Ultrahigh-density linkage map for cultivated cucumber (Cucumis sativus L.) using a single-nucleotide polymorphism genotyping array.
Texto
Imagem

Documentos relacionados
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
A infestação da praga foi medida mediante a contagem de castanhas com orificio de saída do adulto, aberto pela larva no final do seu desenvolvimento, na parte distal da castanha,
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
No campo, os efeitos da seca e da privatiza- ção dos recursos recaíram principalmente sobre agricultores familiares, que mobilizaram as comunidades rurais organizadas e as agências
Este relatório relata as vivências experimentadas durante o estágio curricular, realizado na Farmácia S.Miguel, bem como todas as atividades/formações realizadas
Peça de mão de alta rotação pneumática com sistema Push Button (botão para remoção de broca), podendo apresentar passagem dupla de ar e acoplamento para engate rápido
Duvick (1986), after 45 years of experience in corn breeding, reported witnessing the following major advancements in plant breeding: adopting of new improved machinery and
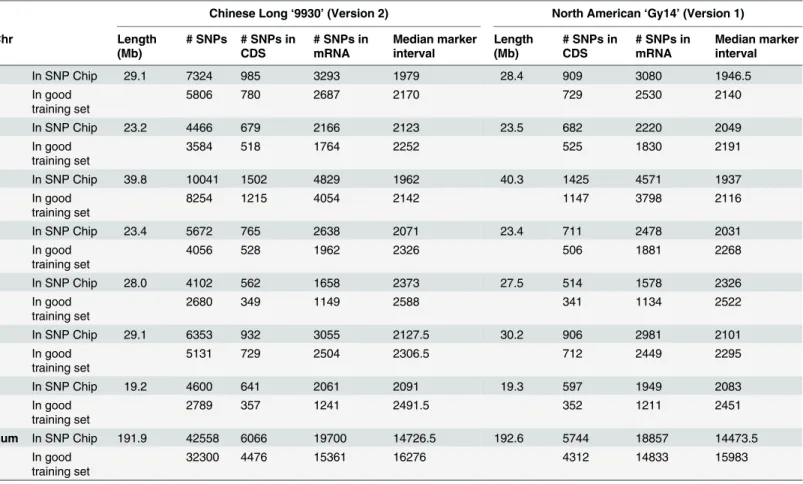
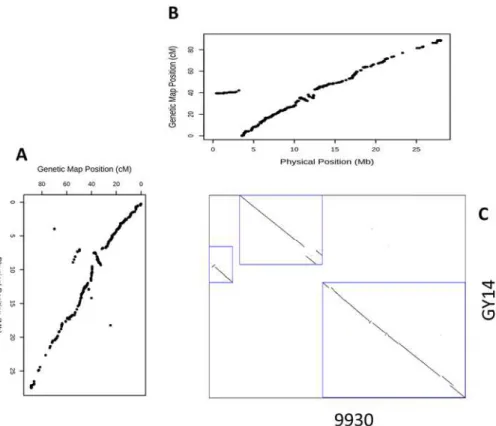
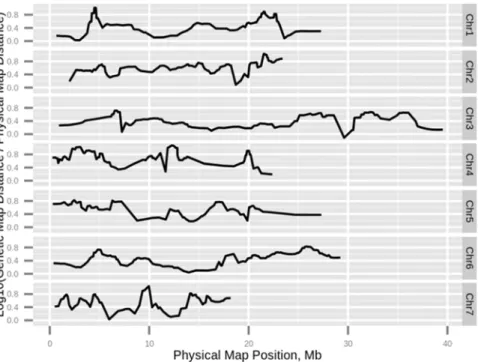
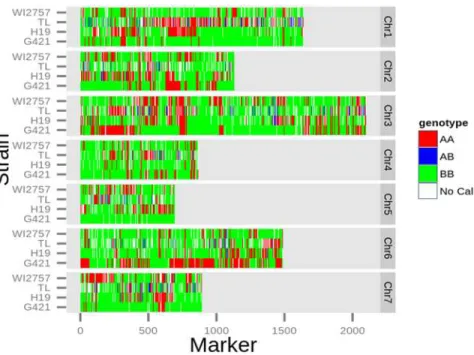
The rapid improvements in high-throughput methods for SNPs genotyping technologies and the development of the SNP arrays accompanied by reduced costs for
