Construção de mutantes em RNases em enterococcus
41
0
0
Texto
(2) Universidade de Lisboa Faculdade de Ciências Departamento de Biologia Vegetal. Construção de mutantes em RNases em Enterococcus. Tiago Maria de Carvalho da Costa Amado. Mestrado em Biologia Molecular e Genética. Dissertação orientada por: Dr. Paulo Emanuel Marujo (IBET/ITQB) Prof. Mário Almeida Santos (FCUL). 2008.
(3) Agradecimentos. Gostaria de agradecer em primeiro lugar à Doutora Fátima, por me ter recebido prontamente, ter dado a oportunidade de realizar este trabalho no seu laboratório e me ter orientado ao longo deste ano, apesar de o seu nome não constar na primeira página; ao Doutor Paulo por ter aceitado ser meu orientador externo e ter sempre "a porta aberta" para as minhas questões e a ambos um sincero obrigado por tudo o que me ensinaram. Aos meus colegas de laboratórios, à Neuza e Teresa por alegrarem os dias, à Tânia, Martas, Renata, Daniela e Frederic pelos concelhos, favores e ajudas e a todos em geral pela vossa simpatia e por me terem mostrado os "cantos à casa".. Agradeço ao Prof. Doutor Mário Santos por ter sido o meu orientador interno e ter feito a revisão da minha tese.. Agradeço também ao ITBQ e IBET por me terem proporcionado as condições de trabalho.. Por último gostaria de agradecer aos meus amigos, porque sim, e às pessoas para mim mais importantes e que enchem a minha vida de cor: os meus pais Ana e Zé, a minha irmã Marta e a minha mais-que-tudo Rita. Um obrigado sentido e carinhoso para vocês.. i.
(4) Sumário. A expressão génica pode ser regulada a diferentes níveis. Nos últimos anos têm surgido imensos exemplos de regulação pós-transcricional quer a nível do processamento, quer da degradação (estabilidade) do RNA, a qual permite uma adaptação rápida a alterações no meio extracelular. No processo de invasão do hospedeiro, as bactérias patogénicas atravessam uma série de condições físico-químicas muito distintas a que têm de fazer frente e se adaptar. Neste contexto, na adaptação a condições de stress, estão envolvidos diversos mecanismos de regulação pós-transcricional. No entanto, em enterococos, bactérias Gram-positivas presentes em diversos ambientes e que tem emergido nos últimos anos como causadores de infecções nosocomiais graves, não foi ainda estudada a função deste tipo de regulação em geral e qual a sua relação com a patogenicidade. Assim, este trabalho teve como objectivos a construção de mutantes em ribonucleases, enzimas cruciais na regulação pós-transcricional, nomeadamente, em duas exoribonucleases, PNPase e RNase R, e em duas endoribonucleases, RNase III e RNase J1. O trabalho efectuado, devido à diversidade das dificuldades encontradas com cada um dos mutantes, abrangeu uma grande gama de técnicas de biologia molecular e de genética, quer em Escherichia coli, quer em Enterococcus faecalis. Assim, o 'mutante' da RNase III encontra-se na última etapa do protocolo, enquanto os 'mutantes' das outras RNases se encontram na fase de confirmação das construções a utilizar para integração em enterococos (PNPase e RNase R) ou ainda na primeira clonagem (RNase J1). Os resultados obtidos até este momento, parecem indicar que a RNase III é essencial em enterococos, tal como acontece em Bacillus subtilis; no entanto, estudos adicionais serão necessários para confirmação deste resultado.. Palavras-chave: Enterococcus, ribonucleases, mutantes, OE-PCR. ii.
(5) Abstract. Gene expression can be regulated at different levels. In the past few years several examples of post-transcriptional regulation have arise regarding the processing and degradation (stability) of RNA molecules, which allows a rapid adaptation to changing environments. When invading the host, pathogenic bacteria have to go through several and very distinct extracellular conditions to which they have to adapt in order to survive. In this adaptation to foreign stress causing factors are involved various mechanisms of post-transcriptional regulation. However in enterococci, Gram-positive cocci present in a large variety of habitats and that have become one of the leading cause of nosocomial infections, the function of this kind of regulation and its relation with pathogenicity has not been studied. Thus the goal of this work was the creation of bacteria mutants on ribonucleases namely on the exoribonucleases PNPase and RNase R genes and on the endoribonucleases RNase III and RNase J1. These RNases are enzymes of vital importance on the post-transcriptional regulation processes. Due to the diverse difficulties encountered during this work, it came to involve a great variety of molecular and genetic techniques in both Escherichia coli and Enterococcus faecalis. At the moment the RNase III 'mutant' is at the final stage of the protocol while the other RNases 'mutants' are still at the construction confirmation stage before transforming enterococci (PNPase and RNase R) or still at the first cloning step (RNase J1). The results obtain so far seem to point RNase III as essencial, similarly to what is described in Bacillus subtilis. Nevertheless further studies will be necessary to prove this hypothesis.. Keywords: Enterococcus, ribonucleases, mutants, OE-PCR. iii.
(6) Lista de abreviaturas. Amp - Ampicilina B. subtilis - Bacillus subtilis DNA - 'deoxyribonucleic acid' dNTPs - 'deoxyribonucleotide triphosphate' dsRNA - 'double-stranded ribonucleic acid' E. - Enterococcus E. coli - Escherichia coli Ery - Eritromicina FCUL - Faculdade de Ciências da Universidade de Lisboa IBET - Instituto de Biologia Experimental e Tecnológica ITQB - Instituto de Tecnologia Química e Biológica kb - kilo pares de base LA - Luria-Bertani agar LB - Luria-Bertani broth M - marcador de massa molecular MCS - 'multiple cloning site' MIC - 'minimum inhibitory concentration' mRNA - 'messenger ribonucleic acid' ncRNA - 'non-coding ribonucleic acid' nt - nucleótidos OE-PCR - 'overlap extension by polymerase chain reaction' ORF - 'open reading frame' pb - pares de base PCR - 'polymerase chain reaction' pGh 9 - pGhost 9 PNPase - 'polynucleotide phosphorylase' pSK+ - pBluescript SK+ RNA - 'ribonucleic acid' RNase - ribonuclease rRNA - 'ribosomal ribonucleic acid' S. - Staphylococcus Ta - Temperatura de 'annealing' tRNA - 'transfer ribonucleic acid' V583 - Enterococcus faecalis V583 V583 ∆ABC - Enterococcus faecalis V583 ∆ABC iv.
(7) Índice Geral. 1. Introdução. 1. 2. Contextualização e Objectivos. 6. 3. Materiais. 7. 4. Metodologia e Discussão. 9. 5. Perspectivas futuras. 24. 6. Bibliografia. 25. 7. Anexos. 29. v.
(8) Índice de figuras. Figura 1 | Ribonucleases identificadas em B. subtilis e E. coli.. Figura 2 | Representação esquemática do protocolo para a. 4 11. obtenção de mutantes em Enterococcus faecalis utilizando o pGhost 9. Figura 3 | Conceito de 'Overlap Entension by Polymerase. 12. Chain Reaction' (OE-PCR). Figura 4 |. Quantificação dos fragmentos resultantes dos. 15. PCRs I e II em gel de agarose. Figura 5 | Obtenção dos fragmentos híbridos.. 15. Figura 6 | Combinações das hibridações no passo de 'Overlap Entension'.. 16. Figura 7 | PCR I+II com baixo rendimento.. 17. Figura 8 | PCR I+II com o rendimento melhorado.. 17. Figura 9 | Obtenção do fragmento híbrido correspondente à RNase III.. 18. Figura 10 | Variabilidade nos tamanhos dos fragmentos da. 18. PNPase e RNase R clonados em pSK+. Figura 11 | Digestão dupla do pSAVE 6.. 20. Figura 12 | Obtenção de transformantes de E. coli com o pSAVE 8.. 20. Figura 13 | Obtenção de transformantes de E. faecalis com o pSAVE 8.. 20. Figura 14 | Representação esquemática dos acontecimentos. 22. de integração e excisão. Figura 15 | Obtenção de um só tipo de integração do. 23. pSAVE 8 no cromossoma de E. faecalis. Figura 16 | Clones em que ocorreu excisão.. 23. Figura Suplementar 1 | Marcadores de massa molecular.. 32. vi.
(9) Índice de tabelas. Tabela Suplementar 1 | Estirpes e plasmídios utilizados neste trabalho.. 29. Tabela Suplementar 2 | 'Primers' utilizados neste trabalho.. 30. Tabela Suplementar 3 | Dimensões dos fragmentos esperadas ao longo. 31. do protocolo.. vii.
(10) Construção de mutantes em RNases em Enterococcus. 1. Introdução. O termo francês “entérocoque” foi utilizado pela primeira vez numa publicação científica em 1899 por Thiercelin para descrever uma nova bactéria Gram-positiva de origem intestinal capaz de formar pares e pequenas cadeias (1). Em 1906, Andrewes e Horna introduziram este microorganismo no género Streptococcus com o nome de Streptococcus faecalis (2). Um segundo microorganismo de origem fecal com características semelhantes a S. faecalis foi isolado e denominado S. faecium por Orla-Jensen no ano de 1919 (3). Em 1933, Lancefield cria um sistema de classificação serológica de Streptococcus com base na composição dos antigénios bacterianos, onde os enterococos são inseridos no grupo D. Posteriormente Sherman, em 1937, salienta o facto do termo enterococos ser utilizado com diferentes significados, desde qualquer estreptococos de origem fecal até organismos idênticos a S. faecalis (4). Cria então um novo sistema para a classificação dos estreptococos que os divide em quatro grupos: piogénicos, lácticos, 'viridans' (ou orais) e enterococos. Em 1970 Kalina propõe que os enterococos deveriam ser transferidos para um novo género: Enterococcus (5). No entanto, isso só vem a acontecer em 1984 após um estudo realizado por Schleifer e Kilpper-Balz em que a hibridação de DNA-DNA e DNArRNA confirmam uma maior distância dos enterococos em relação aos estreptococos, fixando assim a sua localização taxonómica em: Bacteria; Firmicutes; Bacilli; Lactobacillales; Enterococcaceae; Enterococcus (6). Nessa altura apenas faziam parte do novo género E. faecalis e E. faecium mas desde então o número de espécies pertencendo ao género tem vindo a aumentar. Actualmente pertencem ao género Enterococcus quarenta espécies (taxonomicamente. validadas). sendo. E.. faecalis. a. espécie. tipo. (http://www.dsmz.de/microorganisms/bacterial_nomenclature_info.php?genus=Enterococcus &show_all_details=1 acedido a 07/09/2008).. Os enterococos caracterizam-se por serem: bactérias Gram-positivas em forma de cocos, podendo aparecer individualmente, aos pares ou sob a forma de pequenas cadeias, não formarem esporos e terem um conteúdo baixo em G+C no genoma (entre os 37 e 45 mol%). Metabolicamente são: anaeróbios facultativos, catalase e oxidase negativos, realizam fermentação. ácido. láctica,. possuem. resistência. intrínseca. a. vários. antibióticos. (e.g. β-lactâmicos, cefaloesporinas, sulfonamidas) e são capazes de tolerar uma vasta gama de condições de crescimento, nomeadamente a presença de 40% de sais biliares, 6,5% de NaCl, pH entre 4,6 e 9,6 e temperaturas entre os 10 e os 45 ºC, sendo ainda capazes de sobreviver a 60 ºC durante 30 minutos (7, 8). Estas bactérias são parte natural da microbiota intestinal de humanos e animais mas são também frequentemente encontradas em plantas,. 1.
(11) Construção de mutantes em RNases em Enterococcus. alimentos, solo e águas superficiais, provavelmente devido à contaminação fecal e à tolerância intrínseca a condições ambientais adversas (9). Este grupo de bactérias ácido lácticas é importante não só na microbiologia ambiental como também na microbiologia clínica e alimentar. Os enterococos têm um papel benéfico importante, considerado por muitos essencial, na produção de muitos queijos tradicionais, inclusive queijos portugueses, o que se deve provavelmente à sua capacidade proteolítica, lipolítica e de metabolizar o citrato (7). Estão igualmente envolvidos na fermentação de vários outros produtos tradicionais, especialmente de origem mediterrânea, como salsichas e azeitonas (10). Em alguns países estas bactérias são ainda utilizadas na constituição de probióticos aplicados em produtos alimentares, como iogurtes e leites, e em produtos farmacêuticos (10). A importância do uso de estirpes bacterianas como probióticos deve-se às suas diversas acções benéficas, nomeadamente, a inibição de microrganismos patogénicos, o reforço das barreiras mucosas, a estimulação do sistema imunitário e a diminuição dos níveis de colesterol (8). Apesar destes seus papéis benéficos, os enterococos estão também associados ao deterioramento de alimentos, em especial carnes, e mais preocupantemente, a uma enorme variedade de infecções tanto em animais como no ser humano (8). Estes patogénios oportunistas são capazes de provocar infecções ao nível da corrente sanguínea, endocárdio, sistema urinário, sistema nervoso central, abdómen, feridas e queimaduras, entre outros (11). O número de enterococos resistentes a antibióticos, com particular destaque para a vancomicina, tem vindo a aumentar de uma forma preocupante, o que se deve à eficiente capacidade dos enterococos para transferirem e incorporarem material genético e também à incorrecta utilização dos antibióticos (12). Existem várias características envolvidas na patogenicidade dos enterococos (7, 11): −. Síntese de citolisina (Cyl; proteína com actividade simultaneamente hemolítica e bacteriocida);. −. Síntese de substância de agregação (Agg; envolvida na conjugação bacteriana e induzida por feromonas);. −. Síntese de uma proteína de superfície extracelular (Esp; envolvida na adesão e evasão à resposta imune do hospedeiro);. −. Síntese de uma endoprotease (GelE; envolvida na degradação de várias proteínas bioactivas);. −. Síntese de outras enzimas hidrolíticas (e.g. hialuronidase e desoxirribonuclease);. −. Formação de biofilmes;. −. Presença de vários mecanismos de resistência a antibióticos, quer intrínseca (e.g. β-lactâmicos, cefaloesporinas, sulfonamidas) quer adquirida (e.g. ampicilina, vancomicina, eritromicina).. 2.
(12) Construção de mutantes em RNases em Enterococcus. Estas características contribuíram para que, nos últimos anos, os enterococos se tornassem numa das principais causas de infecção nosocomial em todo o mundo. Estas infecções são provocadas em cerca de 80% dos casos pela espécie E. faecalis, sendo a maioria dos restantes 20% causados pela espécie E. faecium (13). Devido ao forte impacto dos enterococos na saúde humana, tem-se vindo a discutir com alguma preocupação a segurança da utilização dos enterococos na fermentação e em produtos probióticos (8). Determinar se há um risco geral ou se o risco se aplica apenas a determinadas espécies ou estirpes e elaborar novas estratégias para combater as infecções são alguns dos objectivos que se tem procurado alcançar. Os enterococos assumem assim uma posição paradoxal pois são benéficos na fermentação de diversos alimentos e na acção como probióticos, mas acarretam uma potencial patogenicidade (14).. Uma das primeiras estirpes de enterococos resistentes à vancomicina foi isolada em 1989 de uma amostra clínica e denominada E. faecalis V583 e conta com o seu cromossoma e plasmídios sequenciados desde 2003 (15, 16). Esta estirpe tem sido alvo de vários estudos e descobertas, nomeadamente a caracterização de uma ilha de patogenicidade (que contém alguns dos factores de virulência acima mencionados), análise do transcriptoma e a descrição de sistemas de dois componentes envolvidos em vários mecanismos celulares (17-20). Foram identificados e estudados até à data dezoito sistemas de dois componentes no genoma de Enterococcus faecalis, alguns dos quais representam potenciais alvos terapêuticos (18, 21, 22). Entre estes encontram-se um sistema envolvido na resistência à vancomicina (VanSB-VanRB) e um sistema envolvido na formação de biofilmes e produção de factores de virulência (FsrC-FsrA) (23-25). Os sistemas de dois componentes são um mecanismo muito utilizado pelos organismos procariotas como forma de responder a sinais ou condições exteriores. Levam tipicamente à regulação da expressão génica, por exemplo, de genes relacionados com o metabolismo ou a virulência. Apesar de nos últimos anos se terem vindo a descobrir muitos genes envolvidos na virulência de enterococos, poucos estudos foram ainda feitos para explicar a sua expressão diferencial e muito pouco se sabe acerca dos mecanismos envolvidos na regulação da sua expressão (26-31).. A expressão de um gene até ao produto final, atravessa vários estádios, em que cada um deles oferece uma oportunidade de regulação. Embora durante muito tempo se tenha pensado que a iniciação da transcrição seria a etapa mais importante no controlo da expressão génica, têm surgido nos últimos anos diversos exemplos importantes de regulação ao nível pós-transcricional (32-34). São estes últimos, por vezes bastante complexos, que comandam o destino e a funcionalidade de uma molécula de RNA através do controlo da sua maturação, estrutura e degradação (estabilidade). Neste tipo de 3.
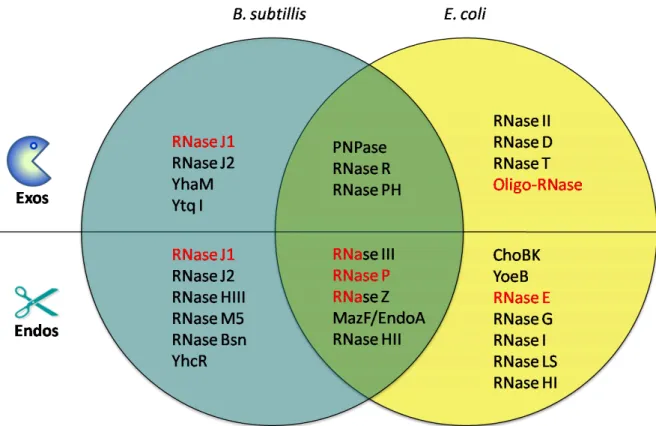
(13) Construção de mutantes em RNases em Enterococcus. regulação estão envolvidas diversas proteínas bem como outras moléculas de RNA denominados RNAs reguladores ou não codificantes (ncRNAs). Entre as várias proteínas envolvidas na regulação pós-transcricional estão as ribonucleases (RNases) e as 'chaperones' de RNA (e.g. Hfq e stpA).. As ribonucleases são enzimas capazes de processar e degradar moléculas de RNA, sendo um dos principais e mais importantes intervenientes na regulação pós-transcricional. As RNases exibem diferentes estruturas e modos de acção e podem ser agrupadas em duas classes consoante o forma como clivam o RNA: Endoribonucleases e Exoribonucleases. As primeiras clivam o RNA internamente (e.g. RNase III, RNase P, RNase J1) enquanto que as segundas clivam o RNA a partir de uma das extremidades, nucleótido a nucleótido (e.g. PNPase, RNase R, RNase J1) (35, 36). Em Escherichia coli e em Bacillus subtilis, bactérias modelo nos estudos de degradação e processamento de RNA, estão identificadas vinte e sete ribonucleases (36, 37) (Figura 1).. Figura 1 | Ribonucleases identificadas em Bacillus subtilis e em Escherichia coli. Algumas das exoribonucleases (em cima) e endoribonucleases (em baixo) são partilhadas por B. subtilis e E. coli (centro), enquanto as restantes foram apenas identificadas numa das espécies (esquerda ou direita respectivamente). A vermelho estão representadas as ribonucleases essenciais.. 4.
(14) Construção de mutantes em RNases em Enterococcus. O papel fundamental das RNases para o correcto funcionamento da célula advém do facto de, para além de todo o RNA ter de ser, mais tarde ou mais cedo, degradado a nucleótidos para permitir a reciclagem dos mesmos, estas proteínas actuarem ao nível dos vários tipos de RNA: tRNA, rRNA, mRNAs e ncRNAs. Assim as RNases acabam por estar directa ou indirectamente envolvidas em todos os processos de que estes RNAs fazem parte, nomeadamente na formação das subunidades ribossomais (rRNA), na tradução (tRNA e mRNAs) e nos variados processos a que estão associados os ncRNAs (38-42). A identificação de novas moléculas pertencentes a este último grupo, ncRNAs, e a determinação das estruturas dos RNAs, tem sido feita a um ritmo alucinante nos últimos anos. Embora a função de muitos destes ncRNAs seja ainda desconhecida, alguns deles comportam-se como reguladores chave em respostas adaptativas face a condições e sinais do meio exterior, como é o caso do stress imposto por iões, produção de luminiscência, resistência a antibióticos e controlo da virulência (25, 43, 44). Como exemplos de RNAs reguladores envolvidos no controlo da virulência bacteriana encontram-se o RNAIII em Staphylococcus aureus, o RsmB' em Erwinia carotovora e o RNAα em Vibrio anguillarum, entre muitos outros (32, 43). Assim, tendo em conta o referido anteriormente é natural que as ribonucleases estejam também envolvidas no controlo da virulência. Na realidade têm sido publicados diversos artigos que relatam o envolvimento das RNases na virulência bacteriana. São exemplos disso a RNase R, necessária para a expressão da virulência em Shigella flexneri, Escherichia coli e Aeromonas hydrophila; a PNPase, que regula a virulência e persistência de Salmonella enterica e a RNAse III envolvida na regulação da virulência em Staphylococcus aureus (45-49). Um profundo conhecimento dos mecanismos e do modo como controlam a expressão de factores de virulência poderá abrir novas perspectivas na quimioterapia antibacteriana.. 5.
(15) Construção de mutantes em RNases em Enterococcus. 2. Contextualização e objectivos. O laboratório onde este trabalho foi realizado, dedica-se ao estudo dos mecanismos de resposta a diferentes stresses (biocidas, iões metálicos e antibióticos), da resistência a antibióticos em enterococos e, de uma forma mais global, ao estudo da virulência e patogenicidade destas bactérias (50-54). Dado não existir, até à data, nenhum estudo sobre a regulação pós-transcricional, bem como a função das RNases na virulência de enterococos, decidiu-se levar a cabo a construção de mutantes em ribonucleases. Estes mutantes constituirão ferramentas essenciais para estudar os efeitos dessas enzimas ao nível do metabolismo global da célula e, em particular, de factores de virulência específicos, da patogenicidade e da capacidade de resistência a antibióticos (11, 32, 55). Neste contexto, o objectivo deste trabalho foi a construção de mutantes em RNases, bem como a familiarização com técnicas de biologia molecular e de genética associadas à construção de mutantes em Enterococcus faecalis. Para este trabalho foram escolhidas quatro RNases: PNPase, RNase R, RNase III e RNase J1.. 6.
(16) Construção de mutantes em RNases em Enterococcus. 3. Materiais. Estirpes e plasmídios - As estirpes bacterianas e os plasmídios utilizados neste trabalho estão listados na Tabela 1.. Conservação de estirpes - E. coli (-80 ºC): 1 ml de cultura bacteriana + 0,2 ml de glicerol 80%. E. faecalis (-20 ºC): 0,8 ml de cultura bacteriana + 0,2 ml de glicerol 80%. E. faecalis (-80 ºC): 0,5 ml de cultura bacteriana + 0,5 ml de glicerol 40%.. Condições de crescimento - Para o crescimento de E. coli foram utilizados os meios Luria-Bertani broth (LB) e Luria-Bertani agar (LA) suplementados, quando apropriado, com antibiótico: [ampicilina] = 100 µg/ml; [eritromicina] = 150 µg/ml. Em alguns casos, em que estava presente o gene repórter lacZ, o meio foi também suplementado com X-GAL (80 µg/ml) e IPTG (0,5 mM), adquiridos à Apollo Scientific e à Promega, respectivamente. Células de E. faecalis foram crescidas em M17 (56) suplementado com glucose (0,5%) e, quando apropriado, com antibiótico: [eritromicina] = 30 µg/ml. A incubação das bactérias foi feita a 27, 37 ou 42 ºC consoante as características das estirpes e o protocolo.. Enzimas, reagentes e materiais de biologia molecular - As enzimas de restrição (PstI, SalI, SmaI, SacI) e a 'T4 DNA ligase' utilizadas foram adquiridas à New England Biolabs, sendo as restrições e ligações feitas de acordo com as instruções do fabricante. Para as reacções de PCR a partir de colónias foi utilizada a MasterMix adquirida à 5Prime (62,5 U/ml 'Taq DNA Polymerase'; 125 mM KCl, ®-CA360 0,5%; 500 µM de cada dNTP; 75 mM Tris-HCl pH 8,3; 3,75 mM Mg(OAc)2 ; 0,25% Igepal e estabilizadores). Para as restantes reacções de PCR foi utilizada a 'VentR DNA polymerase' e respectivo tampão, adquiridos à New England Biolabs, e os dNTPs (dATP, dTTP, dCTP e dGTP) adquiridos individualmente à Promega. Todos os 'primers' utilizados (Tabela Suplementar 2) foram desenhados neste estudo tendo por base o genoma de Enterococcus faecalis V583 publicado (GenBank, número de acesso AE016830) e as sequências dos vectores utilizados: pBluescript SK+ (Stratagene) e pGhost 9 (UBLO, Jouy-en-Jousas). Os 'primers' foram encomendados à MWG. A água utilizada foi sempre ultra-pura. As reacções de PCR foram realizadas no aparelho 'T3000 Thermocycler' da Biometra. Os serviços de sequenciação foram realizados pela BaseClear. O software utilizado para a análise das sequências foi o Vector NTI da Invitrogen. Os marcadores de massa molecular utilizados na migração do DNA em géis de agarose foram o 1 Kb Plus DNA Ladder (Invitrogen) e o GeneRule Express (Fermentas) (Figura Suplementar 1).. 7.
(17) Construção de mutantes em RNases em Enterococcus. Extracção e purificação de DNA - A extracção de DNA plasmídico, a purificação de produtos de PCR e de digestões e a extracção e purificação de DNA a partir de géis de agarose foi efectuada recorrendo a 'kits' da Qiagen e da Macherey-Nigel e de acordo com as instruções do fabricante.. Células electrocompetentes e electroporação - A preparação de células electrocompetentes de E. coli e de E. faecalis foi feita segundo os protocolos de Dower et al. (1988) e de Dunny et al. (1991), respectivamente. A electroporação, quer de E. coli quer de E. faecalis, foi realizada no 'Gene Pulser Xcell' da Bio-Rad seguindo as instruções do fabricante e utilizando o programa pré-definido 'Bacterial; 2. E. coli; Exponential decay; 25 µF; 200 ohm; 2,5 kV; 0,2 cm cuvette; 20-40 µl cell vol'.. 8.
(18) Construção de mutantes em RNases em Enterococcus. 4. Metodologia e Discussão A estirpe em que se pretendeu criar mutações foi a E. faecalis V583 ∆ABC. ∆ABC significa que esta estirpe não possui os três plasmídios que são característicos da estirpe V583 original (pTEF1, pTEF2, pTEF3), isolada em 1989 de uma amostra clínica e descrita como um dos primeiros isolados de enterococos resistentes à vancomicina (15). A estirpe V583 ∆ABC é sensível à eritromicina, uma vez que os genes responsáveis pela resistência à eritromicina em V583 se localizam no plasmídio pTEF1. Esta característica é importante na medida em que a marca de selecção do vector utilizado, o pGhost 9, na transformação desta estirpe é o gene ermC que confere resistência à eritromicina. A escolha desta estirpe deveu-se ao facto de ser a única pertencente ao género com o genoma totalmente sequenciado na altura do início do trabalho, o que permitiu, através da comparação com o genoma de B. subtilis, identificar os genes de ribonucleases presentes no seu genoma (16). Foram escolhidos três genes para se criarem mutantes de delecção: o gene pnpA (EF3064), o gene vacB (EF2617) e o gene rnc (EF3097), que codificam para a PNPase, RNase R e RNase III, respectivamente. Foi também escolhido o gene da RNase J1 (EF2924) mas para se inserir um codão de terminação prematuro através de uma mutação pontual. Escolheram-se estas RNases por abrangerem mecanismos de degradação distintos.. A PNPase, 'polynucleotide phosphorylase', é uma exoribonuclease que remove os nucleótidos de uma molécula de RNA de um modo processivo e no sentido 3' → 5'. A reacção é fosforolítica e dependente de fosfato, uma vez que para haver a quebra de uma ligação fosfodiester e se libertar um nucleótido 5' - difosfato é necessário a presença de uma molécula de fosfato inorgânico (Pi). A PNPase é uma 'cold shock protein' sendo portanto importante para a célula fazer face a temperaturas baixas. A inactivação do seu gene provoca um fenótipo sensível a baixas temperaturas, nomeadamente com paragem do crescimento por volta dos 16 ºC, aumento do tempo de meia-vida (ou semi-vida) do mRNA total, aumento da susceptibilidade à tetraciclina e algumas alterações da morfologia celular (57-59). Em B. subtilis, a PNPase é tida como uma das principais ribonucleases 3' → 5' envolvidas na degradação do RNA, embora a RNase R também tenha um papel muito importante, especialmente no que diz respeito à degradação de RNAs com estruturas secundárias mais estáveis, como as presentes nos tRNAs e rRNAs, as quais a PNPase tem dificuldades em ultrapassar. A RNAse R é, à semelhança da PNPase, uma exoribonuclease 3' → 5' e uma 'cold shock protein'. A degradação do RNA é hidrolítica, feita de um modo processivo e cujo produto final são pequenos oligonucleótidos (58, 60).. 9.
(19) Construção de mutantes em RNases em Enterococcus. Estas duas ribonucleases foram seleccionadas devido ao importante papel que assumem na degradação e maturação de RNA em B. subtilis (organismo modelo no estudo de degradação de RNA em bactérias Gram-positivas) e por, como já referido na introdução, ambas já terem sido implicadas na regulação da virulência bacteriana (45-48).. A RNAse III é uma endoribonuclease que reconhece e cliva RNA em cadeia dupla (dsRNA) de uma forma hidrolítica. A família das RNases III é composta por três classes. A RNAse III bacteriana é a mais estudada e melhor caracterizada até à data e pertence à classe 1 (61). Foi escolhida devido ao facto de ter sido descrita como essencial em B. subtilis e de, juntamente com o RNAIII, regular a virulência em Staphylococcus aureus, cujo sistema de virulência, agr, apresenta grande semelhança com o sistema fsr em Enterococcus faecalis (49, 62, 63).. A RNase J1 foi seleccionada por ser uma RNase sem paralelo pois é simultaneamente uma endoribonuclease e uma exoribonuclease. Foi descoberta em B. subtilis onde está envolvida na maturação do rRNA; juntamente com a RNase J2 tem um impacto profundo na expressão génica e apresenta-se como essencial para a viabilidade celular. Proteínas ortólogas têm sido identificadas em diversos grupos procariotas (e.g. cianobactérias, alfa-proteobactérias e alguns grupos de arqueobactérias). A sua actividade endonucleolítica é comparada à da RNase E de E. coli e a sua actividade exonucleolítica de 5' → 3', única, pois até à data pensava-se que as bactérias apenas eram capazes de degradar exonucleoliticamente RNAs no sentido 3' → 5', faz com que a degradação de moléculas de RNA possa acontecer a partir de ambas as extremidades, quer 5' quer 3', à semelhança do que acontece nos eucariotas (36, 64-69).. A Figura 2 representa esquematicamente o protocolo seguido para a obtenção dos mutantes.. 10.
(20) Construção de mutantes em RNases em Enterococcus. Figura 2 | Representação esquemática do protocolo para a obtenção de mutantes em Enterococcus faecalis utilizando o pGhost 9. Adaptado de Heckman e Pease (2007).. * E. faecalis com pGhost 9 : fragmento mutado livre no citoplasma (transformantes). 'Choque térmico' 27 ºC 2h 42 ºC 2h. E. faecalis com pGhost 9 : fragmento mutado. pSK+. integrado no genoma (integrantes) E. coli. Passagens sucessivas em meio. Sequenciação. sem antibiótico. Identificação colónias sensíveis ao antibiótico. pGh 9. PCR. E. coli. E. faecalis com. E. faecalis com. gene selvagem. gene mutado. pGh 9. *. E. faecalis 11.
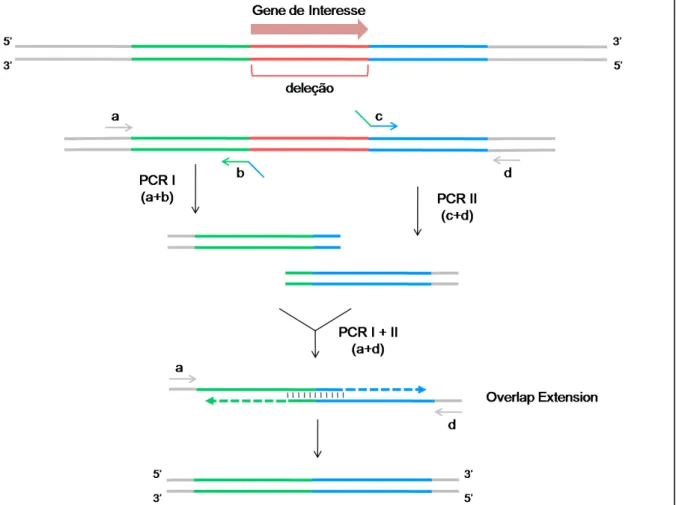
(21) Construção de mutantes em RNases em Enterococcus. O primeiro passo é a construção de um fragmento de DNA recombinante composto pela junção das duas zonas adjacentes ao gene de interesse. O objectivo final é a substituição da zona homóloga no cromossoma, contendo o gene selvagem, pela construção por nós criada, onde parte do gene não se encontra presente, obtendo-se assim um mutante de delecção. Para a construção deste fragmento recorreu-se à técnica de 'overlap extension by polymerase chain reaction' (OE-PCR). Esta técnica inovadora foi descrita pela primeira vez por Horton e colaboradores em 1989 e veio revolucionar a engenharia genética, dado que permite a fusão de duas ou mais moléculas de DNA numa só de um modo preciso, independente da sua sequência nucleotídica e sem o uso de enzimas de restrição ou de ligação (70-73). O fundamento da fusão de duas moléculas de DNA numa só ('splicing') utilizando o OE-PCR está esquematizado na Figura 3.. Figura 3 | Conceito de 'Overlap Entension by Polymerase Chain Reaction' (OE-PCR). Os 'primers' a e c são complementares à cadeia 3' → 5' e os 'primers' b e d são complementares à cadeia 5' → 3'. Os 'primers' a e d são denomidados de 'externos'. Os 'primers' b e c são complementares e denominados de 'internos'. A vermelho está representada a porção de DNA que se pretende eliminar.. 12.
(22) Construção de mutantes em RNases em Enterococcus. Os dois fragmentos a serem unidos são produzidos separadamente numa reacção de PCR (PCR I e PCR II). São utilizados 'primers' internos complementares, c e d, de modo a que os fragmentos. produzidos contenham. nas. extremidades. sequências complementares.. Seguidamente os produtos destes dois PCRs são misturados e utilizados num novo PCR (PCR I+II). No passo de 'annealing', as cadeias com a sequência complementar na sua extremidade 3' podem emparelhar e servir de 'primer' uma à outra, permitindo a extensão das cadeias levada a cabo pela DNA polimerase (Figura 3). A este passo dá-se o nome de 'overlap extension'. A molécula de DNA produzida desta forma pode ser seguidamente amplificada do modo exponencial característico do PCR, através dos 'primers' externos a e d. Estes 'primers' podem conter locais de restrição de modo a permitir a clonagem do fragmento obtido. Assim, o produto final deste PCR será uma molécula de DNA em que as duas sequências originais estão agora concatenadas.. No caso esquematizado na Figura 3, o OE-PCR é utilizado para fundir as duas regiões adjacentes, assinaladas a verde e a azul, a uma região de interesse, assinalada a vermelho, de modo a se obter um fragmento idêntico ao inicial mas sem a região de interesse. Foi com este intuito, o de criar uma delecção sítio-específica, que esta técnica foi utilizada, nomeadamente para a eliminação quase na totalidade da sequência do gene alvo (excepto para o caso da RNAse J onde se recorreu à criação de uma mutação pontual). No entanto esta técnica, mantendo sempre o mesmo fundamento, permite a obtenção de diferentes construções no que diz respeito à zona da fusão. Nesta região de justaposição das duas moléculas é possível realizar vários tipos de mutações sítio-específicas: inserções, substituições ou delecções de um ou vários nucleótidos. Esta técnica pode ser ainda utilizada para a remoção de intrões e para a criação de genes híbridos que vão permitir a criação de proteínas de fusão de interesse, por exemplo na área da imunologia e da biologia molecular e celular (70-72, 74, 75). É na escolha da sequência dos 'primers' internos (b e c) que se define qual das possibilidades referidas nos parágrafos anteriores é aplicada. É importante que estes PCRs sejam realizados utilizando uma DNA polimerase de alta fidelidade, i.e. com baixa taxa de erro, de modo a minimizar-se a probabilidade de introdução de mutações não pretendidas. A DNA polimerase utilizada deverá também ter a característica de não adicionar adeninas no final da polimerização (3'), de modo a que no passo de 'overlap extension' no PCR I+II não haja a introdução destas adeninas na sequência do fragmento final.. Neste trabalho recorreu-se à técnica de OE-PCR para se proceder à eliminação quase completa dos genes da PNPase (pnpA), RNase R (vacB) e RNase III (rnc) e à inserção de um codão de terminação prematuro no gene da RNase J1 (EF2924). Ao escolher a região a 13.
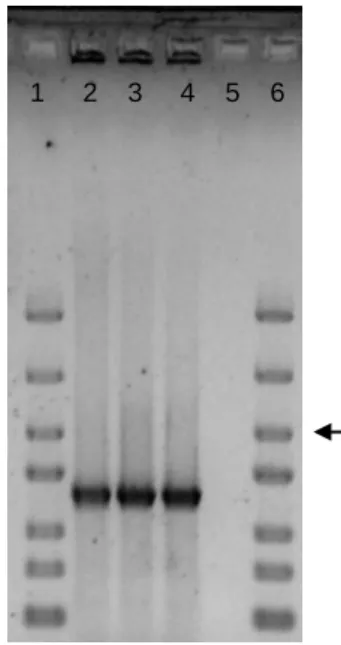
(23) Construção de mutantes em RNases em Enterococcus. eliminar, definida pela sequência dos 'primers' internos b e c, teve-se em conta a presença de outras ORFs e tentou-se ao máximo que as alterações introduzidas não interferissem com a sua expressão. No caso do gene pnpA a delecção feita deixou presentes os tripletos codificantes para os últimos 21 aminoácidos da extremidade carboxilo da proteína devido ao facto de na cadeia complementar e ainda dentro da zona codificante para a PNPase, existir um codão de terminação correspondente a uma outra ORF (EF3063). Na delecção do gene vacB mantiveram-se os nucleótidos correspondentes aos 27 aminoácidos da extremidade carboxilo, devido à sobreposição do seu codão de terminação com a sequência promotora da ORF EF2616, e aos 13 aminoácidos da extremidade amina, devido à sobreposição parcial do codão de iniciação com o codão de terminação da ORF EF2618. No que diz respeito ao gene rnc, e à semelhança do gene vacB, foram mantidos os nucleótidos equivalentes aos 16 aminoácidos da extremidade carboxilo devido a uma sobreposição do codão de terminação com a sequência promotora da ORF EF3096. No gene da RNase J1 foi introduzido um codão de terminação prematuro deixando os tripletos codificantes para os primeiros 57 aminoácidos devido à sobreposição com a região 3' da ORF EF2923. Na escolha da posição dos 'primers' externos a e d teve-se em consideração a sua sequência e, principalmente, a distância ao 'primer' interno correspondente, b e d, respectivamente. Esta distância deve ser aproximadamente 1000 pares de base de modo a permitir a recombinação com a sequência homóloga do cromossoma (Figura 2). A estes 'primers' foi adicionado um local de restrição compatível com ambos os vectores utilizados.. Na Tabela Suplementar 3 estão representados os tamanhos esperados dos fragmentos de DNA nas várias etapas do protocolo. Para facilitar a referência foi atribuída uma letra a cada RNase: A para a PNPase, B para a RNase R, C para a RNase III e D para a RNase J1.. Nos primeiros PCRs, PCR I e PCR II (Figura 3), não foram encontradas dificuldades em obter os fragmentos pretendidos. O DNA molde para cada um destes PCRs foi um fragmento amplificado por PCR a partir do DNA genómico de E. faecalis V583 ∆ABC e utilizando os 'primers' externos (a e d) respectivos a cada RNase (Tabela Suplementar 3). As moléculas obtidas nestes PCRs serviram de DNA molde para o PCR seguinte, o PCR I+II. No entanto aquando da realização deste último PCR, não se verificou a amplificação de nenhum fragmento com o tamanho esperado. Na tentativa de resolver o problema fizeramse variar algumas das condições que normalmente estão associadas à optimização da reacção de PCR:, nomeadamente a quantidade inicial de DNA molde, a temperatura de 'annealing' (Ta), o tempo das várias etapas da reacção: desnaturação, emparelhamento e extensão, o número de ciclos e a quantidade de DNA polimerase. Esta estratégia revelou-se infrutífera quaisquer que fossem as alterações feitas. A segunda abordagem ao problema 14.
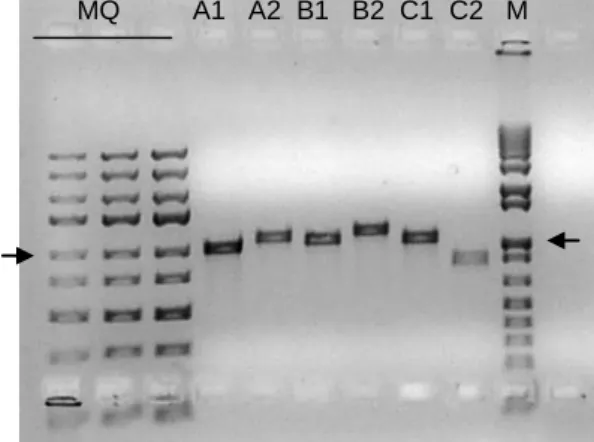
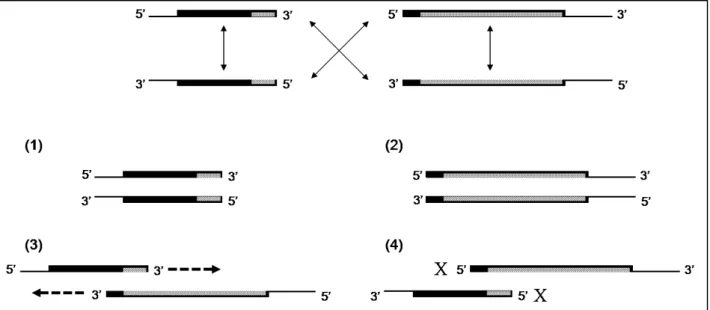
(24) Construção de mutantes em RNases em Enterococcus. consistiu em seguir rigorosamente um protocolo da revista Nature Protocols intitulado: 'Gene splicing and mutagenesis by PCR-driven overlap extension' (75). A quantificação do DNA para o cumprimento deste protocolo foi feita através de leituras no espectrofotómetro e em gel de agarose, utilizando o marcador quantitativo GeneRuler Express (Figura 4 e Figura Suplementar 1). Apesar de muito bem estruturado e parecer bastante promissor, este. protocolo não conduziu à formação do fragmento híbrido desejado. Numa nova tentativa propôs-se a realização do PCR I+II em duas etapas distintas. A primeira com o objectivo de promover o passo de 'overlap extension', i.e. a formação do fragmento híbrido, através de alguns ciclos de PCR na ausência dos 'primers' exteriores. Na segunda etapa os 'primers' seriam adicionados com o objectivo de levar à amplificação exponencial da molécula híbrida formada anteriormente. Este ensaio, à semelhança dos anteriores, não teve os resultados pretendidos. Como tal, e dado a obtenção destes fragmentos híbridos de DNA ser absolutamente fundamental para a construção dos mutantes, tentou-se novamente uma estratégia diferente. Esta consistiu na utilização do método descrito por Warrens et al. em 1997, o que finalmente permitiu a formação da molécula híbrida pretendida (Figura 5) (74).. A MQ. C. B. B. M. A1 A2 B1 B2 C1 C2 M. Figura 4 | Quantificação dos fragmentos. Figura 5 | Obtenção dos fragmentos híbridos.. resultantes dos PCRs I e II em gel de agarose. Fragmentos após purificação por kit: PNPase /. recorrendo ao marcador quantitativo (MQ). RNase III / RNase R / RNase R / Marcador. GeneRuler Express. A seta indica a banda de. (M). A seta indica a banda de 2 kb do. 1 kb dos marcadores de massa molecular.. marcador de massa molecular.. Como ilustrado na figura Figura 6, na fase de 'annealing' do PCR I+II são possíveis quatro combinações distintas. Apenas uma delas, (3), pode servir de molde à DNA polimerase de modo a que se gere o fragmento híbrido completo, e esta combinação é uma das duas menos favorecidas, (3) e (4). Como forma de favorecer esta combinação (3), em relação às 15.
(25) Construção de mutantes em RNases em Enterococcus. outras três, (1),(2),(4), as cadeias nela envolvidas foram produzidas em maior quantidade que as suas complementares nos PCRs I e II. Para tal foi introduzido mais um PCR no protocolo que tem a particularidade de ser assimétrico, isto é, um dos 'primers' está presente numa maior concentração do que o outro, consequentemente originando um produto em que uma das cadeias existe em maior quantidade do que a outra.. Figura 6 | Combinações das hibridações no passo de 'Overlap Entension'. Apenas uma das quatro combinações (3) permite obter o fragmento híbrido de interesse, sendo esta combinação uma das duas menos favorecidas - necessidade de utilização de PCR assimétrico. Adaptado de Warrens et al. (1997).. Assim, foram feitos PCRs I e II assimétricos tendo como DNA molde o resultado dos PCRs I e II não assimétricos, e onde os 'primers' a e d estavam 20x mais concentrados que os b e c respectivamente. Os produtos de amplificação destes PCRs assimétricos vão servir de DNA molde para o PCR I+II. Como se observa na Figura 7, a reacção tem um baixo rendimento e a presença de outras bandas para além da com o tamanho pretendido implica a purificação por gel de agarose do fragmento de interesse. Procurou-se amplificar com os 'primers' a e d o resultado deste PCR mas sempre sem sucesso. A maneira encontrada para aumentar a quantidade de fragmento e poupar tempo foi a de fazer os PCR assimétricos I e II directamente a partir dos fragmentos iniciais ou do DNA genómico, reduzir a diferença da quantidade dos 'primers' de 20x para 8x e aumentar o número de ciclos. O resultado destas alterações pode ser visto na Figura 8. Assim deixa de haver a necessidade de fazer os PCRs I e II não assimétricos e a obtenção de maiores quantidades de DNA facilita os passos subsequentes de restrição e ligação para clonagem. 16.
(26) Construção de mutantes em RNases em Enterococcus. Aparentemente a maior ou menor dificuldade associada à obtenção destas moléculas híbridas parece depender não só do tamanho dos fragmentos e do desenho dos 'primers' mas também da natureza das próprias moléculas. Nos casos em que existe maior dificuldade na obtenção do fragmento híbrido, como neste trabalho, este método revelou ter uma grande eficácia e robustez. A M C. A. A. M. A. Figura 7 | PCR I+II com baixo rendimento. A. Figura 8 | PCR I+II com o rendimento. seta indica a banda de 2 kb do marcador de. melhorado. A seta indica a banda de 2 kb do. massa molecular.. marcador de massa molecular.. Uma vez obtido e purificado o fragmento, este foi digerido com as respectivas enzimas de restrição, clonado no vector pBluescript SK+ (pSK+) e inserido em E. coli JM101TR por electroporação (Tabela Suplementar 1). Foi escolhido este vector porque possui o gene lacZ, o que possibilita a indicação branco/azul (distinção das moléculas recombinantes das não recombinantes, respectivamente) e por ser um vector com elevado número de cópias, o que permite obter grandes quantidades de inserto necessárias para a posterior sequenciação. As colónias resultantes da transformação que apresentavam cor branca foram repicadas para novo meio de cultura sólido e, a partir destas, foram efectuados PCRs de colónia com os 'primers' #92 e #93 (Tabela Suplementar 2), desenhados de modo a amplificar a região do multiple cloning site (MCS) do vector pSK+. Todos os plasmídios com fragmentos com tamanho próximo do esperado foram sequenciados de modo a verificar se a sequência do inserto é a desejada. Os 'primers' para a sequenciação foram desenhados de modo que houvessem sempre zonas de sobreposição entre as várias sequenciações parciais. Para 17.
(27) Construção de mutantes em RNases em Enterococcus. garantir que a sequência obtida era a correcta e estava isenta de mutações, ambas as cadeias de DNA foram sequenciadas.. Esta etapa não apresentou qualquer dificuldade para o fragmento da RNAse III, pois à primeira tentativa foi possível obter clones com o fragmento do tamanho esperado e que a sequenciação viria a demonstrar ser o correcto e não ter nenhuma mutação adicional (Figura 9). M. C. C. Figura 9 | Obtenção do fragmento híbrido correspondente à RNase III. A seta indica a banda de 2 kb do marcador de massa molecular.. No entanto, para os restantes fragmentos, da PNPase, RNase R e RNase J1, a situação foi substancialmente diferente. Para além do facto de a coloração branca das colónias não ser uma indicação muito fidedigna de que existiria de facto um fragmento inserido no vector a interromper o gene da β-galactosidase, foi obtida uma elevada e inesperada variabilidade no que diz respeito à dimensão dos fragmentos inseridos no vector (Figura 10). Não se conseguiu determinar a causa desta diversidade de massas moleculares.. Figura 10 | Variabilidade nos tamanhos dos fragmentos da PNPase e RNase R clonados em pSK+. 18.
(28) Construção de mutantes em RNases em Enterococcus. Devido ao facto de a indicação branco/azul, principal vantagem pela qual se optou pela utilização do pSK+, não se ter mostrado tão útil quanto esperado e ter obrigado à realização de dezenas de reacções de PCR de colónia, propõe-se a obliteração desta etapa do protocolo, procedendo-se à clonagem directamente no vector pGhost 9. Assim, a procura pelos transformantes será feita da mesma forma, recorrendo ao PCR de colónia, e deixa de ser necessário remover o fragmento de interesse do pSK+ para o transferir para o pGhost 9. O problema do pGhost 9 não ser um plasmídio com múltiplas cópias é facilmente contornado através da extracção do plasmídio a partir de volumes superiores de cultura e utilizando kits comerciais próprios para estes casos. Como já foi referido, os fragmentos da RNase III que foram sequenciados não apresentavam qualquer erro, ao contrário dos muitos fragmentos das restantes RNAses que apresentavam sequências muito diferentes das pretendidas e cuja origem não se conseguiu determinar. Foram necessárias numerosas tentativas de transformação para se conseguirem obter os fragmentos correctos correspondentes à PNPase e à RNase R clonados em pSK+. Ainda não foi possível clonar o fragmento da RNase J1, possivelmente devido às suas elevadas dimensões, muito superiores às dos outros fragmentos. Para solucionar este problema vão ser desenhados novos 'primers' de modo a reduzir as dimensões do fragmento.. Uma vez tendo o fragmento sem erros no pSK+, procedeu-se à restrição com as mesmas enzimas utilizadas anteriormente (Tabela Suplementar 3 e Figura 11) e clonou-se o fragmento em pGhost 9. O vector foi depois introduzido em E. coli VE 14188 por electroporação (Tabela Suplementar 1). É neste passo que se encontram os fragmentos correspondentes à PNPase e à RNase R. A estirpe de E. coli transformada com o pGhost 9 tem de codificar no seu genoma a proteína RepA selvagem (genótipo repA+), proteína responsável pela replicação do plasmídio, de modo a permitir a replicação do pGhost 9 a 37 ºC, temperatura óptima de crescimento de E. coli. Este plasmídio é capaz de se replicar a 27 ºC mas é incapaz de o fazer a 37 ou 42 ºC. Esta incapacidade de replicação a estas temperaturas mais altas (37 e 42 ºC) deve-se ao facto de a proteína RepA codificada pelo plasmídio pGhost 9 ser termosensível, não se encontrando funcional a tais temperaturas.. A transformação de E. coli com o pGhost 9 contendo o fragmento de interesse permite obter esta construção em quantidade suficiente para permitir a transformação de E. faecalis V583 ∆ABC por electroporação (para a qual é necessária maior quantidade de plasmídio do que relativamente à transformação de E. coli). Os transformantes foram verificados através de PCR de colónia com os 'primers' #112 e #113 (Tabela Suplementar 2) desenhados de modo a amplificar a região do 'multiple cloning site' (MCS) do vector pGhost 9 (Figura 12).. 19.
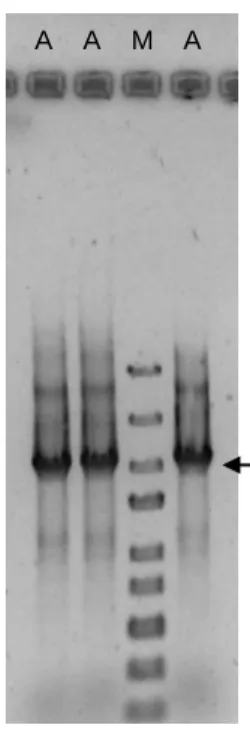
(29) Construção de mutantes em RNases em Enterococcus. Figura 11| Digestão dupla do pSAVE 6. A seta. Figura 12 | Obtenção de transformantes de. indica a banda de 2 kb do marcador de massa. E. coli com o pSAVE 8. A banda inferior é. molecular.. resultado de uma amplificação inespecífica com o cromossoma da estirpe VE 14188. A seta indica a banda de 2 kb do marcador de massa molecular.. O plasmídio pGhost 9 com o fragmento de interesse clonado foi extraído de E. coli e inserido em E. faecalis. A identificação de transformantes foi efectuada da mesma forma do que para E. coli, realizando PCRs de colónia com os 'primers' #112 e #113 (Tabela Suplementar 2 e Figura 13).. Figura 13| Obtenção de transformantes de E. faecalis com o pSAVE 8. A seta indica a banda de 2 kb do marcador de massa molecular. 20.
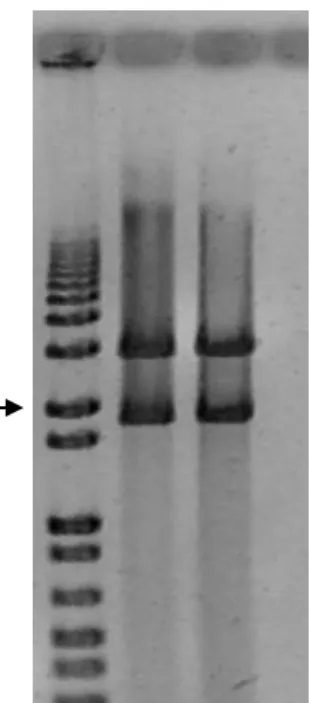
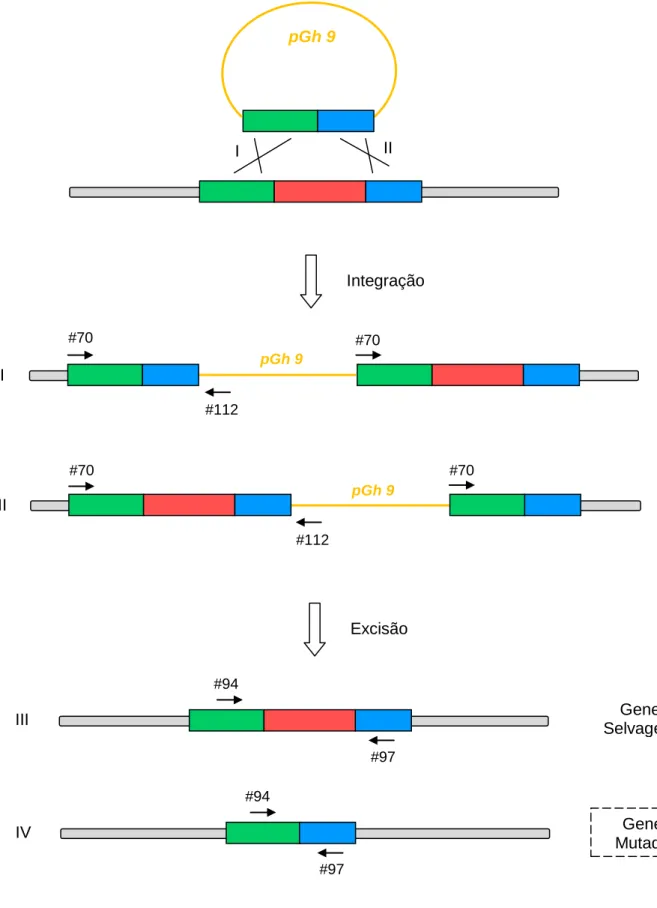
(30) Construção de mutantes em RNases em Enterococcus. Uma vez em E. faecalis o objectivo é a integração do plasmídio no genoma através de recombinação homóloga entre o fragmento de interesse presente no plasmídio e uma das duas regiões de homologia presentes no cromossoma. Para seleccionar os clones onde a integração ocorreu as bactérias foram incubadas durante 2 horas a 27 ºC sem antibiótico (pGhost 9 é capaz de replicar) e posteriormente 2 horas a 42 ºC (pGhost 9 é incapaz de replicar). Após esta variação de temperatura, a cultura foi plaqueada em meio com antibiótico e incubada a 42 ºC. Deste modo as bactérias que conseguirem resistir ao antibiótico fazem-no porque integraram o plasmídio no seu genoma, dado este não ser capaz de se replicar a 42 ºC. Os integrantes foram confirmados através de um PCR de colónia com os 'primers' #70 e #112 (Tabela Suplementar 2). No caso do mutante da RNase III (o único a chegar até este ponto do protocolo) só foi obtido um tipo de integração, a do tipo I relativamente à Figura 14 (Figura 15).. Depois de o fragmento, juntamente com o vector, estar integrado no cromossoma de E. faecalis V583 ∆ABC, procurou-se promover a excisão, por recombinação homóloga, da sequência do vector em conjunto com a sequência a eliminar ladeada pelas duas regiões de homologia de modo a que fique no cromossoma o fragmento com o gene mutado em substituição do gene selvagem. Para tal realizaram-se várias passagens crescendo os enterococos a 27 ºC sem antibiótico. No final plaquearam-se as bactérias em meio com e sem antibiótico e incubaram-se a 42 ºC. Comparando a mesma diluição, o número de colónias na placa sem antibiótico deveria ser consideravelmente superior ao número de colónias na placa com antibiótico. A existência de um razoável número de colónias sensíveis ao antibiótico constitui um bom indicador da ocorrência da excisão do pGhost 9 (o qual confere resistência à eritromicina) do cromossoma e posterior perda do pGhost 9, devido à sua incapacidade de replicação a 42 ºC. Infelizmente o verificado para o caso da RNase III, nos ensaios repetidos várias vezes e para diferentes integrantes, foi um número muito semelhante o que indica uma fraca probabilidade de encontrar o mutante de delecção. Para se identificar um clone onde tenha ocorrido excisão, as colónias das placas sem antibiótico foram repicadas simultaneamente para placas com e sem antibiótico, utilizando uma grelha para identificação. Aqueles que cresceram no meio sem antibiótico e não no meio com antibiótico foram repicados novamente para placas com e sem antibiótico e foi realizado um PCR de colónia com dois dos 'primers' utilizados na sequenciação, no caso da RNase III, #94 e #95, respectivamente (Tabela Suplementar 2 e Figura 14). Após várias tentativas e dezenas de colónias testadas ainda não foi possível encontrar um clone com o gene mutado, somente clones em que a excisão deixou no cromossoma o gene selvagem (Figura 16). A eficácia desta etapa do protocolo pode variar bastante e por vezes ser muito baixa. mas,. dado. o. número. de. tentativas,. somos. levados. a. considerar. a 21.
(31) Construção de mutantes em RNases em Enterococcus. hipótese de que talvez, à semelhança do que acontece em B. subtilis, o gene da RNase III seja essencial em E. faecalis, não sendo possível obter mutantes de delecção para este gene. Figura 14 | Representação esquemática dos acontecimentos de integração e excisão que podem ocorrer durante o protocolo.. pGh 9. II. I. Integração. #70. #70 pGh 9. I #112. #70. #70 pGh 9. II #112. Excisão #94. Gene Selvagem. III #97 #94. Gene Mutado. IV #97. 22.
(32) Construção de mutantes em RNases em Enterococcus. 1. 2. 3. 4. 5. 6. Figura 15 | Obtenção de um só tipo de. Figura 16 | Clones em que ocorreu excisão.. integração do pSAVE 8 no cromossoma de. Na pista 2 e 3 estão clones nos quais ocorreu. E. faecalis. A seta indica a banda de 2 kb do. a excisão e onde o gene selvagem ficou no. marcador de massa molecular.. cromossoma, na pista 4 está o V583 ∆ABC, na pista 5 está o controlo sem colónia e nas pistas 1 e 6 está o marcador. A seta indica a banda de 2 kb do marcador de massa molecular.. Apesar deste processo de construção de mutantes envolver várias etapas (um primeiro 'crossing-over' para integração do plasmídio, seguido de um segundo para excisão do mesmo de forma a substituir o gene selvagem pelo mutante) (Figura 14), esta abordagem foi seleccionada uma vez que é do nosso interesse obter um mutante exclusivamente neste gene deixando o restante genoma inalterado. Por esta razão não se optou por exemplo pela disrupção do gene por inserção de uma cassete de resistência a antibiótico.. 23.
(33) Construção de mutantes em RNases em Enterococcus. 5. Perspectivas futuras Estabelecer se o gene rnc, que codifica para a RNase III, é essencial em Enterococcus faecalis V583 ∆ABC, à semelhança do que acontece em Bacillus subtilis. Para isso ir-se-á clonar o gene rnc selvagem num plasmídio com um promotor indutível, com replicação não sensível à temperatura, com origem de replicação em E. faecalis e E. coli e com uma marca de selecção apropriada. Este plasmídio será depois introduzido numa das estirpes de Enterococcus que já possuem o pGhost 9 com o fragmento mutado integrado no genoma (integrantes) e os transformantes serão submetidos às etapas finais do protocolo de elaboração dos mutantes, nomeadamente as passagens a 27 ºC sem a presença de eritromicina, de modo a promover a excisão do cromossoma do pGhost 9 juntamente com a forma selvagem do gene rnc. Naquelas bactérias em que a excisão pretendida se verificar, e se o gene for essencial, só sobreviverão os clones que possuam o plasmídio não termosensível com o gene rnc selvagem a ser expresso sob a acção do promotor indutível. Posteriormente, estudos poderão ser feitos para observar as consequências da variação na expressão deste gene. Após a obtenção dos mutantes na PNPase, na RNase R e na RNase J1 pretende-se estudar o efeito destas mutações em vários processos, embora no caso da última exista também a possibilidade de ser essencial dado que, à semelhança da RNase III, é essencial em B. subtilis. Serão estudadas as alterações ao nível do crescimento celular, realizando curvas de crescimento à temperatura óptima e a baixas temperaturas (uma vez que a PNPase e a RNase R são 'cold shock proteins'); ao nível da resistência a vários antibióticos, determinando as concentrações mínimas inibitórias (MIC) e ao nível da morfologia celular, por observação ao microscópio. Em paralelo estes mutantes serão utilizados para a análise do processamento e estabilidade das moléculas RNA em geral, e dos transcritos do operão fsr em particular (sistema em estudo no laboratório). Pretende-se estudar também o comportamento dos mutantes no que diz respeito à produção de factores de virulência (e.g. teste da gelatinase) e à sua patogenecidade recorrendo a organismos modelos, nomeadamente Galleria mellonella, macrófagos e Drosophila melanogaster. Por último, será também interessante, recorrendo ao protocolo aqui apresentado, efectuar os mesmos mutantes na estirpe E. faecalis V583 selvagem (que possui os três plasmídios: pTEF1, pTEF2, pTEF3) ou noutras que se revelem de especial interesse.. Assim, com o trabalho desenvolvido nesta tese, pretendeu-se melhorar os protocolos para a obtenção de mutantes em Enterococcus faecalis e abrir caminho para uma nova área, criando ferramentas que irão permitir estudos futuros sobre as complexas relações entre RNAses, RNAs e virulência em enterococos. 24.
(34) Construção de mutantes em RNases em Enterococcus. 6. Bibliografia. 1. 2. 3. 4. 5. 6.. 7. 8. 9. 10. 11. 12. 13. 14. 15.. 16.. 17.. 18. 19.. 20.. Thiercelin ME (1899) Sur un diplocoque saprophyte de l'intestin susceptible de devenir pathogen. C R Soc Biol 5:269-271. Andrewes FW, Horder TJ (1906) A study of the streptococci pathogenic for man. Lancet ii:708-713. Orla-Jensen S (1919) The lactic acid bacteria. Mémoires de l’Académie Royale des Sciences et des Lettres de Danemark, Section des Sciences 8iéme Série 5:81–197. Sherman JM (1937) The streptococci. Bacteriol Rev 1:3-97. Kalina AP (1970) [The position of enterococci in the system of microorganisms]. Zh Mikrobiol Epidemiol Immunobiol 47:20-21. Schleifer KH, Kilpper-Balz R (1984) Transfer of Streptococcus faecalis and Streptococcus faecium to the Genus Enterococcus nom. rev. as Enterococcus faecalis comb. nov. and Enterococcus faecium comb. nov. Int J Syst Bacteriol 34:31-34. Foulquie Moreno MR, Sarantinopoulos P, Tsakalidou E, De Vuyst L (2006) The role and application of enterococci in food and health. Int J Food Microbiol 106:1-24. Franz CM, Stiles ME, Schleifer KH, Holzapfel WH (2003) Enterococci in foods--a conundrum for food safety. Int J Food Microbiol 88:105-122. Giraffa G (2002) Enterococci from foods. FEMS Microbiol Rev 26:163-171. Franz CM, Holzapfel WH, Stiles ME (1999) Enterococci at the crossroads of food safety? Int J Food Microbiol 47:1-24. Jett BD, Huycke MM, Gilmore MS (1994) Virulence of enterococci. Clin Microbiol Rev 7:462-478. Endtz HP, van den Braak N, Verbrugh HA, van Belkum A (1999) Vancomycin resistance: status quo and quo vadis. Eur J Clin Microbiol Infect Dis 18:683-690. Huycke MM, Sahm DF, Gilmore MS (1998) Multiple-drug resistant enterococci: the nature of the problem and an agenda for the future. Emerg Infect Dis 4:239-249. Ogier JC, Serror P (2007) Safety assessment of dairy microorganisms: The Enterococcus genus. Int J Food Microbiol. Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B (1989) In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 33:1588-1591. Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM (2003) Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071-2074. Shankar N, Baghdayan AS, Gilmore MS (2002) Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417:746750. Hancock L, Perego M (2002) Two-component signal transduction in Enterococcus faecalis. J Bacteriol 184:5819-5825. Aakra A, Vebo H, Snipen L, Hirt H, Aastveit A, Kapur V, Dunny G, Murray BE, Nes IF (2005) Transcriptional response of Enterococcus faecalis V583 to erythromycin. Antimicrob Agents Chemother 49:2246-2259. Solheim M, Aakra A, Vebo H, Snipen L, Nes IF (2007) Transcriptional responses of Enterococcus faecalis V583 to bovine bile and sodium dodecyl sulfate. Appl Environ Microbiol 73:5767-5774. 25.
(35) Construção de mutantes em RNases em Enterococcus. 21.. 22.. 23.. 24.. 25. 26.. 27.. 28. 29. 30. 31.. 32. 33. 34.. 35. 36. 37.. 38. 39.. 40.. Le Breton Y, Boel G, Benachour A, Prevost H, Auffray Y, Rince A (2003) Molecular characterization of Enterococcus faecalis two-component signal transduction pathways related to environmental stresses. Environ Microbiol 5:329-337. Hancock LE, Perego M (2004) Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol 186:7951-7958. Evers S, Courvalin P (1996) Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J Bacteriol 178:1302-1309. Hancock LE, Perego M (2004) The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol 186:56295639. Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P (2007) Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev 20:79-114. Shepard BD, Gilmore MS (2002) Differential expression of virulence-related genes in Enterococcus faecalis in response to biological cues in serum and urine. Infect Immun 70:4344-4352. Hew CM, Korakli M, Vogel RF (2007) Expression of virulence-related genes by Enterococcus faecalis in response to different environments. Syst Appl Microbiol 30:257-267. Romby P, Wagner EG (2008) Exploring the complex world of RNA regulation. Biol Cell 100:e1-e3. Khodursky AB, Bernstein JA (2003) Life after transcription - revisiting the fate of messenger RNA. Trends Genet 19:113-115. Arraiano CM, Maquat LE (2003) Post-transcriptional control of gene expression: effectors of mRNA decay. Mol Microbiol 49:267-276. Kimata K, Tanaka Y, Inada T, Aiba H (2001) Expression of the glucose transporter gene, ptsG, is regulated at the mRNA degradation step in response to glycolytic flux in Escherichia coli. Embo J 20:3587-3595. Johansson J, Cossart P (2003) RNA-mediated control of virulence gene expression in bacterial pathogens. Trends Microbiol 11:280-285. Murphy ER, Payne SM (2007) RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect Immun 75:3470-3477. Jude F, Kohler T, Branny P, Perron K, Mayer MP, Comte R, van Delden C (2003) Posttranscriptional control of quorum-sensing-dependent virulence genes by DksA in Pseudomonas aeruginosa. J Bacteriol 185:3558-3566. Nicholson AW (1999) Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol Rev 23:371-390. Condon C (2007) Maturation and degradation of RNA in bacteria. Curr Opin Microbiol 10:271-278. Mechold U, Fang G, Ngo S, Ogryzko V, Danchin A (2007) YtqI from Bacillus subtilis has both oligoribonuclease and pAp-phosphatase activity. Nucl Acids Res 35:45524561. Schiffer S, Rosch S, Marchfelder A (2002) Assigning a function to a conserved group of proteins: the tRNA 3'- processing enzymes. Embo J 21:2769-2777. Redko Y, Bechhofer DH, Condon C (2008) Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol 68:1096-1106. Ow MC, Kushner SR (2002) Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes & Dev 16:1102-1115. 26.
(36) Construção de mutantes em RNases em Enterococcus. 41.. 42.. 43.. 44.. 45.. 46.. 47.. 48.. 49.. 50.. 51.. 52.. 53.. 54.. 55. 56.. Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM (2007) Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35:7651-7664. Bardey V, Vallet C, Robas N, Charpentier B, Thouvenot B, Mougin A, Hajnsdorf E, Regnier P, Springer M, Branlant C (2005) Characterization of the molecular mechanisms involved in the differential production of erythrose-4-phosphate dehydrogenase, 3-phosphoglycerate kinase and class II fructose-1,6-bisphosphate aldolase in Escherichia coli. Mol Microbiol 57:1265-1287. Geissmann T, Possedko M, Huntzinger E, Fechter P, Ehresmann C, Romby P (2006) Regulatory RNAs as mediators of virulence gene expression in bacteria. Handb Exp Pharmacol 9-43. Grundy FJ, Henkin TM (2006) From ribosome to riboswitch: control of gene expression in bacteria by RNA structural rearrangements. Crit Rev Biochem Mol Biol 41:329-338. Clements MO, Eriksson S, Thompson A, Lucchini S, Hinton JC, Normark S, Rhen M (2002) Polynucleotide phosphorylase is a global regulator of virulence and persistency in Salmonella enterica. Proc Natl Acad Sci U S A 99:8784-8789. Ygberg SE, Clements MO, Rytkonen A, Thompson A, Holden DW, Hinton JC, Rhen M (2006) Polynucleotide phosphorylase negatively controls spv virulence gene expression in Salmonella enterica. Infect Immun 74:1243-1254. Cheng ZF, Zuo Y, Li Z, Rudd KE, Deutscher MP (1998) The vacB gene required for virulence in Shigella flexneri and Escherichia coli encodes the exoribonuclease RNase R. J Biol Chem 273:14077-14080. Erova TE, Kosykh VG, Fadl AA, Sha J, Horneman AJ, Chopra AK (2008) Cold shock exoribonuclease R (VacB) is involved in Aeromonas hydrophila pathogenesis. J Bacteriol 190:3467-3474. Huntzinger E, Boisset S, Saveanu C, Benito Y, Geissmann T, Namane A, Lina G, Etienne J, Ehresmann B, Ehresmann C, Jacquier A, Vandenesch F, Romby P (2005) Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24:824-835. de Fatima Silva Lopes M, Ribeiro T, Abrantes M, Figueiredo Marques JJ, Tenreiro R, Crespo MT (2005) Antimicrobial resistance profiles of dairy and clinical isolates and type strains of enterococci. Int J Food Microbiol 103:191-198. Lopes Mde F, Ribeiro T, Martins MP, Tenreiro R, Crespo MT (2003) Gentamicin resistance in dairy and clinical enterococcal isolates and in reference strains. J Antimicrob Chemother 52:214-219. Lopes Mde F, Simoes AP, Tenreiro R, Marques JJ, Crespo MT (2006) Activity and expression of a virulence factor, gelatinase, in dairy enterococci. Int J Food Microbiol 112:208-214. Ribeiro T, Abrantes M, Lopes Mde F, Crespo MT (2007) Vancomycin-susceptible dairy and clinical enterococcal isolates carry vanA and vanB genes. Int J Food Microbiol 113:289-295. Semedo T, Santos MA, Lopes MF, Figueiredo Marques JJ, Barreto Crespo MT, Tenreiro R (2003) Virulence factors in food, clinical and reference Enterococci: A common trait in the genus? Syst Appl Microbiol 26:13-22. Shepard BD, Gilmore MS (2002) Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect 4:215-224. Terzaghi BE, Sandine WE (1975) Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol 29:807-813.. 27.
Imagem
+7

Documentos relacionados
De seguida, vamos adaptar a nossa demonstrac¸ ˜ao da f ´ormula de M ¨untz, partindo de outras transformadas aritm ´eticas diferentes da transformada de M ¨obius, para dedu-
9º, §1º da Lei nº 12.513/2011, as atividades dos servidores ativos nos cursos não poderão prejudicar a carga horária regular de atuação e o atendimento do plano de metas do
6 Consideraremos que a narrativa de Lewis Carroll oscila ficcionalmente entre o maravilhoso e o fantástico, chegando mesmo a sugerir-se com aspectos do estranho,
(grifos nossos). b) Em observância ao princípio da impessoalidade, a Administração não pode atuar com vistas a prejudicar ou beneficiar pessoas determinadas, vez que é
Na primeira década da existência do Estado de Minas Gerais, a legislação educacional mineira estabeleceu o perfil de professor estadual, imputando como formação
O modelo conceitual procura mostrar quais são os elementos de informação tratados pelo sistema, para que mais adiante se possa mostrar ainda como essa informação é transformada pelo
Nesse contexto, a análise numérica via MEF de vibrações sísmicas induzidas por detonações se mostra como uma metodologia que pode contribuir significativamente
Nessa situação temos claramente a relação de tecnovívio apresentado por Dubatti (2012) operando, visto que nessa experiência ambos os atores tra- çam um diálogo que não se dá
