CRAQUEAMENTO TERMOCATALÍTICO DE ÓLEO DE GIRASSOL
NA PRESENÇA DA PENEIRA MOLECULAR SAPO-5
Ricardo Miguel de Oliveira
_______________________________________
Dissertação de Mestrado
Natal/RN, dezembro de 2010
CRAQUEAMENTO TERMOCATALÍTICO DE ÓLEO DE GIRASSOL NA
PRESENÇA DA PENEIRA MOLECULAR SAPO-5
Dissertação apresentada ao Programa de Pós-Graduação em Química da Universidade Federal do Rio Grande do Norte, como parte dos requisitos para a obtenção do título de Mestre em Química.
Orientador: Prof. Dr. Antonio Souza de Araujo Co-orientador: Prof. Dr.Valter José Fernandes Junior
NATAL-RN
Divisão de Serviços Técnicos
Catalogação da Publicação na Fonte. UFRN / Biblioteca Central Zila Mamede
Oliveira, Ricardo Miguel de.
Craqueamento termocatalítico de óleo de girassol na presença da peneira molecular SAPO-5 / Ricardo Miguel de Oliveira. – Natal, RN, 2010.
79 f. : il.
Orientador: Antonio Souza de Araújo. Co-orientador: Valter José Fernandes Júnior.
Dissertação (mestrado) – Universidade Federal do Rio Grande do Norte. Centro de Ciências Exatas e da Terra. Programa de Pós-Graduação em Química.
1. Catálise – Dissertação. 2. Craqueamento catalítico – Dissertação. 3. Óleo de girassol – Craqueamento – Dissertação. 4. Óleos vegetais – Dissertação. I. Araújo, Antonio Souza de. II. Fernandes Junior, Valter José. III. Universidade Federal do Rio Grande do Norte. IV. Título.
Dedico este trabalho aos meus pais
Manoel e Maria Dalva que me
propiciaram uma vida digna onde eu
pude crescer, acreditando que tudo é
possível, desde que sejamos honestos
A Deus, pela saúde, paz e disposição presenteada a mim para poder realizar este trabalho.
A minha família, especialmente aos meus pais Manoel e Maria Dalva, meus irmãos Daniel e Daniele, minha esposa Ligiane e minha filha Larissa, pela compreensão, apoio e incentivo, que sempre me deram. Sem vocês com certeza eu não seria o que sou.
Aos colegas que me ajudaram diretamente no trabalho – Ana Claudia e Geraldo – em análises, experimentos, sínteses, estudos e em tantas ocasiões que precisei de apoio.
Ao Prof. Dr. Antonio Souza e ao Prof. Dr. Valter que me orientaram, não só na pós-graduação, mas em praticamente todo a minha trajetória na ciência até aqui.
A todos os amigos do LCP que, felizmente são muitos, não caberiam nesta seção do trabalho em que destaco, sem ordem de relevância, pelo companheirismo, união e/ou contribuição para realização dessa etapa: Marcela, Késia, Maria (Dedeia), Regineide, Adalgisa, Vinícius, João Paulo, Thiago, Larissa, Anne, Tarcísio, Mirna, João Paulo, Marcílio, Breno e Aline.
Ao Laboratório de Combustíveis e Lubrificantes (LCL), principalmente Camila e Jilliano, pelas análises realizadas.
Aos membros das bancas de qualificação e defesa pela disponibilidade em avaliar e contribuir para a melhora deste trabalho.
Ao Programa de Pós-Graduação em Química da UFRN, pela oportunidade de realizar este trabalho.
À Agência Nacional do Petróleo, Gás Natural e Biocombustíveis (ANP) por financiar meus estudos.
A todos os professores que passaram na minha vida.
O craqueamento catalítico de triacilglicerídeos se apresenta como uma alternativa viável para a produção de biocombustíveis com baixa emissão de poluentes. Neste trabalho foi sintetizado o SAPO-5, o catalisador foi utilizado na reação de craqueamento do óleo de Girassol. O sólido foi caracterizado por difração de raios -X, análise termogravimétrica, espectroscopia no infravermelho. As análises indicaram que através do método de síntese empregado foram obtidos materiais com elevada área específica e alta acidez. A partir do craqueamento térmico e termocatalítico do óleo de girassol, realizado desde a temperatura ambiente até 450 oC em um sistema de destilação simples, foram obtidas duas frações líquidas, cada uma contendo duas fases, uma aquosa e outra orgânica. A primeira fração do líquido orgânico obtido apresentou alto índice de acidez, no processo de craqueamento térmico. Os produtos obtidos no craqueamento de óleo de Girassol foram analisados por destilação, índice de acidez, espectroscopia no infravermelho, densidade, viscosidade e determinação do índice de cetano. A análise dos produtos obtidos na presença e na ausência do SAPO-5, sintetizados permitiu concluir que todos os sólidos testados apresentaram atividade catalítica para a desoxigenação dos produtos finais apenas na segunda etapa do processo de craqueamento.
The catalytic cracking of triglycerides presents itself as a possible alternative to the production of biofuels with low emission of pollutants. In this work were synthesized the SAPO-5, the catalysts for the cracking reaction of soybean oil is presented. The solids were powder X-ray diffraction (XRD), thermogravimetric analysis (TG/DTG) and infrared spectroscopy (FTIR). The analyses indicated that the synthesis method has employed to obtain materials with high surface area and high acid. The soybean oil thermal and thermal catalytic cracking, realized from the room temperature to 450 ºC in a simple distillation system, has allowed obtaining two liquid fractions, each consisting of two phases, one aqueous and another organic, organic liquid (OL). The OL obtained from first fractions has shown high acid index, even in the thermal catalytic process. The products obtained in the cracking of soybean oil were analyzed by distillation, acid number, infra-red spectroscopy, density, viscosity, carbon residue, cetane number determination and characterization. The analysis of the products obtained in the presence and in the absence of the SAPO-5 permitted to conclude that all the solids tested presented catalytic activity in the deoxygenation of final products only at the second step of the cracking process.
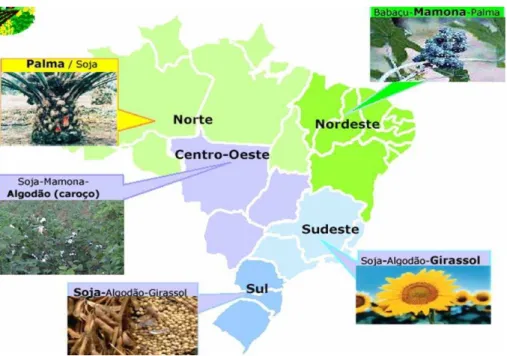
Figura 1.1- Diversidade de oleaginosas presentes por região no Brasil ... 16
Figura 3.1- Cadeia do Ácido Linoléico e oléico... 22
Figura 3.2- Rotas de obtenção de bicombustíveis ... 23
Figura 3.3- Reação de Transesterificação ... 24
Figura 3.4 Degradação do triglicerídeo formando ácido carboxílico, 2-propenal e cetenos.. 25
Figura 3.5- Reação entre aldeídos e cetenos ... 27
Figura 3.6- Representação esquemática do processo de descarbonilação ... 27
Figura 3.7- Produtos do Craqueamento térmico de triacilglicerídeos ... 28
Figura 3.8- Esquema representativo para Lei de Bragg ... 30
Figura 3.9- Classificação das Isotermas Segundo a IUPAC ... 33
Figura 3.10- Estrutura do AlPO4 ... 36
Figura 3.11- Mecanismo de Incorporação de Silício nos ALPO´s ... 38
Figura 3.12- Proposta de mecanismo mistos nos ALPO´s ... 38
Figura 3.13- Esquema planar de Si, Al e P distribuídos na rede do SAPO: (a) Si isolados (b) 5 Si Isolados (c) Ilha contendo 11 Si (d) Si-Al em uma ilha ... 39
Figura 3.14- Esquema representativo dos anéis do SAPO-5 ... 40
Figura 3.15- Difratograma padrão da Fase AFI ... 40
Figura 4.1 - Fluxograma geral da síntese da amostra de SAPO-5... 42
Figura 4.2- Esquema de calcinação. ... 43
Figura 4.3- Perfil de aquecimento das amostras calcinadas. ... 43
Figura 4.4- Esquema do sistema utilizado no craqueamento ... 48
Figura 5.1- Difratograma do SAPO-5 calcinado ... 54
Figura 5.2- Microscopia Eletrônica de Varredura da amostra de SAPO 5 ... 55
Figura 5.3- Isoterma de adsorção de Nitrogênio do SAPO-5. ... 56
Figura 5.4- Curva Termogravimétrica da amostra de SAPO-5 não calcinado ... 57
Figura 5.5- Curva Termogravimétrica do SAPO-5 calcinado ... 58
Figura 5.6- Curva Termogravimétrica do Óleo de Girassol ... 59
Figura 5.7 – Curva Termogravimétrica do SAPO-5 + Óleo de Girassol ... 59
Figura 5.10- Comparação entre os espectros obtidos a partir do craqueamento do óleo de girassol e do diesel oriundo do petróleo. ... 65 Figura 5.11- Acidezdo bicombustível por porcentagem de catalisador ... 66 Figura 5.12-Cromatogramas dos LO oriundos dos craqueamentos térmico e termocatalíticos
ABNT- Associação Brasileira de Normas Técnicas.
AFI - Topologia da estrutura da peneira molecular SAPO-5
AlPO - Aluminofosfato
ANP - Agência Nacional de Petróleo, Gás Natural e Biocombustíveis.
AOCS - American Oil Chemists Society.
ASTM- American Society for Testing and Materials.
BET - Braunauer, Emmett e Teller
CG- Cromatografia gasosa.
DTG- Termogravimétrica derivada.
DRX - Difratograma de Raios - X
EMBRAPA- Empresa Brasileira de Pesquisa Agropecuária
FID - Detector de Ionização de Chama
IA-Índice de acidez.
IC- Índice de cetano.
IZA - International Zeolite Association
1 INTRODUÇÃO ... 16
2 OBJETIVOS ... 18
2.1 OBJETIVO GERAL ... 18
2.2 OBJETIVOS ESPECÍFICOS ... 18
3 FUNDAMENTAÇÃO TEÓRICA ... 19
3.1 ÓLEOS VEGETAIS ... 19
3.2 CARACTERÍSTICAS FÍSICO-QUÍMICAS DOS ÓLEOS VEGETAIS ... 19
3.2.1 Ponto de Fusão ... 19
3.2.2 Viscosidade ... 20
3.2.3 Densidade ... 20
3.2.4 Acidez.... ... 20
3.3 ÓLEO DE GIRASSOL ... 20
3.3.1 Composição Química e estrutura do óleo de girassol ... 21
3.4 O USO DIRETO DE ÓLEOS E GORDURAS PARA OBTENÇÃO DE BIOCOMBUSTIVEIS ... 23
3.4.1 O uso direto de óleos e gorduras ... 23
3.4.2 O uso de gorduras e óleos vegetais para obtenção de combustíveis ... 23
3.5 REAÇÃO DE TRANSESTERIFICAÇÃO ... 24
3.6 CRAQUEAMENTO TÉRMICO ... 25
3.7 CRAQUEAMENTO CATALÍTICO ... 29
3.8 CARACTERIZAÇÕES FÍSICO-QUÍMICAS DOS CATALISADORES ... 29
3.8.1 Difratometria de Raios X ... 29
3.8.2 Espectroscopia no Infravermelho ... 31
3.8.3 Adsorção de Nitrogênio ... 32
3.8.4 Termogravimétria ... 34
3.9 PENEIRAS MOLECULARES ... 35
3.9.1 Aluminofosfatos (AlPO´s) ... 35
3.9.2 Silicoaluminofosfatos (SAPO´s) ... 36
4.2 CARACTERIZAÇÃO DOS SÓLIDOS ... 44
4.2.1Análise Térmica via TG/DTG ... 44
4.2.2 Calcinação ... 44
4.2.3 Difratometria de Raios X ... 45
4.2.4 Determinação da Acidez Superficial ... 45
4.2.5 Determinação de Área Específica ... 46
4.2.6 Microscopia Eletrônica de Varredura ... 46
4.3 REAÇÃO DE CRAQUEAMENTO ... 46
4.3.1 Craqueamento térmico ... 46
4.3.2 Craqueamento termocatalítico ... 47
4.4 CARACTERIZAÇÕES DOS PRODUTOS OBTIDOS NO CRAQUEAMENTO TÉRMICO E TERMOCATALÍTICO ... 48
4.4.1 Espectroscopia na região do Infravermelho ... 49
4.4.2 Cromatografia CG-FID ... 49
4.4.3 Índice de Acidez ... 50
4.5 ANÁLISES FÍSICO-QUÍMICAS DOS BIOCOMBUSTÍVEIS OBTIDOS ... 51
4.5.1 Destilação Atmosférica ... 51
4.5.2 Viscosidade Cinemática ... 51
4.5.3 Ponto de Fulgor ... 51
4.5.4 Densidade ... 52
4.5.5 Índice de Cetano ... 52
5. RESULTADOS E DISCUSSÃO ... 54
5.1 DIFRAÇÃO DE RAIOS X (DRX) ... 54
5.2 MICROSCOPIA ELETRÔNICA DE VARREDURA ... 55
5.3 DETERMINAÇÃO DA AREA SUPERFICIAL ... 56
5.4 ANÁLISE TERMOGRAVIMETRICA ... 57
5.5 DETERMINAÇÃO DA ACIDEZ SUPERFICIAL ... 60
5.6 REAÇÃO DE CRAQUEAMENTO ... 61
5.7 ESPECTROSCOPIA NO INFRAVERMELHO ... 64
5.8 CROMATOGRAFIA ... 66
6. CONCLUSÕES ... 74
1 INTRODUÇÃO
Em virtude do aumento na demanda cada vez maior de combustíveis, uma maior consciência ecológica e uma provável escassez das fontes fosseis, tem levado os olhares da comunidade científica para os recursos renováveis que até então são pouco explorados, como é o caso dos óleos vegetais, pois o Brasil é um país rico em oleaginosas, porém restringe as suas culturas para fins alimentícios.
Dentre as várias espécies vegetais no Brasil das quais se podem produzir biocombustível, como pode ser observado na Figura 1.1, destacam-se a mamona, dendê (palma), girassol, babaçu e soja, dentre outras. Além da diversificada disponibilidade de oleaginosas, há de se considerar as gorduras animais, como o sebo bovino, os óleos de peixe, o óleo de mocotó e a banha de porco, entre outros. Outra fonte de matéria potencial para produção de biocombustíveis no país consiste nos óleos e gorduras residuais, resultantes de processamento doméstico, comercial e industrial (VILAS, 2005).
A idéia da utilização de óleos vegetais, como fonte de matéria-prima, para a geração de energia não vem de hoje; há cem anos, Rudolf Diesel testou óleo vegetal como combustível para o motor a diesel. Com o advento do petróleo barato, frações de petróleo adequadas foram refinadas para servir como combustível e de combustíveis para motores a diesel. Na década de 1930 e 1940 óleos vegetais foram usados como combustíveis em motores a diesel de vez em quando, só em situações de emergência.
Com o aumento no preço do petróleo e estes recursos cada vez mais limitados, aliado a preocupação ambiental têm provocado um foco renovado em óleos vegetais e gorduras animais para fazer bicombustíveis (SHAY, 1993).
Devido a estas referidas razões, vários estudos têm sido feitos para obter fontes alternativas de combustível. Neste aspecto, a fermentação, transesterificação e pirólise de biomassa têm sido propostos como alternativas para solucionar o aumento da procura de energia e consciência ambiental. Entre estes, a pirólise parece ser um método simples e eficiente para a produção de combustíveis, contudo, diferentes abordagens têm sido feitas, por exemplo, plásticos (JALIL, 2002), óleos automotivos usados (NER´IN, 2000), óleos de pirólise da madeira (VITOLO et al., 2001).
Desde então, vários estudos sobre o craqueamento do óleo vegetal, como um método alternativo, para a obtenção de produtos químicos e de combustíveis, têm sido relatados na literatura (SCHWAB et al., 1988). Pirólise, assistido de catalisadores sólidos, também tem sido relatada e se tem observado que a seletividade do produto é fortemente afetada pela presença e natureza heterogênea de catalisadores, bem como a presença de vapor de água e alimentação de gás (DA COSTA et al., 1998).
2 OBJETIVOS
2.1 OBJETIV O GERAL
O objetivo geral deste trabalho é o estudo do processo de craqueamento térmico e termocatalítico do óleo de girassol para obtenção de biocombustíveis, além da síntese e caracterização do SAPO-5 e sua utilização na reação de craqueamento do óleo de girassol visando à desoxigenação e a diminuição da acidez.
2.2 OBJETIVOS ESPECÍFICOS
Sintetizar o SAPO-5 pelo método hidrotérmico e caracterizá-lo por diferentes técnicas;
Estudar a presença do silicoaluminofosfato (SAPO-5) na reação do craqueamento do óleo de girassol;
Determinar as propriedades físico-químicas dos bicombustíveis obtidos;
Caracterizar os produtos da reação de craqueamento;
Testar a atividade catalítica do SAPO-5;
3 FUNDAMENTAÇÃO TEÓRICA
3.1 ÓLEOS VEGETAIS
Óleos vegetais são formados principalmente por misturas de ácidos graxos: Estes, por sua vez são ácidos orgânicos que diferem entre si pelo número de átomos de carbono que formam a cadeia carbônica e também pelo número de insaturações. Os ácidos graxos que não apresentam duplas ligações são conhecidos como saturados, e os que apresentam são chamados de insaturados, estas duplas podem gerar isômeros cis (Z) e trans (E), sendo os ácidos graxos encontrados na natureza apenas na forma cis (Z).
Os ácidos graxos podem ser encontrados livres ou associados, sendo sua principal ocorrência associadas a um tri-álcool conhecido como glicerina, formando ésteres de ácidos graxos na forma de triacilglicerídeos. É importante salientar que os triglicerídeos podem ser formados por ácidos graxos iguais ou diferentes.
3.2 CARACTERÍSTICAS FÍSICO-QUÍMICAS DOS ÓLEOS VEGETAIS
3.2.1 Ponto de Fusão
3.2.2 Viscosidade
A explicação para a viscosidade é semelhante, pois ela evidencia a dificuldade de um líquido escoar. Porém, a viscosidade diminui com o aumento das insaturações e amplia-se com o aumento das cadeias dos ácidos graxos, isto ocorre pois aumenta as forças de interação de Wander walls.
3.2.3 Densidade
Essa grandeza sempre é menor que a unidade, permitindo concluir que óleos são menos densos que a água. A variação é pequena, mas vale ressaltar que a densidade de ácidos graxos cresce com o aumento do número de átomos de carbono na cadeia e decrescem com o número de insaturações, podendo ser realizadas considerações semelhantes às feitas para o ponto de fusão para explicar este comportamento.
3.2.4 Acidez
3.3 ÓLEO DE GIRASSOL
O girassol é uma planta oleaginosa que apresenta características interessantes como a resistência ao calor e ao frio, além de seu rendimento ser pouco influenciado pela altitude, podendo assim ser cultivada em diversas regiões do Brasil.
Entre todos os óleos vegetais, o óleo de girassol ocupa lugar de destaque pelas suas excelentes características físico-químicas e nutricionais, sendo recomendado para o tratamento de doenças como a aterosclerose e doenças cardiovasculares.
O óleo possui uma alta concentração de ácidos graxos poli-insaturados, resultado da sua concentração de ácido linoléico. Esse ácido graxo é essencial para o desempenho de determinadas funções fisiológicas do organismo humano, o que faz do óleo de girassol um dos óleos de maior qualidade nutricional no mundo.
Além da alta concentração de ácido linoléico, o óleo de girassol possui outros compostos que são importantes do ponto de vista da qualidade e de estabilidade dos óleos vegetais (OLIVEIRA e VIEIRA, 2004).
3.3.1 Composição Química e estrutura do óleo de girassol
Tabela 3.1- Composição química do óleo de Girassol
Número de Carbonos Ácidos graxos Concentração (%)
C16:0 Palmítico 6,4
C16:1 Palmitoléico 0,1
C18:0 Estereático 2,9
C18:1 Oléico 17,7
C18:2 Linoléico 72,9
C18:3 Linolênico 0,0
Adaptado de (Leite et al., 2005)
Onde:
Cno é o número de carbonos presentes na cadeia carbônica do ácido graxo: :no é o número de insaturações presente no acido graxo:
Com relação à estrutura, os ácidos graxos insaturados presentes no óleo de girassol são mais comumente encontrados nos ácidos oléico e linoléico. As estruturas dos ácidos graxos mais abundantes no óleo de girassol podem ser observadas na Figura 3.1.
3.4 O USO DIRETO DE ÓLEOS E GORDURAS PARA OBTENÇÃO DE BIOCOMBUSTÍVEIS
3.4.1 O uso direto de óleos e gorduras
O uso direto de óleos vegetais como combustível gera uma série de desvantagens como depósitos de carbono no motor, obstrução dos filtros e bicos injetores, e o custo na manutenção do motor. Além de possuir uma alta viscosidade contém uma série de ácidos graxos livres, o que gera características inviáveis para o seu uso direto.
Para minimizar esses problemas, os óleos vegetais requerem uma modificação química que pode ser realizada através do craqueamento térmico ou catalítico (MEHER
et al., 2006).
3.4.2 O uso de gorduras e óleos vegetais para obtenção de combustíveis
A idéia de usar óleos e gorduras para obtenção de biocombustíveis vem se intensificando cada vez mais, devido à razão do bicombustível ser renovável e biodegradável e acelerado pela alta demanda do consumo de combustíveis fósseis, além da conscientização ambiental tem levado o governo a investir cada vez mais em pesquisas relacionadas a esse tema.
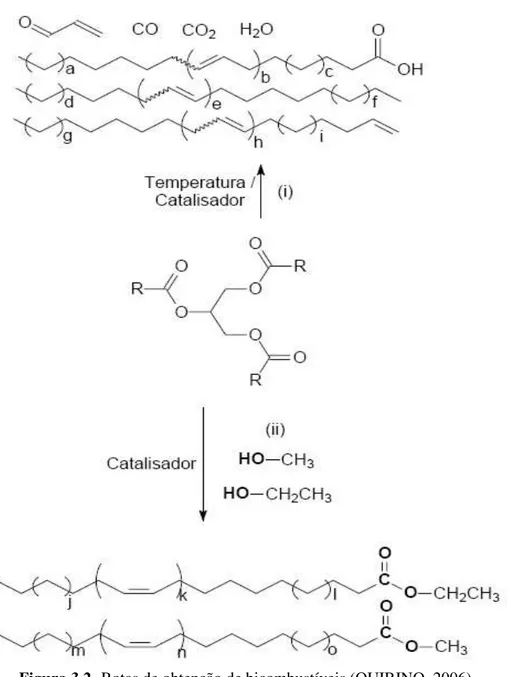
Figura 3.2- Rotas de obtenção de bicombustíveis (QUIRINO, 2006)
3.5 REAÇÃO DE TRANSESTERIFICAÇÃO
Entre todos os processos utilizados para produção de biodiesel, o que emprega a reação de transesterificação é o mais importante e preferencialmente utilizado (MEHER
A transesterificação ou alcoólise é a reação na qual os óleos vegetais ou gorduras animais ou residuais e um álcool na presença de um catalisador reagem, formando ésteres de ácidos graxos e glicerol, como pode ser observado na Figura3.3.
Figura 3.3- Reação de Transesterificação (Leite et al., 2005)
Onde os radicais R1, R2 e R3 representam cadeias carbônicas ou cadeias de
ácidos graxos, e R* representa uma cadeia carbônica que vai identificar qual tipo de álcool de cadeia curta será utilizado.
3.6 CRAQUEAMENTO TÉRMICO
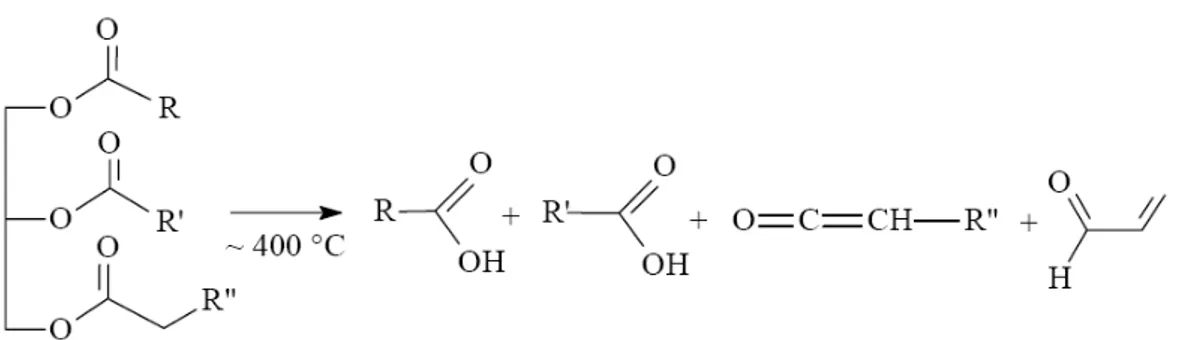
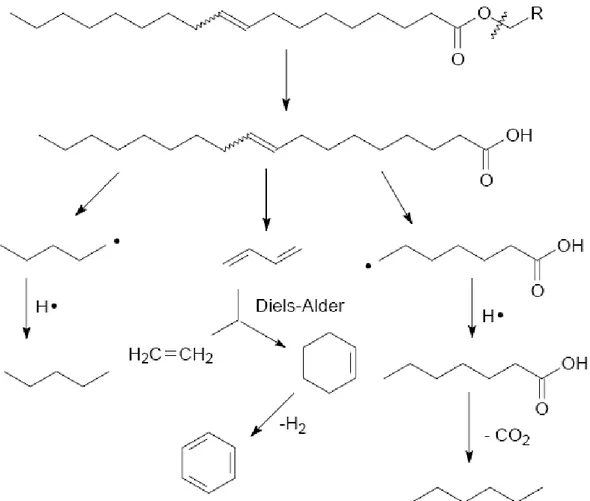
Segundo Suarez (2006), o craqueamento térmico é um processo que provoca a quebra de moléculas de óleos vegetais por aquecimento a altas temperaturas (temperaturas superiores a 350°C), formando uma mistura de compostos químicos com propriedades muito semelhantes às do diesel de petróleo, podendo este processo ocorrer na presença ou não de catalisadores. Observando a Figura 3.4 mostra que, além das cadeias longas de carbono, as quais apresentam características semelhantes às do diesel, são formados também o ácido propiônico (ou propanóico), monóxido de carbono, dióxido de carbono e água.
por mais de 100 anos, especialmente em áreas do mundo em que há falta de depósitos de petróleo (SONNTAG, 1979).
A reação de craqueamento ocorre a temperaturas superiores a 350 °C, na presença ou ausência de catalisadores, em diversas etapas distintas e consecutivas (KATIKANENI, 1996). Na primeira etapa, mostrada na reação 1 da Figura 3.4,
que é chamada craqueamento primário, são formadas duas moléculas de ácidos carboxílicos, uma molécula de ceteno e outra de acroleína.
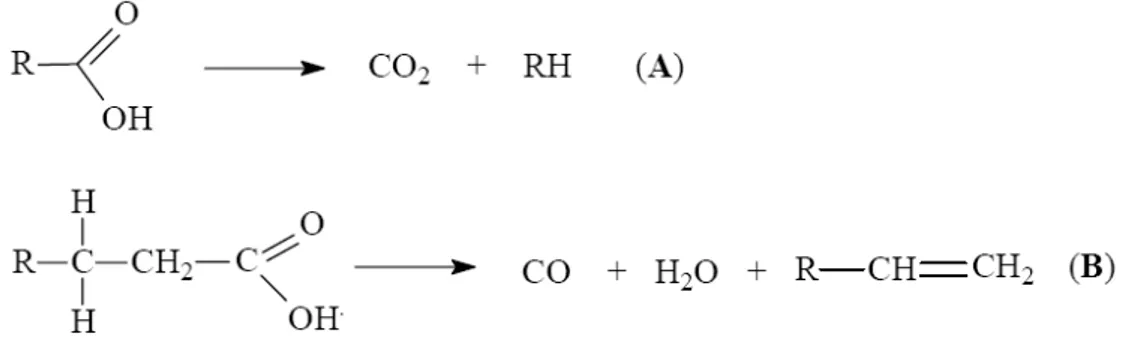
A mistura formada é extremamente instável nas condições reacionais e reage rapidamente, conforme mostrado na reação da Figura 3.5, formando novos ácidos carboxílicos, hidrocarbonetos, aldeídos e cetonas. Numa segunda etapa de craqueamento, chamada de craqueamento secundário, verifica-se a desoxigenação dos produtos formados no craqueamento primário, principalmente os ácidos carboxílicos. De fato, os ácidos carboxílicos formados durante o craqueamento primário e reações de rearranjo são desoxigenados no craqueamento secundário, que pode ocorrer por duas rotas distintas: descarboxilação e descarbonilação, conforme mostrado nas reações (A e B) da Figura 3.6. Posteriormente, podem ocorrer inúmeras reações consecutivas, tais como rearranjos, craqueamento das cadeias carbonílicas, acoplamentos radicalares, entre outras.
Figura 3.5- Reação entre aldeídos e cetenos (QUIRINO, 2006)
Existem trabalhos na literatura que demonstram que os produtos finais do craqueamento dependem de diversos fatores, tais como a composição química da matéria-prima utilizada, temperatura do processo, tempo de residência, presença de catalisadores e vapor (UZUN et al., 2006).
Figura 3.6- Representação esquemática do processo de descarbonilação (QUIRINO, 2006)
Figura 3.7- Produtos do Craqueamento térmico de triacilglicerídeos (QUIRINO, 2006)
Ocorre, nesse contexto, também reações via radicais, principalmente quando o ácido graxo apresenta insaturações, devido às duplas ligações que conferem uma maior estabilização aos radicais formados, a formação destes se deve a saída de um hidrogênio ligado ao carbono alfa da ligação dupla.
3.7CRAQUEAMENTO CATALÍTICO
Por outro lado, a estratégia do uso de catalisadores heterogêneos vem demonstrando que catalisadores sólidos que apresentam acidez de Lewis, como aluminossilicatos e óxidos, são eficientes para promover a desoxigenação dos produtos, sendo a seletividade dos produtos dependente da natureza do sólido utilizado. Por exemplo, a desoxigenação dos produtos da decomposição térmica de óleo de soja é favorecida, quando a reação é realizada na presença de diferentes zeólitas, (KATIKANENI, 1997), sendo também observado que reações de craqueamento da cadeia dos hidrocarbonetos formados, ciclizações e aromatização levam à formação de um produto próximo à faixa da gasolina (8-10 carbonos).
Já aluminossilicatos não estruturados e (γ-alumina) (DANDIK et al., 1998)são
também eficientes na desoxigenação dos produtos formados, diminuindo as reações de craqueamento da cadeia carbônica e de ciclização e aromatização, gerando um biocombustível com perfil de destilação próximo ao do diesel. Por outro lado, foi relatado que alguns catalisadores, como óxidos básicos MgO e CaO59ou Nb2O5e alguns derivadosestabilizam os ácidos carboxílicos formados, levando a produtos com acidez superior a verificada no craqueamento térmico.
3.8 CARACTERIZAÇÕES FÍSICO-QUÍMICAS DOS CATALISADORES
3.8.1 Difratometria de Raios X
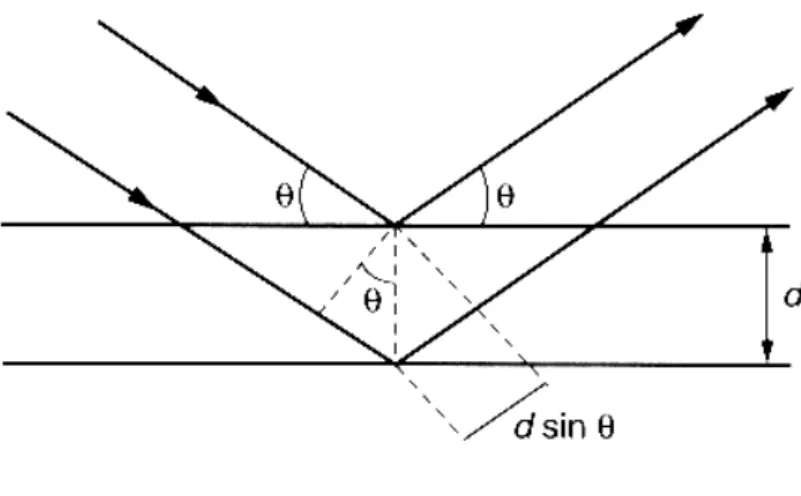
Figura 3.8- Esquema representativo para Lei de Bragg.
A equação de Bragg.
n = 2d sen θ
(3.1)
Onde n é a ordem de reflexão (n = {1,2,3,...}.
é o comprimento de onda, d é a distância interplanar. θ é o ângulo de incidência entre os planos reticulados.
Nesta ótica, a técnica foi sugerida inicialmente pelo físico alemão Max Von Laue, hoje em dia há diversas modificações do método empregado por Laue. Cada uma dessas modificações fornece dados que podem ser utilizados para determinar a orientação dos planos em um cristal.
O material pode ser analisado na forma de sólidos em pó, monocristais, matrizes, folhas e fibras. As amostras consistem em monocristais de 0,1 a 0,5mm de lado e pós (da ordem gramas). Apesar de ser bastante empregada em catálise, principalmente na determinação da estrutura cristalina de zeólitas e peneiras moleculares, a técnica apresenta também suas limitações, dentre elas:
Usada apenas em materiais cristalinos. Materiais amorfos geralmente não reproduzem difração proveitosa;
Picos sobrepostos podem atrasar a identificação na análise quantitativa;
Amostras fluorescentes podem elevar a linha de difração ou podem causar saturação em certos tipos de detectores.
3.8.2 Espectroscopia no Infravermelho
A Espectroscopia na Região do Infravermelho por Transformada de Fourier (FT-IR) é uma das mais comuns técnicas de caracterização mais comuns existentes, que permite caracterizar uma larga faixa de compostos inorgânicos e orgânicos. Esta se baseia fundamentalmente em medir a absorção em frequências de infravermelho de uma amostra posicionada na direção do feixe de radiação infravermelha. As radiações infravermelhas apresentam comprimentos de onda típicos que variam 0,78 a 1000 μm e números de onda variando de 13000 a 10 cm-1. O número de onda pode ser definido como o inverso do comprimento de onda.
Os espectros de infravermelho são gráficos apresentados sob a forma de número de onda ou comprimento de onda (eixo das abscissas) versus absorbância ou transmitância (eixo das ordenadas). A absorbância e a transmitância estão relacionadas entre si pela Eq. 3.2.
Ab = log10(1/Tr) (3. 2)
As principais aplicações para esta técnica são:
a) identificação de todos os tipos de compostos orgânicos e muitos tipos de compostos inorgânicos;
b) determinação de grupos funcionais em substâncias orgânicas; c) determinação quantitativa de compostos em misturas;
d) identificação de componentes de reação e estudo cinético das reações.
μL e para gases cerca de 50 ppm são requeridos. Essas quantidades são padrões e podem variar, dependendo do tipo do equipamento (SETTLE, 1997).
3.8.3Adsorção de Nitrogênio
A Adsorção é o termo usado para descrever o fenômeno no qual moléculas de um fluido se concentram espontaneamente sobre uma superfície sólida. De um modo geral, a adsorção ocorre tipicamente como um resultado de forças não balanceadas na superfície que atraem as moléculas de um fluido em contato por um tempo finito.
Denomina-se adsorvente o sólido sobre o qual ocorre o fenômeno de adsorção; adsorbato a(s) espécie(s) química(s) retida(s) pelo adsorvente; e adsortivo o fluido em contato com o adsorvente. Classificam-se os fenômenos adsortivos quanto às forças responsáveis em dois tipos: adsorção química e adsorção física. A adsorção química é assim denominada por que neste processo ocorre efetiva troca de elétrons entre o sólido e a molécula adsorvida, ocasionando as seguintes características: formação de uma única camada sobre a superfície sólida, irreversibilidade e liberação de uma quantidade de energia considerável (da ordem de uma reação química). A adsorção física, que constitui o princípio da maioria dos processos de purificação e separação é um fenômeno reversível, em que se observa normalmente a deposição de mais de uma camada de adsorbato sobre a superfície adsorvente. As energias liberadas são relativamente baixas.
O fenômeno da adsorção é a base da medição das propriedades superficiais de diversos materiais, como área específica, volume e distribuição de poros. Segundo a IUPAC, a maioria dos sólidos obedece a um dos seis tipos de isotermas de adsorção existentes, contudo quatro tipos de isotermas (I, II, IV e VI) são comumente encontrados em caracterização de catalisadores (ROQUEIROL et al., 1994). A Figura
Figura 3.9- Classificação das Isotermas segundo a IUPAC. (ROQUEIROL et al., 1994)
Assim, as isotermas de adsorção de nitrogênio para cada material específico, apresentadas na Figura 3.9, podem ser descritas como:
Tipo I: Isoterma típica de materiais microporosos, onde a adsorção se dá a baixas pressões devido à forte interação entre as paredes porosas e o adsorbato. Podem ser obtidas por adsorção química. Quando a adsorção física produz isotermas do tipo I, indica que os poros são microporosos, e, que a superfície exposta reside somente dentro dos microporos, os quais, uma vez cheios com o adsorbato, deixam pouca ou nenhuma superfície para adsorção adicional.
Tipo II: nesse tipo de isoterma a baixas pressões relativas ocorre a formação de uma monocamada de moléculas adsorvidas. São encontradas quando a adsorção ocorre em materiais porosos ou com poros de grande diâmetro. O ponto de inflexão ocorre, quando a primeira camada de cobertura ficar completa. Com o aumento da pressão relativa, o sólido ficará coberto de diversas camadas até que na saturação seu número será infinito.
Tipo IV: ocorrem em materiais mesoporosos. Neste caso ocorre inicialmente a cobertura de uma monocamada. O segundo degrau de adsorção indica a adsorção na faixa dos mesoporos. Normalmente, esse tipo de isoterma apresenta um “loop” de histerese, ou seja, a isoterma não segue o mesmo caminho para a adsorção e dessorção.
Tipo V: ocorrem quando existe pouca interação entre o adsorvente e o adsorbato, como no tipo III. Entretanto, o tipo V está associado a estruturas porosas que produzem o mesmo degrau que nas isotermas de tipo IV.
Tipo VI: ocorrem em materiais ultramicroporosos. A pressão na qual a adsorção é efetivada depende fundamentalmente da interação entre a superfície e o adsorbato. Se for energeticamente uniforme, o processo ocorre com uma pressão bem definida. Porém se a superfície contém poucos grupos de sítios energeticamente uniformes, uma isoterma com degraus pode ser esperada. Cada degrau na isoterma corresponde a um grupo específico de sítios.
3.8.4Termogravimetria
3.9 PENEIRAS MOLECULARES
Este termo foi empregado pela primeira vez por McBain em 1932 para definir materiais sólidos microporos, com a propriedade de adsorver moléculas seletivamente. A estrutura porosa de uma peneira molecular baseia-se em uma extensa rede de íons oxigênio, contendo átomos coordenados tetraedricamente (URBINA, 1997).
As peneiras moleculares apresentam estruturas cristalinas ordenadas de três dimensões, que proporcionam uma uniformidade no seu sistema de poros, além de selecionar moléculas com tamanho e dimensões específicas para dentro dos seus sistemas de microporos.
A capacidade de selecionar componentes está diretamente relacionada com a estrutura cristalina ordenada, que confere uniformidade às dimensões de seus microporos. Por essa razão, são capazes de selecionar (peneirar) as moléculas que podem ter acesso ao espaço intracristalino (SILVA, 2000).
3.9.1 Aluminofosfatos (AlPO´s)
No início da década de 1980, pesquisadores da Union Carbide descobriram uma nova família de materiais microporosos baseados em aluminofosfatos, conhecidos genericamente por AlPO. Estes materiaispossuem propriedades estruturais semelhantes às zeólitas, e, por este motivo,têm sido denominados de zeolitoides. Diferentemente das zeólitas, os aluminofosfatos são formados por uma rede cristalina de tetraedros alternados [AlO4]- e [PO4]+, resultando numa rede tridimensional neutra, não
Figura 3.10- Estrutura do AlPO4 (URBINA, 1997)
Existem mais de vinte estruturas de aluminofosfatos microporosos com tamanhos de poros pequenos, médios, grandes e supergrandes. Entre estas, nove estruturas são análogas àquelas zeólitas conhecidas, enquanto as demais são estruturas completamente novas, sem similares nem na natureza, nem entre as zeólitas previamente sintetizadas (URBINA, 1997).
3.9.2 Silicoaluminofosfatos (SAPO´s)
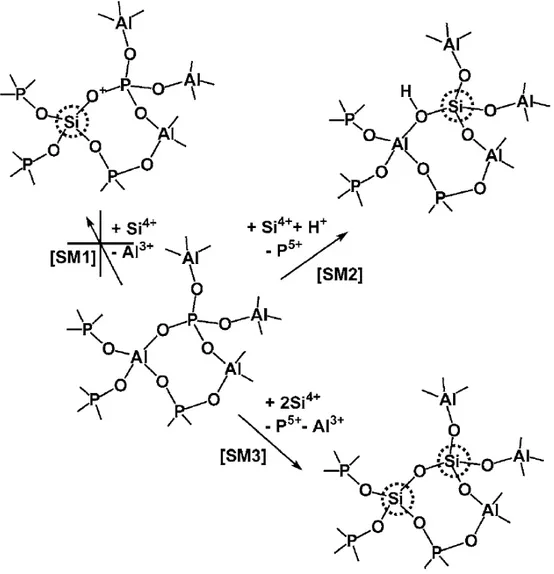
A introdução de átomos de silício na rede dos ALPO`s leva ao aparecimento de acidez de Brönsted relevante para reações catalisadas por ácido, como a
methanol-to-olefins (MTO) (STOCKER, 1999), craqueamento e hidrocraqueamento de n-alcanos (WANG et al., 2003). A origem da acidez de Brönsted em SAPO´s é a inserção de Si no sítio de fósforo, que leva à formação de carga negativa que é compensado por prótons ligados ao Si-O-Al em pontes, o mecanismo é denotado SM2. A substituição de silício por alumínio, SM1, é outro possível mecanismo de inserção de Si, porém, isso levaria à formação de pontes Si-O-P, que são energeticamente desfavoráveis, como sugerido por um estudo computacional sobre SAPO-5 (SASTRE et al., 1996) e nunca foram observados experimentalmente.
Figura 3.11- Mecanismo de Incorporação de Silício nos ALPO´s (PASTORE et al., 2005)
Um passo fundamental para a compreensão da acidez dos sapos e zeólitas veio do elegantwork de Barthomeuf, que racionalizou o mecanismo de inserção de silício na rede ALPO´s e as tendências que regem o número e a força da Bronsted dos sítios ácidos produzidos dessa maneira (Barthomeuf, 1994). Uma abordagem topológica, que levou em conta tanto a topologia da estrutura e do teor de Al, foi o primeiro desenvolvido para várias zeólitas (BARTHOMEUF, 1993) e, posteriormente, aplicada a SAPO´s.
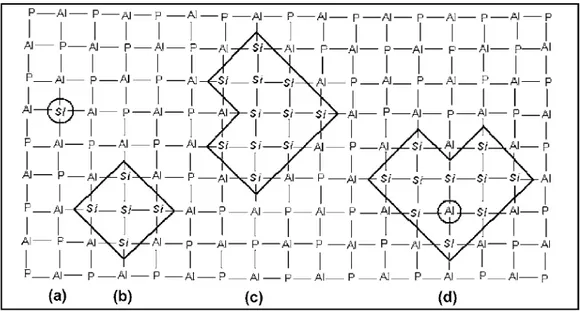
Com a ajuda de sistemas planares para representar a distribuição de Si e Al em zeólitas e Si, Al, P e nas redes de SAPO´s na Figura 3.13, (BARTHOMEUF, 1993, 1994) mostrou que a ocupação por Al, Si, P e átomos da primeira e segunda camada de átomos T em torno de um Si central regula a força do ácido.
Figura 3.13: Esquema planar de Si, Al e P distribuídos na rede do SAPO: (a) Si isolados (b) 5 Si Isolados (c) Ilha contendo 11 Si (d) Si-Al em uma ilha. (BARTHOMEUF, 1994)
3.9.3 Silicoaluminofosfato 5 (SAPO-5)
entre os da zeólita e dos ALPO´s (URBINA, 1997). Foi descoberto pela union carbide em 1984 e apresenta estrutura AFI com grupo de simetria P6cc com 12 canais retos.
A estrutura AFI apresenta simetria hexagonal com parâmetros de cela unitária a=13,73Å e c=8,4Å, (MURTHYet al., 2000), sendo que a sua cela unitária possui 24
tetraedros, como podemos observar na Figura 3.14.
Figura 3.14- Esquema representativo dos anéis do SAPO-5 (IZA)
O difratograma de raios-x característico da estrutura AFI pode ser visto na Figura 3.15, com indicações dos índices de Miller dos planos cristalográficos da sua estrutura.
4 EXPERIMENTAL
4.1 SÍNTESE DO SAPO-5
O catalisador utilizado neste estudo foi sintetizado em meio bifásico baseado na seguinte composição molar: 0.46SiO2, P2O5,Al2O3 ,(C2H5)3N ,0,072CTMABr,
4.4hexanol, 40H2O. A síntese começou pela adição 16,2mL de ácido fosfórico (85%)
em um Becker contendo 18,58g de pseudobohemita dispersa em 68,2mL de água destilada, esta mistura permaneceu em agitação por um período de 2h, em seguida adicionou-se 22,4mL de trietilamina, ficando em agitação por mais 2h, dando seguimento, preparou-se uma solução contendo: 12,2mL de Tetraortosilicato (TEOS) como fonte de silício, 65,3mL de n-Hexanol e 3,1g de Brometo de cetiltrimetilamônio (CTMABr), conservando a mistura resultante em agitação por mais 2h como pode ser observado na Figura 4.1. Por fim, o gel resultante foi colocado em uma autoclave e levado à estufa a 170 °C, permanecendo por 18h para cristalização do material.
Figura 4.1 Fluxograma geral da síntese da amostra de SAPO-5
Para a remoção do direcionador dos poros das peneiras moleculares foi utilizada a técnica de calcinação. Neste procedimento, a amostra foi submetida a uma razão de aquecimento de 10 °C/min, partindo da temperatura ambiente até 550 °C, sob atmosfera
dinâmica de N2 com fluxo de 100 mL/min com uma razão de aquecimento de
Trietilamina
Ácido Fosfórico
diluído em água dissolvida empseudobohemita água
Agitação por 2h
Agitação por 2h
Hexanol CTMABr
TEOS
Agitação por 2h
Autoclave de teflon a 170 oC por 18h
Filtrar e lavar com água a 50oC
10 °C/min. Ao chegar a 550 °C, o material permaneceu por 2 horas sob fluxo de nitrogênio. Após esse tempo, o gás foi trocado para ar sintético, ficando a amostra por mais 11 horas a 550 °C com fluxo de 100 mL/min. Pode ser visto na Figura 4.2 o sistema de calcinação horizontal empregado e a Figura 4.3 mostra um gráfico do perfil de aquecimento da amostra.
Figura 4.2- Esquema de calcinação
0 200 400 600 800 0
100 200 300 400 500 600
T
em
pe
ra
tu
ra
(
o C
)
Tempo(minutos)
Troca de N2 por Ar Sintetico
4.2 CARACTERIZAÇÃO DOS SÓLIDOS
4.2.1 Análise Térmica via TG/DTG
As curvas termogravimétricas do SAPO-5 foram obtidas em uma termobalança com forno horizontal, modelo TA/SDTA 951 da METTLER. As curvas padrão das amostras não calcinadas foram obtidas aquecendo a amostra da temperatura ambiente até 900 °C em atmosfera dinâmica de nitrogênio a uma taxa de aquecimento de 10 °C/min. Para cada ensaio foram utilizados cadinhos de alumina com massa em torno de 10 mg. Foram obtidas também curvas adicionais em taxas de aquecimento de 10 e 20 °C/mim com o objetivo de se realizar um série de estudos cinéticos referente às melhores condições de remoção das espécies dos poros dos materiais e estabelecer assim as melhores condições de calcinação.
Através da TG também foi possível estudar as melhores condições de calcinação do material microporoso. Em todos os casos, cerca de 10 mg de cada amostra foi aquecida, partindo da temperatura ambiente até 900 °C a uma razão de aquecimento de 10 °C /min, a fim de avaliar as melhores condições de calcinação para decompor o direcionador na forma não calcinada. As curvas foram obtidas em atmosfera dinâmica de ar sintético de 25 mL/minpartindo da temperatura ambiente até 900 °C com uma razão de aquecimento de 10 °C min.
4.2.2 Calcinação
A peneira molecular foi calcinada a 550 oC sendo 2h em fluxo de nitrogênio de
100mL/min e 11h em ar sintético com um fluxo de 100mL/min(BRITO et al., 2007) . Posteriormente, o catalisador foi ativado em fluxo de N2 e submetido às análises de
Determinação de acidez com adsorção de n-Butilamina seguida de curva termogravimétrica.
4.2.3 Difratometria de Raios X
A análise por difração de raios X foi aplicada na caracterização do SAPO-5,
utilizando um equipamento Shimadzu modelo XRD 6000 com varredura de 2θ= 5 a 45o.
Esta técnica foi utilizada para observar a cristalinidade do material obtido.
4.2.4 Determinação da Acidez Superficial
As medidas de acidez das amostras de silicoaluminofosfato foram realizadas pelo método de adsorção de base seguido de dessorção por aumento de temperatura. A base utilizada como molécula sonda foi a n-butilamina e a determinação da quantidade de base retida em cada faixa de temperatura foi realizada em um analisador termogravimétrico modelo METTLER TOLEDO TGA 851.
O procedimento utilizado para a saturação dos centros ácidos dos materiais com n-butilamina, consistiu em submeter a amostra previamente calcinada, a uma ativação da temperatura ambiente a 400 °C, sob fluxo de nitrogênio de 30mL/min durante 2h. Após esta ativação, a temperatura foi reduzida para 95 °C e os vapores de n-butilamina foram continuamente direcionados para a amostra pelo fluxo de N2 por 40 minutos, para
uma completa saturação dos sítios ácidos presentes no material. Em seguida, as amostras saturadas foram purificadas com nitrogênio puro na mesma temperatura de saturação, por 40 minutos, para a remoção da base fisicamente adsorvida.
termodissolvida (mmol/g de amostra). Este procedimento tem sido bastante utilizado para determinar a acidez de materiais zeolíticos (ARAUJO e JARONIECK, 1999).
4.2.5 Determinação de Área Específica
A determinação da área específica foi realizado pelo método BET (Brunauer, Emmet e Teller). O volume microporoso foi determinado pelo método t-plot. O equipamento utilizado foi um Quantachrome Instruments, modelo NOVA 1200e usando nitrogênio como adsorbato. A amostra foi submetida a um pré-tratamento a 300 °C por
3 horas para remoção de umidade e eventuais impurezas nelas adsorvidas.
4.2.6 Microscopia Eletrônica de Varredura
As análises eletrônicas de varredura do suporte microporoso SAPO-5 foram obtidas em um equipamento Philipps modelo XL30-ESEM. Antes das análises, as amostras foram aderidas à porta por meio de uma fina fita de carbono, e submetidas a um pré-tratamento que consistiu na deposição de uma fina camada de ouro, com o objetivo de tornar a amostra boa condutora de elétrons e assim poder dar uma excelente qualidade e resolução de imagem.
4.3 REAÇÃO DE CRAQUEAMENTO
4.3.1 Craqueamento Térmico
proporções de 0, 5, 1 e 2% com o intuito de otimizar a quantidade de catalisador, o aquecimento foi realizado por meio de uma manta aquecedora desde a temperatura ambiente até 450 °C por uma razão de aquecimento de 10 °C, quando a temperatura no fundo do balão chegou próximo de 370 °C os vapores começaram a entrar no condensador com uma temperatura entre 150 e 250 °C e foram coletados em uma proveta,. Foi dividido em duas frações, uma coletada em temperaturas inferiores a 190 °C e a outra sendo coletada em temperaturas superiores a 190 °C. As duas frações apresentaram uma fase orgânica que foi denominada de líquido orgânico (LO), as duas fases foram separadas por decantação, as massas das frações foram pesadas, como também foi determinada a massa do resíduo, a massa dos gases foi determinada por diferença estequiométrica. Em seguida os líquidos orgânicos foram armazenados em frascos de vidro de cor âmbar e guardados em abrigos na ausência de luz para posterior caracterização.
4.3.2 Craqueamento Térmocatalítico
frascos de vidro de cor âmbar e guardados em abrigos na ausência de luz para posterior caracterização.
Figura 4.4- Esquema do sistema utilizado no craqueamento
Como foi observado que a primeira fração do líquido orgânico apresentou uma acidez acima de 200mg KOH /g de líquido orgânico, somente será analisada a segunda fração que apresenta valores bem inferiores de índice de acidez em torno de 10mg KOH/g de líquido orgânico.
4.4 CARACTERIZAÇÕES DOS PRODUTOS OBTIDOS NO CRAQUEAMENTO TERMICO E TERMOCATALÍTICO
4.4.1 Espectroscopia na região do Infravermelho
Com o intuito de verificar as variações de intensidades das bandas características dos ácidos carboxílicos e demais produtos oxigenados, Os LO obtidos a partir do craqueamento térmico e termocatalítico foram analisados por espectroscopia na região do infravermelho com transformada de Fourier. Os espectros de infravermelho dos LO´s foram obtidos na faixa de 400 – 4000 cm-1 com resolução de 4 cm-1, com auxilio de uma janela de KBr, onde os líquidos orgânicos não poderiam conter água, já que o KBr é solúvel em água, e os espectros foram gerados em um equipamento Bomem MB102.
4.4.2 Cromatografia CG-FID
A composição dos hidrocarbonetos foi realizada em um equipamento CG-2010 da Shimadzu equipado com uma coluna de polidimetilsiloxano, modelo CBPI PONA – M50-042 (50 m, 0,42 μm, 0,15 mm), e detector de ionização de chama (FID). Para tanto, os seguintes parâmetros foram utilizados: temperatura de injeção de 250 ºC, razão de split igual a 200, fluxo na coluna de 0,50 mL H2/min, aquecimento da coluna de 35 a
250 ºC, a 2 °C/min., mantendo a 250 °C por 30 min.
4.4.3 Índice de Acidez
O índice de acidez foi determinado por titulação ácido-base, utilizando como titulante uma solução metanólica de hidróxido de potássio 0,1mol/L, de acordo com o método da AOCS Cd3d63. A titulação foi realizada em triplicata, colocando alíquotas de 1g de combustível e diluindo em uma solução de 10g de uma mistura de 1:1 de tolueno e isopropanol, como indicador foi utilizado a fenolftaleína. A titulação foi realizada com agitação até observar o ponto de viragem.
A partir do volume de titulante gasto foi determinado o índice de acidez utilizando a Equação 4.1.
I A= V.C.56.1/m (4.1)
Onde V é o volume de titulante gasto; C é a concentração do titulante e m é a massa de combustível.
4.5ANÁLISES FÍSICO-QUÍMICAS DOS PRODUTOS OBTIDOS
4.5.1 Destilação Atmosférica
A destilação atmosférica das amostras foi realizada em um destilador automático Hezorg modelo HDA 627. No procedimento 100 mL da fração na faixa referente ao diesel foram submetidos à destilação segundo a norma ASTM D86. Este processo forneceu as temperaturas de destilação e registrou os dados no computador.
4.5.2 Viscosidade Cinemática
A viscosidade cinemática foi determinada utilizando um viscosímetro TANAKA, modelo AKV-202 Auto Kinematic Viscosity, onde o combustível foi escoado através de um capilar. A viscosidade é determinada através do produto do tempo de escoamento pela constante do capilar segundo a norma ASTM D445.
4.5.3 Ponto de Fulgor
O ponto de fulgor das amostras foi determinado em um equipamento manual
4.5.4 Densidade
As amostras foram mantidas as temperaturas de 15 e 20 °C segundo a norma ASTM D4052 com o intuito de determinassem suas densidades nessas temperaturas foi utilizado um densímetro digital de bancada da Mettler Toledo, modelo DE40. O procedimento foi realizado em triplicata para cada amostra e o resultado foi obtido diretamente no seu visor.
4.5.5 Índice de Cetano
O índice de cetano (IC) da amostra foi determinado por meio da Equação 4.3, de acordo com a norma ASTM D 4737, fazendo-se uso das temperaturas de destilação (10, 50 e 90 %) e da massa específica da amostra a 15 °C.
IC= 45,2 + (0,0892)(T10N) + [0,131 +(0,901)(B)[T50N]+[0,0523(0,420)(B)][T90N]
+(0,00049)[(T10N)2 - (T90N)2]+(107)(B) + (60)(B)2 (4.3)
Onde:
IC = Índice de cetano.
T10 = Temperatura a 10 % de líquido destilado, em °C, determinada pelo método certificado
ASTM D 86 e corrigido para pressão barométrica padrão, T10N = T10 – 215. T50 = Temperatura a 50 % de líquido destilado, em °C, determinada pelo método certificado
ASTM D 86 e corrigido para pressão barométrica padrão, T50N = T50 – 260. T90 = Temperatura a 90 % de líquido destilado, em °C, determinada pelo método certificado
5 RESULTADOS E DISCUSSÃO
5.1 DIFRAÇÃO DE RAIOS X (DRX)
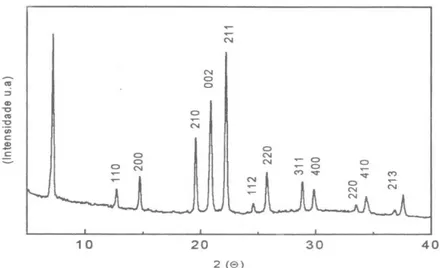
A difração de raios X com reflexões características da topologia do SAPO-5 na região 2θ de 5 a 45°, marcado por uma combinação de picos estreitos, tem semelhante à topologia do ALPO-5 , consistindo em poros unidimensionais com canais cilíndricos ligados por anéis de 12 membros, com diâmetro de 0,8 nm. Estes dados indicam que o método de síntese empregado neste trabalho foi efetivo para a produção do silicoaluminofosfato. (Figura 5.1)
0 10 20 30 40 50
0 200 400 600 800 1000 1200 In te n si d a d e 2 Theta SAPO-5 (110) (200) (210) (002) (211) (112) (220) (311) (400) (220) (410) (213)
Figura 5.1- Difratograma do SAPO-5 calcinado
No difratograma pode ser visto que os picos apresentam alta intensidade, isto é um indício de alta cristalinidade, que foi calculado pelo DRX por medida da intensidade dos picos e foi observada uma ótima cristalinidade.
5.2 MICROSCOPIA ELETRÔNICA DE VARREDURA
A microscopia eletrônica de varredura é uma técnica que permite a caracterização da morfologia dos materiais nanoestruturados sintetizados. As micrografias da amostra do material silicoaluminofosfato estão apresentadas na Figura 5.2.
Figura 5.2- Micrografias Eletrônicas de Varredura da amostra de SAPO-5
As micrografias eletrônicas de varreduras da peneira molecular SAPO-5 são observados na Figura 5.2, a partir destas é possível observar alta cristalinidade da peneira molecular bem como sua porosidade, geralmente os silicoalumino fosfato apresentam cristais com simetria ortorrômbica (HÖCHTL et al., 2001). Notou-se que, a morfologia da amostra apresenta formas esféricas bem definidas, mostrando um aspecto poroso visível. Esta morfologia é ideal, pois um maior número de sítios ativos estão disponíveis para a reação de craqueamento.
5.3 DETERMINAÇÃO DA ÁREA ESPECÍFICA
As isotermas de adsorção e dessorção de nitrogênio estão apresentadas na Figura 5.3 e pode-se observar que a isoterma da amostra de SAPO-5 é do tipo IV que é característica de materiais mesoporosos semelhantes aos encontrados por (UTCHARIYAJIT e
WONGKASEMMJIT, 2010), observa-se também histerese, fenômeno que é associado à
condensação e evaporação capilar de gases nos poros. A área superficial do material microporoso foi relativamente alta para o padrão do silicoaluminofosfato, sendo esta de 306m2/g que foi determinada pela isoterma de adsorção de Nitrogênio a -196 °C pelo método BET na faixa de P/P0 de 0,05 a 0,3. A vantagem de se ter uma área elevada é uma alta densidade de sítios ácidos.
0,0 0,2 0,4 0,6 0,8 1,0
80 100 120 140 V o lu m e ad so rv id o ( m L g -1 ) P/P0 SAPO-5
5.4 ANÁLISE TERMOGRAVIMÉTRICA
A calcinação é uma etapa muito importante para obtenção de silicoaluminofosfato de alta qualidade, pois nesta etapa ocorre a remoção do direcionador da estrutura, ou seja, a remoção de todo o material orgânico e preservação da estrutura do material bem ordenada foi utilizada a termogrametria, como pode ser visto na Figura 5.4, onde foram observadas três perdas de massa, sendo a primeira perda referente à água fisissorvida nos poros do material que ocorreu entre 30 e 150 °C, ou ainda, a saída de moléculas do direcionador a baixas temperaturas, o que explicaria a intensidade da perda; a segunda refere-se a moléculas do direcionador nos poros do material que ocorre entre 150 e 350 °C e a terceira perda de massa é devido a moléculas do direcionador protonadas que ocorreu na faixa de 350 a 550 °C, como foi relatado por (Brito et al., 2007).
0 200 400 600 800 1000
0 200 400 600 800 1000
65 70 75 80 85 90 95 100 105 110 Ma ssa (% )
Temperatura (oC)
TG DTG -0,40 -0,35 -0,30 -0,25 -0,20 -0,15 -0,10 -0,05 0,00 0,05
As curvas TG e DTG do SAPO-5 calcinado podem ser vistos na Figura 5.5. Como pode ser observado, ocorre apenas uma perda de massa, que está relacionada com a saída de água adsorvida, que acontece a aproximadamente a 150 °C.
0 100 200 300 400 500 600
-0,005 -0,005 -0,005 -0,004 -0,004 -0,003 -0,003 -0,002 -0,002 -0,001 -0,001 0,000 0,001
0 100 200 300 400 500 600
75 80 85 90 95 100 105 110 -0,005 -0,004 -0,003 -0,002 -0,001 0,000 Ma ssa (% )
Temperatura (oC)
DTG
TG
Figura 5.5- Curva Termogravimétrica da amostra de SAPO-5 calcinada
0 100 200 300 400 500 600 0 20 40 60 80 100 M as sa ( % ) Temperatura (°C) DTG TG -0.018 -0.016 -0.014 -0.012 -0.010 -0.008 -0.006 -0.004 -0.002 0.000 0.002
Figura 5.6- Curva Termogravimétrica do Óleo de Girassol
0 100 200 300 400 500 600
0 100 200 300 400 500 600
0 15 30 45 60 75 90 105 Ma ssa (% )
Temperatura (oC)
DTG TG -0,012 -0,010 -0,008 -0,006 -0,004 -0,002 0,000
5.5 DETERMINAÇÃO DA ACIDEZ SUPERFICIAL
A amostra foi previamente saturada com n-butilamina em seguida foi submetida a uma dessorção numa termobalança, utilizando aproximadamente 15 mg de SAPO-5 na análise, o
experimento foi realizado com o objetivo de quantificar a quantidade a massa de n-butilamina adsorvida nos sítios ácidos do material.
A termodessorção de n-butilamina tem sido utilizada para determinação relativa da distribuição de força dos sítios ácidos de diversos catalisadores, decompondo-se o perfil de
dessorção em sítios “arbitrariamente” definidos como fracos médios e fortes e medindo-se a
área dos picos, ao invés da utilização da temperatura máxima do pico. A área do perfil completo permite obter o número de sítios ácidos totais. A curva termogravimétrica da termodessorção de n-butilamina é mostrada na Figura 5.8 para os materiais microporosos.
0 200 400 600 800 1000 0 200 400 600 800 1000 75 80 85 90 95 100 105 DTG TG D T G (u .a .) Ma ssa (% )
Temperatura (oC)
I II III -0,08 -0,07 -0,06 -0,05 -0,04 -0,03 -0,02 -0,01 0,00
FIGURA5.8- Curvas TG/DTG mostrando a termodessorção de n-butilamina sobre a peneira molecular SAPO-5.
fortes, responsáveis pela maior parte das reações de craqueamento (KUEHNE et. al, 1997; NARAYANAN et. al, 1996).
TABELA 5.1- Faixas de temperatura e suas respectivas perdas de massa e densidade dos centros ácidos das amostras obtidas a partir das curvas TG/DTG, para a termodessorção de n-butilamina.
Amostra Faixa de Temperatura (°C) Perda de Massa (%)
Eventos I II III I II III
SAPO-5 105-390 390-535 535-710 11.4763 4.4902 4.2036
Centros Ácidos
SAPO-5 Fracos Médios Fortes
1.77mmol/g 0.647 mmol/g 0.606 mmol/g
Os dados referentes apresentados na Tabela 5.1 foram usados para calcular as quantidades de n-butilamina retida nos estágios I, II e III por massa de catalisador. Estes estágios são importantes por se tratarem das perdas referentes aos centros ácidos médios e fortes, responsáveis pela maior parte das reações de craqueamento (KUEHNE et al., 1997; NARAYANAN et al., 1996).
Pode-se observar uma acidez moderada e forte, indício que a incorporação de silício ocorreu com sucesso e que esta teve a função de formar unidades Si-(OH)-Al, e em menor quantidade nas ilhas de silício, no qual os átomos de Al se encontram na interface entre elas.
5.6 REAÇÕES DE CRAQUEAMENTO TERMOCATALITICO DO ÓLEO DE GIRASSOL
apresentava maior fluidez que a segunda. Isto se deve a ela ter sido coletada a temperaturas menores que 190 °C e como conseqüência apresentar compostos mais leves que os da segunda fração. A primeira fração deve ser rica em compostos na faixa da gasolina e do querosene e uma grande porção de compostos oxigenados leves, como mostra o elevado índice de acidez desta fração próximo de 200 mg KOH/g líquido orgânico, mesmo quando realizado na presença dos catalisadores. Isto sugere a rápida destilação no início do craqueamento e a possível ausência do craqueamento secundário. Contudo o índice de acidez da segunda fração diminui significativamente, uma evidência a esse fato foi à desoxigenação do produto orgânico pelo catalisador. Em virtude da elevada acidez apresentada pela primeira fração, não foi possível obter as propriedades físico-química do mesmo.
A partir deste ponto os resultados mencionados no processo de craqueamento térmico e termocatalítico serão referente à segunda fração, aquela que apresenta menores valores de acidez próximo de 10mg KOH/g de líquido orgânico, mencionadas como líquido orgânico (LO).
Figura: 5.9- Imagem das duas frações líquidas coletadas no craqueamento termocatalítico do óleo de girassol sobre o SAPO-5 Primeira fração, à direita; segunda fração, à esquerda.
sugere um aumento nas reações de descarbonilação, outro aspecto observado é uma maior quantidade de líquido orgânico.
TABELA 5.2. Resultados quantitativos (% em massa) do craqueamento térmico e do craqueamento termocatalítico do óleo de girassol sobre as amostras sólidas de SAPO-5, nas proporções em massa de
0,5; 1 e 2% a 450 ºC.
Amostra LO Total LO 1ª Fração LO 2ª Fração Gases Água Resíduo
Térmico 75,643 42,328 33,315 14 1 9,783
SAPO-5 (0,5%)
76,544 42,323 34,221 8 2 13,181
SAPO-5 (1%)
74,123 38,714 35,286 11 2 12,459
SAPO-5 (2%)
76,756 37,413 39,343 7 3 13.345
Comparando os dados relativos aos processos realizados sobre as amostras de SAPO-5 com aqueles relativos ao craqueamento térmico, nota-se que estas amostras provocaram aumento na quantidade de líquido orgânico (LO) e diminuição na quantidade de gases (DANDIK et al., 1998), sugerindo uma maior atuação do craqueamento secundário. Este comportamento pode estar relacionado ao aumento do número de sítios ácidos, nestas amostras, como indicado pela caracterização das mesmas. Esta característica facilita a adsorção e posterior desoxigenação dos compostos oxigenados decorrentes do craqueamento primário, bem como as reações de ciclização, condensação e aromatização (TWAIQ et al., 2003a) cujos produtos podem constituir o líquido orgânico.
5.7 ESPECTROSCOPIA DE INFRAVERMELHO
Comparando os espectros do diesel oriundo do petróleo e do LO obtido a partir do craqueamento térmico com os espectros daqueles obtidos pelo processo termocatalítico, percebe-se que as bandas características de ácidos carboxílicos têm menor intensidade nas amostras obtidas pelo craqueamento termocatalítico. Fato que evidencia a ação desoxigenante dos catalisadores no craqueamento secundário do óleo. Fato que ressalta a acidez apresentada pelo catalisador e sua estrutura organizada. De acordo com (Williams e Horne, 1995) a ação desoxigenante está relacionada aos dois fatores citados anteriormente.
A intensidade da banda referente ao estiramento C = O, em 1715 cm-1, característico
de ácidos carboxílicos que é confirmado pelo estiramento de H-O acima de 3000 cm-1 até
próximo de 3500 cm-1, é menor no espectro de infravermelho do LO obtido sobre o sólido que aquela observada para semelhante banda no espectro do LO oriundo do craqueamento térmico, indicando uma menor ação desoxigenação dos ácidos graxos provenientes do craqueamento primário do óleo.
O espectro de FTIR pode observar os picos na região de 1750 cm-1 e na região entre 2800 a 3100 cm-1 indicando, respectivamente, as presenças carbônicas e estiramentos simétricos e assimétricos de (CH3 e CH2).
4000 3500 3000 2500 2000 1500 1000 500 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 In te ns id ad e (u a) Térmico SAPO 5 0.5% SAPO 5 1% SAPO 5 2% Diesel
Numero de Onda
Figura 5.10- Comparação entre os espectros obtidos a partir do craqueamento do óleo de girassol e do diesel oriundo do petróleo.
Dentre as diferentes razões de catalisador adicionadas ao óleo, àquela que apresenta menor absorção no infravermelho e menor índice de acidez, como pode ser observada na Figura 5.11 é a de 2% de SAPO-5.
0,0 0,5 1,0 1,5 2,0 0 10 20 30 40 50 60 70 80 A ci de z (m g de K O H / g d e li qu id o or ga ni co )
Porcentagem da Catalisador (%)
Figura 5.11- Acidezdo bicombustível por porcentagem de catalisador
5.8 CROMATOGRAFIA
Figura 5.12 Cromatogramas dos LO´s oriundos dos craqueamentos térmico (a), termocatalíticos, SAPO-5 0,5% (b), SAPO-5 1% (c) e SAPO-5 2% (d) do óleo de girassol sobre a peneira molecular
SAPO-5.
As intensidades dos picos referentes aos produtos de maior massa molar (maior tempo de retenção) são maiores nos cromatogramas dos LO´s obtidos a partir dos processos termocatalíticos , quando comparada à dos mesmos picos no cromatograma do LO oriundo do processo térmico, uma possível explicação para esse fato é a descarbonilação ou descarboxilação sem uma quebra anterior.
Este comportamento reflete a composição, em ácidos graxos, do óleo de girassol, rico em ácido linoléico, oléico (C18:1) e palmítico (C16:0). A partir do ácido linoléico, obtem-se hidrocarboneto C4, C5 e C8 a C14 por quebra das ligações C – C beta (β) à insaturação antes da desoxigenação, posterior mente a descarboxilação podem ser obtidos por desproporção C4 e C6, também podem ser obtidos hidrocarbonetos cíclicos e aromáticos a partir de fragmentos olefínicos formados na quebra da cadeia carbônica antes da desoxigenação do acido linoléico. Partindo do acido oléico (LUIZ, 2010), é possível se obter hidrocarbonetos C5, C7 e C9 a C14 por quebra das ligações C – C beta (β) à insaturação, antes da desoxigenação (VONGHIA et al., 1995; MAHER et al., 2007), seguida por reações radicalares de desproporção, fissão beta (β) e condensação entre radicais de iguais (dimerização) ou diferentes número de átomos de carbono. Também podem ser obtidos, por meio destas mesmas reações, C6 e C8 a C13, após a desoxigenação do fragmento oxigenado decorrente da quebra das ligações beta (β) à insaturação da cadeia carbônica do ácido oléico. Hidrocarbonetos cíclicos e aromáticos também podem ser obtidos a partir dos fragmentos olefínicos oriundos da citada cisão da cadeia carbônica do ácido oléico.
Ainda de acordo com os cromatogramas, o aumento das cadeias carbônicas foi mais evidenciado no craqueamento termocatalítico que no térmico, este fato pode se relacionar com a presença dos microporos e a maioria das reações acontecerem na superfície.
0 20 40 60 80 100 120 -20000 0 20000 40000 60000 80000 100000 120000 140000 160000 180000 In te n si d a d e (u .a .)
Tempo de Retençao (min)
Térmico SAPO-5 0,5% SAPO-5 1,0 % SAPO-5 2,0%
Figura 5.13 - Todos os Cromatogramas dos produtos do craqueamento do óleo de Girassol sobrepostos.
5.9PROPRIEDADES FÍSICO-QUÍMICAS DOS PRODUTOS
5.9.1 Destilação Atmosférica dos Produtos do Craqueamento do Óleo de Girassol.
0 20 40 60 80 100 120 0 50 100 150 200 250 300 350 400 450 T e mp e ra tu ra
Porcentagem de Destilado (%)
Diesel
SAPO-5 0,5% SAPO-5 1%
SAPO-5 2% Térmico
Figura 5.14- Curva de Destilação Atmosférica do Líquido Orgânico obtido e de uma amostra de Diesel Comercial
Um ponto interessante é que as temperaturas iniciais de destilação são maiores no craqueamento térmico que no, termocatalítico, isto ocorre porque o líquido orgânico produzido no craqueamento termocatalítico apresenta em sua maior parte hidrocarbonetos que possui forças intermoleculares fracas, já no LO oriundo do processo térmico apresenta um número alto de ácidos carboxílicos que apresenta pontes de hidrogênio sendo esta muito forte. Como podemos observar a Figura 5.15, apresenta a sobreposição de boa parte das duas curvas de destilação, como os dois combustíveis apresentam algumas semelhanças nas propriedades químicas, supõe que os dois apresentem semelhanças também na composição química pelo menos no intervalo que compreende de 40 a 100% da curva de destilação. No início da destilação nota-se que a curva do LO obtido com 1% de SAPO-5 fica abaixo da curva do diesel mineral, isto reflete a concentração de hidrocarbonetos na faixa de C5 a C9 na curva do LO, isto explica as baixas temperaturas no ponto de fulgor e o baixo índice de cetano.