Ana Catarina da Cruz Rodrigues da Silva
Texto
Imagem

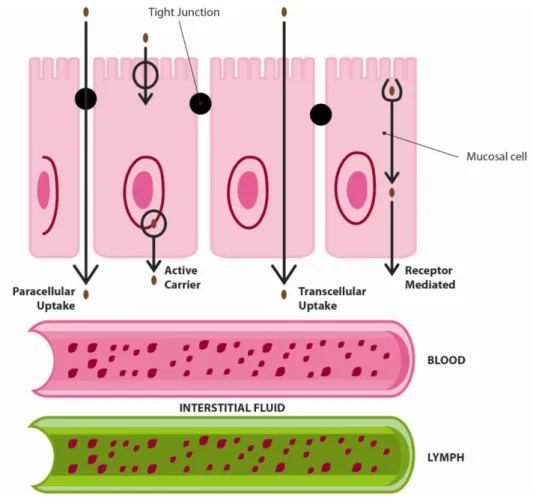
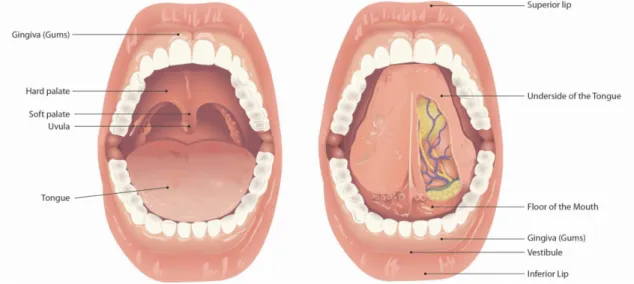
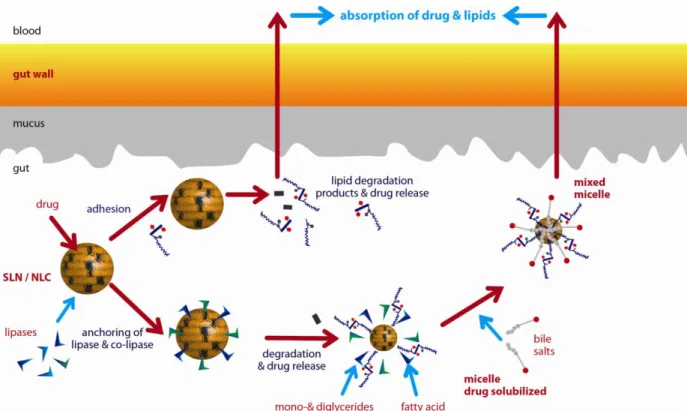

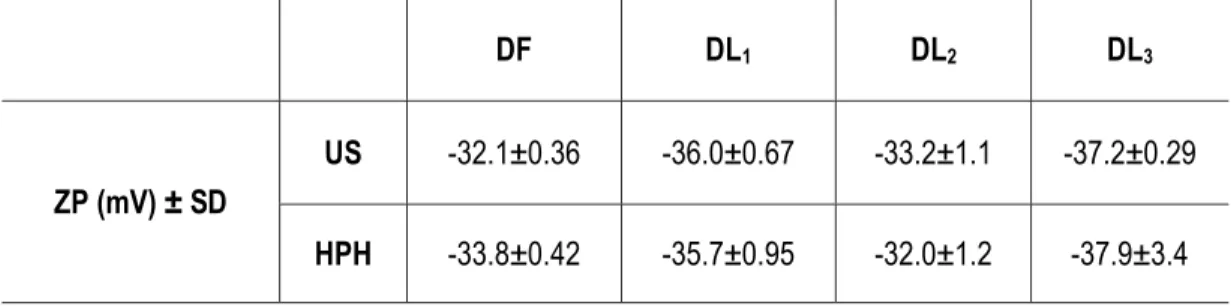



Outline
Documentos relacionados
Poder ter assistido a diferentes aulas de piano, desde Outubro de 2013 a Maio de 2014, de diferentes alunos, incluindo vários que acabaram por não vir a participar no
Recovery of Campylobacter jejuni from fresh and frozen meat and poultry collected
In clonal mini-hedges young plant tissue, such as expanding leaves, is always present because of pruning, which increases pathogen infection. In this case, it
Dipping fresh guaco leaves in ethanol immediately prior to drying led to faster evaporation and consequently shorter drying times - a key factor reducing degradation of
The higher content of extractives, holocellulose, volatile materials, low ash content values, rate of mineral contaminants and high performance for combustion test were observed only
A new optimized formulation of cationic solid lipid nanoparticles intended for gene delivery: Development, characterization and DNA binding efficiency of TCERG1 expression
At the calcination temperature 550 o C, the Cu-MCM-41 catalyst possessed higher surface area, smaller pore diameter, stronger metal-support interaction, and better CuO
Samarium-doped Fe3O4 nanoparticles with improved magnetic and supercapacitive performance: a novel preparation strategy and characterization. Journal of
