNA ou de um único cromossoma não associado ao fuso mitótico são suficientes para causar uma paragem no ciclo. Por exemplo, quando células normais são sujeitas a tratamentos que danificam o DNA, como radiação ionizante, a sua progressão ao longo do ciclo celular pára enquanto o dano é reparado. A irradiação de uma célula durante a fase G1 do ciclo celular atrasa a progressão até à fase S; de modo semelhante, células irradiadas durante a fase S atrasam a continuação da síntese do DNA, enquanto que células irradiadas durante G2 atrasam a entrada em mitose. Um atraso em G1 impede a replicação de DNA danificado (mutado por exemplo) e uma paragem em G2 evita que a célula segregue cromossomas defeituosos.
O conceito de ponto de controlo (checkpoint) foi uma ajuda valiosa para a compreensão do ciclo celular (Hartwell e Weinert, 1989).
Os pontos de controlo mais estudados são os que monitorizam a existência de danos no DNA e alterações na sua replicação, bloqueando a mitose
quando o DNA se encontra danificado ou a sua replicação está incompleta. Estes pontos de controlo têm mecanismos de vigilância que detectam quer estruturas específicas de DNA quer a presença de complexos proteicos envolvidos na replicação e reparação do DNA, que indicam que alguns desses processos está em curso. Em consequência é activada uma via de transmissão de sinal que ou bloqueia a entrada em mitose, mantendo a Cdc2 numa forma fosforilada inactiva, ou bloqueia a mitose numa fase mais avançada através de outros mecanismos. Existe ainda um outro ponto de controlo, que bloqueia a fase S após danos do DNA, e que em células de mamífero depende da presença do gene supressor de tumores p53 (ver abaixo, secção “Terapias de combate ao cancro”).
O outro ponto de controlo ocorre durante a mitose, parando a progressão da mitose se o fuso mitótico não estiver correctamente formado, ou se a ligação e orientação de todos os cromossomas ao fuso não estiver assegurada. Este checkpoint foi descoberto a partir da observação de que a remoção de um cromossoma do fuso provoca um bloqueio da progressão mitótica, e que certos mutantes em levedura continuam o processo de divisão mesmo na ausência de um fuso funcional (Hoyt et al 1991; Li e Murray, 1991;
Nicklas, 1997). Pensa-se que este checkpoint funciona monitorizando a funcionalidade do fuso e a sua ligação aos cromossomas. Na ausência dessa ligação, a coesão entre as cromátidas-irmãs é mantida e os microtúbulos não encolhem, e como consequência as cromatides não ascendem aos polos do fuso. Este checkpoint assegura que a uma replicação precisa do DNA ao nível molecular se segue uma segregação precisa do DNA replicado ao nível celular.
começarem a diferenciar as células saem normalmente do ciclo celular e param de se dividir, pensando-se que esta descontinuação do ciclo é induzida pela síntese dos inibidores das Cdks. Observou-se que os ratinhos “knockout” que não possuem o gene p27 são maiores que os ratinhos normais, e certos dos seus orgãos, tais como o timo e o baço, têm um número significativamente maior de células que os de um animal normal. Em ratinhos normais, as células destes orgãos específicos sintetizam quantidades relativamente elevadas de p27, e presume-se que a ausência desta proteína nos ratinhos deficientes para a p27 permite que as células continuem a dividir-se para além do seu ponto de diferenciação. Em alguns casos esta proliferação descontrolada tem efeitos nefastos: por exemplo, o gene que codifica a p16, um inibidor de Cdks, encontra-se muitas vezes suprimido em determinados tumores humanos. Tal como seria previsível, ratinhos “knockout” que não possuem o gene do p16 exibem uma predisposição elevada para desenvolverem tumores.
O ciclo celular e o
desenvolvimento de
tumores
Antes dos anos 80 pouco se sabia sobre o modo como as células tumorais adquirem as suas propriedades letais de crescimento e disseminação descontrolados. Não surpreendentemente, sabe-se hoje que muitos dos defeitos moleculares
responsáveis pela transformação das células normais em malignas consistem em mutações em genes codificantes para proteínas que regulam o ciclo celular.
Por exemplo, a ataxia-telangiectasia (AT) é uma doença hereditária recessiva caracterizada por uma variedade de sintomas, incluindo um risco aumentado para certos tipos de cancro. Descobriu-se nos anos 60 – após a morte de diversos indivíduos que estavam a ser sujeitos a terapia por radiação – que os pacientes com AT, ou células destes pacientes, são extremamente sensíveis a radiação ionizante. Sabe-se agora que mutações no gene responsável pela AT (gene ATM) conduzem a problemas no checkpoint que detecta a ocorrência de danos no DNA, permitindo-se assim que células com o DNA danificado continuem o ciclo e que entrem em mitose sem que o DNA seja reparado.
Muitas vezes a alteração do padrão de determinadas proteínas é suficiente para provocar defeitos graves na célula. Por exemplo, o complexo ciclina E/Cdk2 catalisa a transição da fase G1 para a fase S. A ciclina E tem um nível baixo no início de G1, que aumenta até atingir um pico na fase G1 tardia, activando a Cdk2 na transição G1-S, e decaindo outra vez durante as fases S, G2 e M. Este perfil de expressão reflecte a importância do complexo ciclina E – Cdk2 na promoção do início da replicação do genoma durante a fase S. Um controlo rigoroso do nível da ciclina E e da actividade da cinase a ela associada é essencial para o
início, na altura correcta, da síntese do DNA. Descobriu-se pois que uma quantidade insuficiente de ciclina E resulta numa paragem em G1, ao passo que o aumento do nível desta proteína provoca uma entrada prematura na fase S, instabilidade genómica e formação de tumores. Para que o nível da ciclina E diminua, segundo o padrão normal, durante a fase S, é necessária que seja destruída tendo-se descoberto recentemente que uma proteína pertencente à família das proteínas F-box, chamada hCDC4 em humanos, é a responsável pela marcação da ciclina E para destruição. As proteínas F-box são adaptadores que formam pontes entre o substrato (neste caso a ciclina E) e o SCF, uma ligase de ubiquitina que faz parte das vias de degradação pelo proteossoma. A hCdc4 liga a ciclina E ao SCF e este último, em conjunto com a conjugase de ubiquitina (E2), cataliza a adição de cadeias de ubiquitina à ciclina E. A destruição da ciclina E ubiquitinada tem como consequência a inibição do complexo cinásico ciclina E/Cdk2. Certas mutações no hCdc4 inactivam a sua capacidade de reconhecer a ciclina E ou de interagir com o SCF, gerando uma acumulação de actividade CiclinaE/Cdk2 quando esta já não deveria existir. Esta acumulação de ciclina E resulta numa proliferação celular excessiva, tendo-se verificado que mutações no hCDC4 também parecem predispor os indivíduos para cancro da mama ou dos ovários (Strohmaier et al., 2001; Moberg et al., 2001).
integridade do DNA DNA não replicado
centrossomas não separados ?
cinétocoros livres
cromossomas não alinhados
fuso mal posicionado
G2 profase metafase anafase telofase
citocinese G1
A B C D
Terapias de combate ao
cancro
Descobriu-se que algumas proteínas descritas como supressoras de tumores (eg. a p53 e a retinoblastoma (Rb)) são proteínas envolvidas no controlo do ciclo celular, sendo centrais na decisão final da via seguida pela célula – progressão/ paragem do ciclo celular e entrada ou não em apoptose – no caso de ocorrência de problemas. Por exemplo, a proteína p53 é responsável pela paragem em G1 quando o DNA contém lesões, impedindo a ocorrência da replica-ção, atrasando a progressão do ciclo celular até à sua reparação e promovendo a apoptose (suicídio celular) nos casos em que tal não é possível. Mutações no p53, por outro lado, permitem a sobrevivência e reprodução de células contendo lesões no DNA. Desta forma, as células alteradas passam as mutações que contêm à sua descendência, que terá depois a oportunidade de acumular mutações adicionais necessárias para o desenvolvimento de neoplasias. Compreende-se assim facilmente que grande parte dos tumores possuam alterações nesta proteína, uma vez que a sua inactivação é muitas vezes um dos primeiros passos da progressão da célula para um estado maligno.
Relativamente à terapia de tumores contendo mutações nestes genes, pensa-se actualmente que o restabelecimento funcional de um gene supressor de tumor, como o p53 ou o Rb, poderá ser suficiente para induzir a apoptose celular e parar o crescimento tumoral, tal como é sugerido pelo facto de a introdução de genes p53 e Rb normais em células tumorais ser capaz de inibir o seu crescimento (Cai et al., 1993). Por estes motivos, a proteína p53 é um dos alvos terapêuticos actual-mente mais estudados, tentando re-estabelecer a sua actividade nas células tumorais em que se encontra inactiva. De uma forma mais geral, o restabelecimento de vias de checkpoint alteradas para o seu
O 100º prémio Nobel para a Fisiologia ou Medicina reconheceu, em 2001, o trabalho dos investigadores Timothy Hunt, Paul Nurse e Leland Hartwell, que os conduziu à identificação das moléculas-chave que regulam o ciclo celular em todos os organismos eucariotas, incluindo leveduras, plantas e animais.
Leland Hartwell (1939-), actualmente director do Fred Hutchinson Cancer Research Center, Seattle, foi distinguido pelos trabalhos que conduziram à identificação de genes com funções-chave na regulação do ciclo celular e pela definição do conceito e mecanismo de checkpoint celular. Utilizando a levedura
Saccharomyces cerevisae como modelo para a análise genética do ciclo celular identificou, no início dos anos 70, mutações em mais de 100 genes especificamente envolvidos no controlo da divisão celular e a que chamou genes CDC (do inglês Cell division control), provando assim que o controlo do ciclo celular era determinado geneticamente. No seguimento deste trabalho, o gene
CDC28 foi identificado como responsável pelo controlo da progressão do ciclo celular através de G1, e posteriormente denominado “start” quando se estabeleceu que a sua expressão marca um ponto crucial no ciclo celular em que ocorre a integração do estado de proliferação celular com os sinais intra- e extra-celulares. Foi também o responsável pela introdução e definição do conceito de checkpoint
celular, que constituiu um marco fundamental no desenvolvimento dos conhecimentos sobre a regulação do ciclo celular (Hartwell e Weinert, 1989). As experiências que conduziram a este conceito pretenderam estudar o efeito da irradiação de células de levedura sobre a progressão do ciclo celular, tendo demonstrado que as células que contêm danos no DNA, provocados por irradiação, sofrem uma paragem do ciclo celular.
Paul Nurse (1949-), Director-Geral do Imperial Cancer Research Fund, Londres, identificou o gene cdc2 numa outra espécie de levedura – Schizosacharomyces pombe - e demonstrou que a proteína que codifica é responsável pela transição da fase G2 para mitose. O gene cdc2 viria posteriormente a ser identificado como homólogo do gene CDC28 de S. cerevisiae isolado por Hartwell, e como envolvido na regulação de diferentes fases do ciclo celular. O seu homólogo humano, conhecido como CDK1, foi identificado por Nurse em 1987 a partir de ensaios de complementação genética utilizando uma biblioteca de cDNAs humanos para complementar a mutação do cdc2 em S. pombe (Lee e Nurse, 1987). Nurse mostrou ainda que a regulação da actividade cinásica das CDK depende do seu estado de fosforilação.
No início dos anos 80 Timothy (Tim) Hunt (1943-), hoje director do Cell Cycle Control Laboratory Imperial Cancer Research Fund, Londres, com base em estudos da divisão celular em embriões de ouriços-do-mar (Arbacia) descreveu uma nova classe de proteínas cuja abundância varia de forma cíclica ao longo do ciclo celular e que, por esse motivo, denominou ciclinas (Evans et al., 1983). Demonstrou ainda que a oscilação do nível de ciclinas é regulada através da sua degradação periódica em pontos específicos do ciclo celular, mecanismo que tem uma importância fundamental para o controlo do ciclo celular, uma vez que a actividade das CDK depende da sua associação com ciclinas.
Em resumo, estes três investigadores premiados com o Nobel, em 2001, descobriram os produtos e mecanismos moleculares básicos de regulação do ciclo celular. As suas descobertas abriram caminho a uma importante e fascinante área de investigação, que inicialmente utilizou como modelo organismos muito mais simples que o Homem. O facto de se ter vindo a demonstrar que os mecanismos básicos que regulam a progressão do ciclo celular foram extraordinariamente conservados ao longo da evolução permitiu um grande avanço no conhecimento do controlo do ciclo celular nas células humanas. Por outro lado, o facto de os mecanismos responsáveis pelo desenvolvimento de processos tumorais resultarem, em última análise, de alterações a nível do controlo da progressão do ciclo celular, faz com que todos estes conhecimentos adquiram extrema importância a nível de implicações terapêuticas.
funcionamento normal poderá restabelecer a resposta apoptótica das células malignas e assim aumentar a sua sensibilidade a agentes que provocam danos a nível do DNA.
Conceptualmente, a estratégia mais simples para conseguir este objectivo seria substituir o gene mutado pelo seu equivalente normal (terapia génica) sendo esta uma linha actual de investigação intensa. Os métodos que se encontram actualmente mais avançados relativamente à terapia génica para o gene p53 tentam re-introduzir o gene p53 normal nos tecidos tumorais, ou inocular o tumor com um adenovírus citolítico que se replica, selectivamente, em células que não possuem a proteína p53 normal. Esta última é particular-mente atractiva dada a sua especificidade para células tumorais. Especificamente, sabe-se que a proteína viral Eb1 do adenovírus “normal” se liga à p53 celular, inactivando-a, o que permite a replicação viral nas células que possuem p53 normal. Logo, a infecção de células normais com uma forma mutada do adenovírus que não possua a proteína Eb1, originará uma infecção não-produtiva porque a p53 normal é activada em consequência da infecção. Por outro lado, a infecção de células tumorais que têm a proteína p53 mutada com o mesmo adenovírus alterado resultará numa infecção produtiva (o vírus pode replicar-se), que causará a morte das células tumorais (revisto por Beaudry et al., 1996 e Gibbs, 2000).
Uma segunda estratégia consiste no desenvolvimento de pequenas moléculas biológicas capazes de interagir com as formas alteradas da proteína p53 presentes em tumores, e assim re-estabelecer a sua função normal. Isto porque em cerca de 75% dos casos descritos, a forma oncogénica da p53 é, predominante-mente, uma proteína com uma substituição num único aminoácido no seu domínio de ligação ao DNA, resultando numa alteração conforma-cional que torna a proteína inactiva. Por este motivo, a pesquisa de pequenas moléculas que consigam
interactuar com a proteína mutada re-estabelecendo a sua conformação funcional é actualmente objecto de grande interesse (Foster et al., 1999; revisto recentemente por Beaudry et al., 1996 e Bullock e Ferhst, 2001).
A possibilidade de explorar vias alternativas de checkpoint, que não se encontrem inactivas nas células tumorais, constitui uma outra possibilidade terapêutica. Os taxanos – drogas actualmente utilizadas com bastante sucesso no tratamento do cancro da mama - foram originalmente descobertas de forma empírica, com base na toxicidade para células tumorais em cultura. O seu mecanismo de acção foi posteriormente elucidado - os taxanos estimulam a polimerização da tubulina, induzindo a apoptose tumoral via um checkpoint G2/M, independente da presença de p53 normal (Lanni et al., 1997 e Sorger et al., 1997). Isto permitiu a identificação de novas drogas, como as epotilonas, descobertas em rastreios na procura de compostos que estimulassem a polimerização da tubulina, e que vieram a revelar-se eficazes em casos de cancros resistentes a outros medicamentos.
Uma outra possibilidade, mais extrema, e que representa uma estratégia conceptualmente oposta, consiste em tentar tirar partido do facto de a alteração de vias de checkpoint constituir uma diferença
fundamental entre as células tumorais e as células normais. Neste caso, a ideia base é a de provocar nas células tumorais uma acumulação tal de mutações que as torne totalmente inviáveis. Uma forma de provocar essa acumulação de mutações nas células tumorais poderá consistir na inibição de vários mecanismos de checkpoint simultaneamente. De acordo com dados obtidos em leveduras, a acumulação de mutações em dois ou mais genes de checkpoint em simultâneo torna as células hipersensíveis à radiação, de tal forma que mesmo doses mínimas de radiação são suficientes para lhes provocar a morte. Com base nestas observações, a pesquisa de produtos capazes de inibir a função de proteínas de checkpoint constitui uma linha actual de investigação na área da terapêutica contra o cancro, que permitiu já a obtenção de alguns resultados promissores em células em cultura.
Para além das proteínas directamente envolvidas nos checkpoints celulares, foram já identificadas alterações, em células tumorais, de outras proteínas que desempenham funções essenciais no controlo do ciclo celular, e que poderão constituir potenciais alvos terapêuticos. Neste sentido, estas proteínas ocupam um lugar importante na investigação actual que tem como objectivo o desenvolvimento de substâncias que funcionem como inibidores
Endereços de Páginas WEB com Informações Úteis
Prémio Nobel da Fisiologia ou Medicina 2001
http://www.nobel.se/medicine/laureates/2001/ http://almaz.com/nobel/medicine/medicine.html
Ciclo Celular
http://www.new-science-press.com/cellc-primer.asp
http://www.ultranet.com/~jkimball/BiologyPages/C/CellCycle.html http://www.nature.com/celldivision/milestones/index.html
Ciclo Celular e Cancro
http://www.sciam.com/0996issue/0996weinberg.html
Proteínas do ciclo celular e terapêutica do cancro
específicos, quer sejam compostos químicos ou péptidos com actividade biológica, anticorpos específicos (que alteram a conformação ou interacção das proteínas mutadas) ou tratamentos baseados em ácidos nucleicos (por exemplo utilização de oligonucleótidos antisense). A identificação de inibidores específi-cos para algumas Cdks constitui um exemplo deste tipo de produtos. Uma das linhas de investigação actualmente em curso tem como base a ideia de desenvolver pequenos péptidos (com menos de 10 amino-ácidos) que simulem o efeito dos inibidores de Cdk que ocorrem normalmente nas células. Neste sentido, a optimização de um péptido de 8 amino-ácidos a partir da sequência da proteína p21 conduziu ao desenvolvimento de um produto, denominado CYC103, que exibe actividade inibidora específica contra a Cdk2 simulando a actividade da proteína p21 endógena normal. O fármaco CYC202 constitui um exemplo de um outro tipo de inibidor, que demonstrou actividade contra células tumorais num vasto painel de células tumorais humanas e se encontra actualmente na fase Ib de ensaios clínicos. CYC202 é uma purina tri-substituída que compete com o ATP para o local activo da Cdk2, apresentando-se como um inibidor altamente específico da actividade Cdk2/ciclina E e induzindo a apoptose de forma selectiva em linhas celulares neoplásicas. O inibidor CYC202 é activo em linhas celulares resistentes à quimioterapia convencional, e a sua actividade é independente da presença das vias p53 e Rb activas.
Por último, novos métodos para a detecção de potenciais alvos terapêuticos podem envolver a utilização de leveduras na realização de rastreios baseados em “letalidade sintética”, na tentativa de identificar identificar proteínas que quando inibidas de forma selectiva, matam as células que contêm defeitos genéticos frequentemente encontrados em tumores humanos.
Referências Bibliográficas
Beaudry GA, Bertelsen AH e Sherman MI (1996) Therapeutic targeting of the
p53 tumor suppressor gene. Current
Opinion in Biotechnology 7:592-600
Bullock AN e Fersht, AR (2001) Rescuing the function of mutant p53. Nature Reviews Cancer 1: 68-76 Cai DW, Mukhopadhyay T, Liu Y,
Fujiwara T, Roth JA (1993) Stable
expression of wild-type p53 gene in human lung cancer cells after
retrovirus-mediated gene transfer. Human Gene
Therapy 4: 617-624
Evans T, Rosenthal ET, Youngblom J,
Distel D e Hunt T (1983) Cyclin: a
protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33:389-96.
Foster BA, Coffey HA, Morin MJ e Rastinejad F (1999) Pharmacological rescue of mutant p53 conformation and function. Science 286:2507-2510 Gatti M e Goldberg M (1991) Mutations affecting cell division in Drosophila. Methods Cell Biol. 35:543-586.
Gibbs JB (2000) Mechanism-based target identification and drug discovery in cancer research. Science 287: 1969-1973 Hartwell L e Weinert T. (1989) Checkpoints: Controls that ensure the order of cell cycle events. Science 246: 629-634
Hartwell LH, Culotti J e Reid B (1970) Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. USA 66:352-359. Hoyt MA, Totis L e Roberts BT (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66:507-17.
Hunt T (1991) Cell biology. Cell cycle
gets more cyclins. Nature 350:462-3.
Johnson DG, Walker CL (1999) Cyclins
and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 39:295-312. Labbe JC, Picard A, Karsenti E, Doree M
(1988) An M-phase-specific protein
kinase of Xenopus oocytes: partial purification and possible mechanism of its periodic activation. Dev Biol. 127:157-69.
Lanni JS, Lowe SW, Licitra EJ Liu JO e Jacks T (1997) p53-independent apoptosis induced by paclitaxel through
an indirect mechanism. Proc Natl Acad
Sci U S A 94:9679-9683
Lee MG e Nurse P (1987) Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327: 31-35 Levine K, Kiang L, Jacobson MD, Fisher RP, Cross FR (1999) Directed evolution to bypass cyclin requirements for the Cdc28p cyclin-dependent kinase. Mol Cell. 4:353-63.
Li R e Murray AW (1991) Feedback
control of mitosis in budding yeast. Cell 66:519-31.
Lohka MJ, Hayes MK, Maller JL (1988) Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc Natl Acad Sci U S A. 85:3009-13.
Masui Y, Markert CL (1971) Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 177:129-45. Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK (2001) Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413:311-6.
Nasmyth K (1996) At the heart of the
budding yeast cell cycle. Trends Genet. 12:405-12.
Nicklas RB (1997) How cells get the
right chromosomes Science 275: 632-7
Nigg EA (2001) Mitotic kinases as
regulators of cell division and its checkpoints Nat Rev Mol Cell Biol.2:21-32
Nurse P, Thuriaux P, Nasmyth K. (1976) Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 146:167-78 Paulovich AG, Toczyski DP e Hartwell
LH (1997) When checkpoints fail. Cell
88: 315-321
Pines J (1999) Four-dimensional control of the cell cycle. Nat Cell Biol. 1:E73-9. Sorger PK, Dobles M, Tournebize R e Hyman AA (1997) Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol 9:807-814