The Mitochondrial Genomes of Aquila fasciata and Buteo lagopus (Aves, Accipitriformes): Sequence, Structure and Phylogenetic Analyses.
Texto
Imagem



Documentos relacionados
Por outro lado, também fica evidenciada uma carência de estudos voltados para a compreensão da noção de liberdade na fenomenologia de Merleau-Ponty, para a
A number of commonly used factors were analysed to compare and contrast the two methods: (1) variation in species count of sessile organisms (often used as a measure of
In Table 12, we have a SAM for Portugal in 2009 with a disaggregation that is appropriate for the identification of the informal aspects of the activity of countries,
O ponto de partida no desenvolvimento da ontologia consistiu na busca da informação necessária para dar resposta às inevitáveis questões sobre o domínio e
species, excluding the grouped species of subgenus Drosophila (Table 1 ) and tripunctata group species (Table 2 ), with their absolute abundances, from collections in two
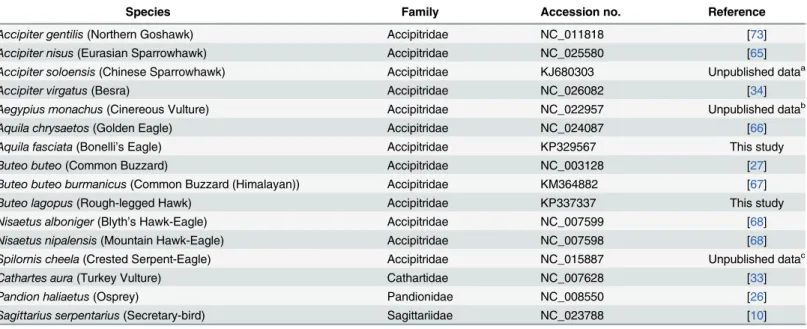
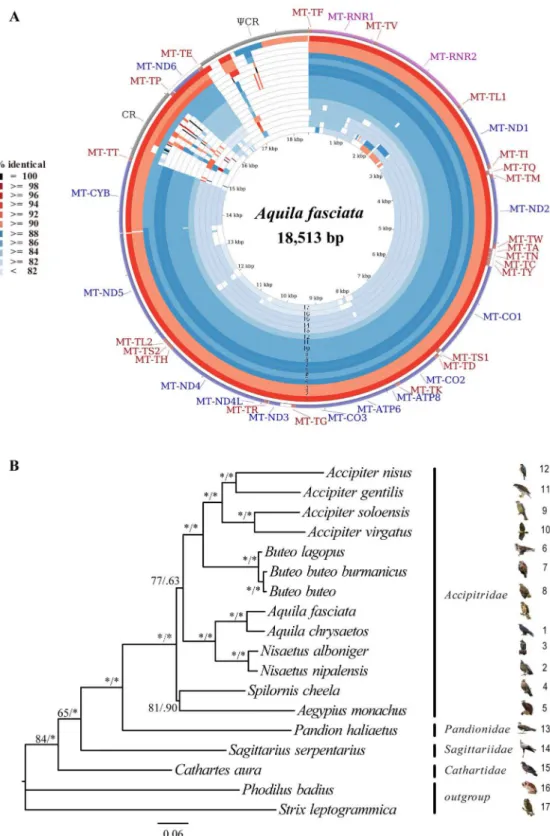
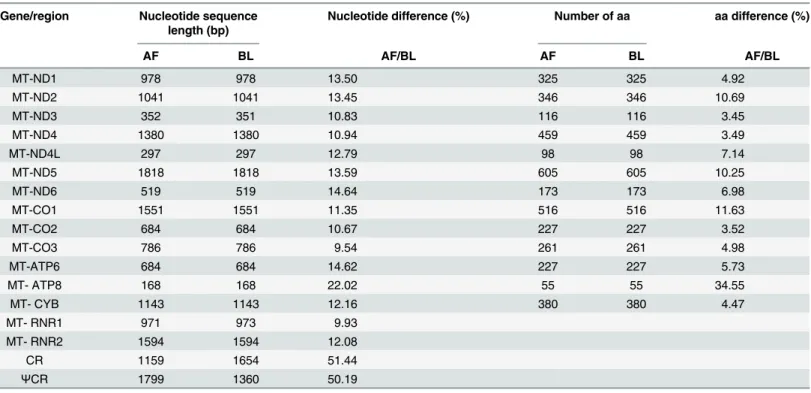
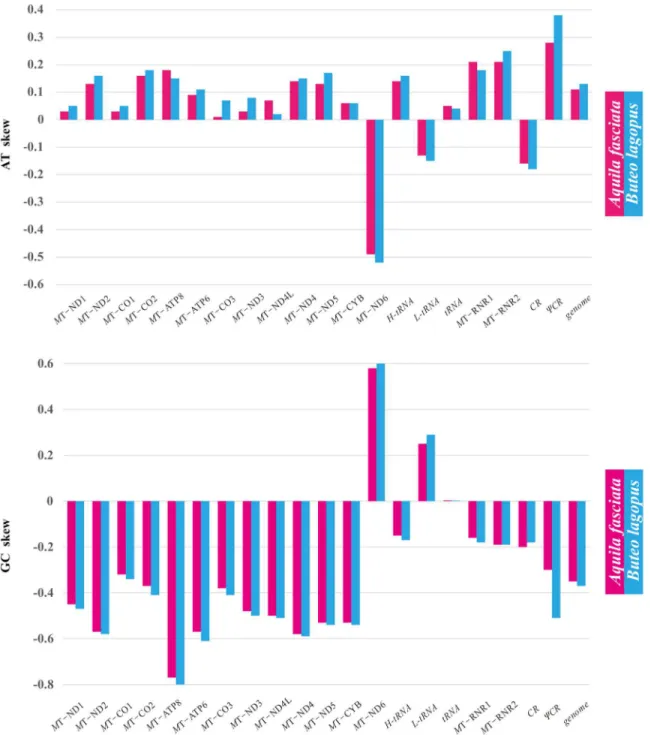
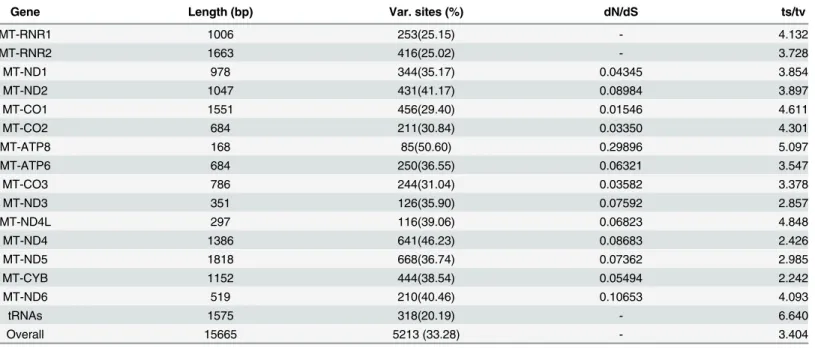
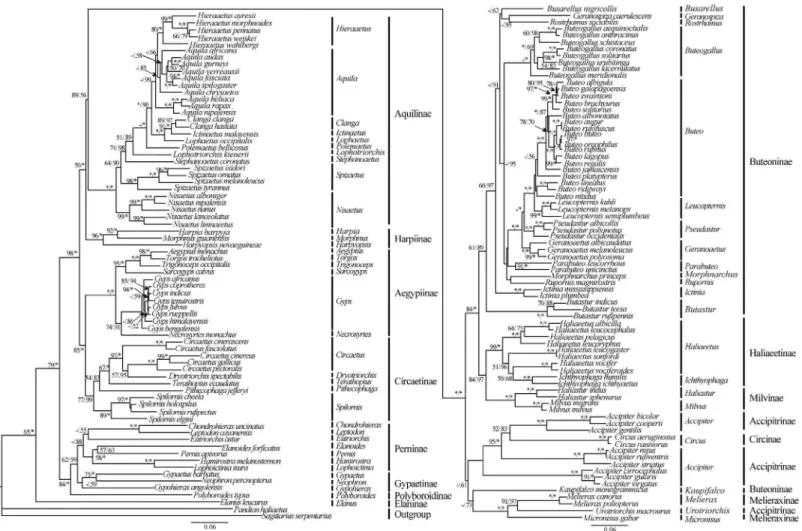
and the inner relationships of the order Hypocreales, phylo- genetic trees were constructed using the nucleotide sequences of 13 PCGs from 20 complete mitochondrial genomes that
Com a atual problemática que se passa em sala de aula de nível médio, onde alunos desmotivados sem respeito á imagem do professor, gerando docentes sem
Através do trabalho proposto foi possível verificar nos ensaios de fotodegradação que quanto menor a concentração inicial da solução maior a remoção de cor
