UNIVERSIDADE DA BEIRA INTERIOR
Ciências da Saúde
Purification and characterization of a serine
protease from Alternaria alternata
Bruno Fernandes dos Santos
Dissertação para obtenção do Grau de Mestre em
Ciências Biomédicas
(2º ciclo de estudos)
Orientador: Prof. Doutora Cândida Ascensão Teixeira Tomaz
Co-orientador: Mestre Diana Faria Bicho
iii
v
Acknowledgments
Primeiramente, quero agradecer a toda a minha família em especial aos meus pais pelo incondicional apoio, amor e motivação que me têm dado, não só nesta fase mas também ao longo da vida! Quero agradecer aos meus avós Júlio e Maria por todas as palavras de motivação, por toda a preocupação e por todo amor e carinho que me proporcionam em todos os momentos da minha vida.
Quero deixar também um profundo agradecimento à Professora Cândida Tomaz pela orientação e conhecimento científico que me permitiu adquirir, ao longo destes últimos anos e pela confiança que sempre demonstrou depositar em mim.
Agradeço também a Universidade da Beira Interior, em especial ao Centro de Investigação em Ciências da Saúde por ter proporcionado as melhores condições que permitiram a realização desta dissertação de mestrado.
Agradeço a todas a pessoas do meu grupo de investigação, em especial a Diana Bicho, Catarina Nunes e Fátima Santos por toda a ajuda, amizade e conhecimento que me disponibilizaram ao longo deste ano académico.
Agradeço também aos meus colegas por todos os bons momentos que me proporcionaram ao longo da minha vida académica. Um agradecimento especial ao meu grande amigo Diogo Tomé, decerto uma das melhores pessoas que conheci até hoje e um dos amigos que guardarei para a vida.
Por fim, mas decerto um dos mais importantes, queria agradecer Carina por todo o apoio, amor e paciência, que tens tido para comigo ao longo destes anos, sem ti nada seria possível…
vi
Resumo alargado
Nos últimos anos, tem vindo a aumentar a prevalência das doenças alérgicas em especial nos países industrializados. Estudos indicam que cerca de um terço da população ocidental sofre de uma ou mais formas de doenças alérgicas. De uma maneira simples, a doença alérgica pode ser definida como uma reação exacerbada do sistema imunitário contra determinados antigénios presentes no meio ambiente (alergénios), mas que na verdade não representam qualquer perigo para indivíduos não sensibilizados. Este tipo de doença está normalmente associada a uma produção aumentade de IgE específica para um determinado alergénio, sendo que a tendência pessoal ou familiar para a produção exacerbada deste tipo de imunoglobulina é denominada de atopia. De entre as fontes alergénicas ambientais capazes de induzir reações de hipersensibilidade, os fungos surgem como um dos principais responsáveis pelo desenvolvimento, persistência e severidade de doença alérgica respiratória, nomeadamente da asma e da rinite. A Alternaria alternata é uma das principais espécies fúngicas associadas a doenças alérgicas tendo, até a data, sido identificados 17 alergénios diferentes para esta espécie. A prevalência de alergias devido à Alternaria varia em diferentes regiões geográficas. Um estudo multicêntrico realizado num grupo de quase 900 doentes com alergias mostrou que 3-20% dos indivíduos (3% em Portugal e 20% em Espanha) apresentava hipersensibilidade a A. alternata. Em estudos recentes foi verificado que um dos alergénios da A. alternata, o Alt a 15, apresentava um homólogo para a Curvularia lunata, C l 4. Indivíduos sensíveis a um destes alergénios apresentava uma resposta alérgica quando exposta ao outro alergénio, ou seja, apresentavam uma resposta alérgica cruzada. O Alt a 15 é uma serina protease descoberta por clonagem do mRNA de A. alternata, não sendo conhecida ainda a sua forma nativa. Apesar de se conhecer a sua massa molecular e de se tratar de uma glicoproteína, a caracterização da Alt a 15 ainda está muito incompleta, sendo difícil estudar esta proteína e assim conseguir mais informação sobre possíveis reações alérgicas cruzadas com outros fungos.
Assim, este trabalho tem como objetivo purificar e caracterizar a forma nativa desta serina protease da A. alternata. A purificação desta proteína foi realizada utilizando um processo constituído por duas etapas cromatográficos. Na primeira foi utilizada uma coluna de Concanavalina A (Con A) Sepharose. Esta coluna cromatográfica apresenta afinidade específica para moléculas glicosiladas, tais como glicoproteínas e glicolípidos, tendo sido utilizada anteriormente na purificação da serina protease da C. lunata. A coluna foi inicialmente equilibrada e lavada com tampão Tris-HCl a pH 7.2 contendo CaCl2 e MgCl2. Após
a remoção de todas as moléculas que não apresentavam afinidade para a Con A, as glicoproteínas foram eluídas da coluna com o tampão metil α-D manopiranosido. Após esta primeira fase de purificação, procedeu-se à segunda etapa cromatográfica de purificação, utilizando um coluna de troca iónica (DEAE-Sepharose). A eluição foi realizada em quatro
vii
passos, utilizando o tampão Tris-HCl, pH 8 com 0%, 40%, 80% e 100% de NaCl. Desta forma, foram obtidas duas frações distintas, sendo que a segunda apresentava uma elevada actividade específica (144.33 U/mg), um factor de purificação de 28.03 e um rendimento de 26.58 %. A análise por SDS-PAGE confirmou a existência de apenas uma banda proteica com massa molecular aproximada de 40 KDa. Após a purificação procedeu-se à caracterização da protease purificada. Para tal, foi estudado o efeito do pH, temperatura, de inibidores (PMSF, EDTA e EGTA) e de iões metálicos na sua atividade e foram determinados os parâmetros cinéticos (KM a Vmax). Desta forma, verificou-se que a protease apresenta uma atividade ótimaa pH 8, e a uma temperatura de 50º C. Já na presença dos inibidores verificou-se que o PMSF inibia a atividade em cerca de 90%, enquanto o ião Mn2+ inibia por completo a atividade da
protease, o que permitiu concluir que se tratava de uma serina protease. No cálculo dos parâmetros cinéticos verificou-se que a serina protease apresentava um KM igual a 0.51 mg/ml
e um Vmax de 7.38 µmol/min/ml. De uma forma geral, pode concluir-se que os objetivos deste
trabalho foram alcançados. Pela primeira vez foi purificada e caracterizada uma serina protease num extrato de A. alternata. Apesar dos resultados apresentados, não se pode concluir que a serina protease purificada é na verdade o alergénio Alt a 15, sendo ainda necessária a realização de teste imunológicos com soro de indivíduos sensibilizados ao Alt a 15. Contudo, este trabalho representa um ponto de partida para um melhor conhecimento dos alergénicos da A. Alternata, podendo contribuir para o avanço no diagnóstico e tratamento das doenças alérgicas provocadas por espécies fúngicas.
Palavras-chave
viii
Abstract
Alternaria alternata is a common environmental allergenic fungi. The prevalence of Alternaria allergies varies in different geographic regions. A multicenter study performed in a
group of almost 900 patients with allergies found hypersensitivity to Alternaria in 3–20% of the individuals (3 % in Portugal to 20 % in Spain). To date, 17 allergens of A. alternata have been identified. Different studies demonstrated the existence of A. alternata serine proteases that can induce lung inflammation and airway epithelial cell activation via PAR2. In 2014, the Alt a 15 (recombinant A. alternata serine protease) was added to allergens list of the official Allergen nomenclature subcommittee. In the present project, we aimed to purify the native serine protease from the A. alternata extract since this form is very important for carrying out characterization studies and to provide a good foundation for improving the accuracy of the diagnosis and the understanding and management of IgE-mediated fungal diseases.
The purification protocol involved a first step of affinity chromatography using a Concanavalin A Sepharose column and a second purification step of ion exchange chromatography using a DEAE-Sepharose column. A global 28.03-fold purification of serine protease was obtained with a specific activity of 144.33 U/mg. The molecular weight of the enzyme was determined as 40 kDa by using SDS-PAGE, and the optimum pH and temperature were 8.0 and 50º C, respectively. The kinetic parameters KM and Vmax, determined against substrate casein, were
0.51 mg/mL and 7.38 μmol/min/mL, respectively. The protease was strongly inhibited by PMSF (phenylmethyl sulfony fluorid) indicating it is a serine protease.
The purification strategy applied in this work could be of great importance to open a new possibility for obtaining native A. alternata serine proteases for application in diagnosis or clinical purposes.
Keywords
ix
Contents
1. Introduction 1 1.1. Hypersensitivity reactions 2 1.1.1. Allergy reactions 3 1.2. Alternaria alternata 5 1.2.1. Allergens from Alternaria alternata 61.3. Purification of fungal serine protease 10 1.3.1 Purification based on ion exchange chromatograph 11
1.3.2 Purification based on ion exchange and hydrophobic interaction chromatograph 14 1.3.3 Purification based on gel filtration chromatography 16
1.3.4 Purification based on affinity chromatography 17
1.4. Aim of the present study 19 2. Materials and Methods 20 2.1. Materials 21 2.1.1. Chemicals 21
2.1.2. Materials and Equipments 23
2.2. Methods 24 2.2.1. Strains and fungal extracts 24
2.2.2. Enzyme-activity determination 24
2.2.3. Protein determination 25
2.2.4. Protease purification 25
2.2.5. Biochemical characterization of purified enzyme 26
2.2.5.1. Electrophoretic analysis 26 2.2.5.2. Effect of pH on purified enzyme activity 26 2.2.5.3. Effect of temperature on purified enzyme activity 27 2.2.5.4. Effect of protease inhibitors on purified enzyme activity 27 2.2.5.5. Effect of metal ions on purified enzyme activity 27 2.2.6. Determination of kinetic parameters 27
3. Results and Discussion 28
3.1. Protease purification 29
3.2. Biochemical characterization of purified enzyme 36
4. Conclusion 41
5. Future Prespectives 43
6. References 45
7. Annexes 52
x
List of Figures
Figure 1. Types of hypersensitivity reactions 3
Figure 2. Immunopathology of allergic reactions 4
Figure 3. A. tenuis description by Christian von Esenbeck (1817) 6
Figure 4. Separation principles in ion exchange chromatography 11
Figure 5. Separation principles in hydrophobic interaction chromatography 14
Figure 6. Separation principle in gel filtration chromatography 16
Figure 7. Separation principles in affinity chromatography 17
Figure 8. Multiple Sequence Alignment used Clustal Omega 1.2.1. 29
Figure 9. Chromatographic profile obtained for the A. alternata extract 30
Figure 10. Two dimensional gel electrophoresis analysis. A) A. alternata stratum;
B) Con A bound fraction collected in the chromatographic assay using Con A Sepharose column
32
Figure 11. Chromatographic profile obtained for Con A bound fraction using a
DEAE-Sepharose column 33
Figure 12. Chromatographic profile obtained for Con A bound fraction using a
DEAE-Sepharose column 34
Figure 13. A) SDS-PAGE electrophoretic analysis of the peak II fraction collected
in the chromatographic assay using DEAE sepharose column. B) Molecular weight determination of curve MW Vs RM
35
Figure 14. Effect of pH on activity of A. alternata protease 36
Figure 15. Effect of temperature on activity of A. alternata protease 37
Figure 16. Effect of protease inhibitors on activity of protease purified 38
Figure 17. Effect of metalions on activity of protease purified 38
Figure 18. Lineweaver–Burk plot for serine protease from A. alternata 40
Figure 19. Straight calibration used to calculate the concentration of L-tyrosine 53 Figure 20. Straight calibration used to calculate the concentration of protein 53
xi
List of Tables
Table 1. The allergens of A. alternata 7
Table 2. Physicochemical properties of the protein exploited by different
chromatographic methods 10
Table 3. Purification of fungal serine protease using IEX 12
Table 4. Purification of fungal serine proteases using IEX and HIC 14
Table 5. Purification of fungal serine protease using gel filtration 16
Table 6. Reagents used in the experimental work 21
Table 7. Materials and equipments used in the experimental work 23
Table 8. Guide to prepare of BSA standards 25
Table 9. Yields and protease activities towards casein of two fractions obtained
by a chromatographic step using Con A column 31
Table 10. Yields and protease activities towards casein of two fractions obtained
by a chromatographic step using DEAE column 34
xii
List of Acronyms
A. alternata Alternaria alternata
A. tenuis Alternaria tenuis
ABPA Allergic bronchopulmonary aspergillosis
AEX Anion exchange chromatography
ALDH Aldehyde dehydrogenase
APCs Antigen-presenting cells
BCA Bicinchoninic acid
BSA Bovine serum albumin
CD4+ Cluster of differentiation 4
CD8+ Cluster of differentiation 8
CEX Cation exchange chromatography
CM Carboxymethyl
Con A Concanacalin A
CTL Cytotoxic T lymphocytes
DEAE Diethylaminoethyl
EDTA Ethylenediamine tetraacetic acid
EGTA ethylene glycol-bis (β-aminoethylether)-tetraacetic acid
ELISE Enzyme-linked immunosorbent assay
FcεRI High affinity receptors
FPLC Fast protein liquid chromatography
GF Gel filtration
GST Glutathione-S-transferase
HIC Hydrophobic interaction chromatograph
IEX Ion exchange chromatography
IgE Immunoglobulin E IgG Immunoglobulin G IgM Immunoglobulin M IL-13 Interleukin-13 IL-4 Interleukin-4 IL-5 Interleukin-5 IL-6 Interleukin-6 IP Isoelectric point KDa Kilodalton KDa kilodalton
KERAB Keratinolytic serine alkaline proteinase
KM Michaelis constant
MALDI-TOF Matrix-Assisted Laser Desorption Ionization- Time of Flight
MHC class II Major histocompatibility complex class II
min minute
MnSOD Manganese-dependent Superoxide dismutase
mRNA messenger Ribonucleic acid
MtDH Mannitol dehydrogenase
MW Molecular weight
xiii
NADP Nicotinamide adenine dinucleotide phosphate
NADPH Nicotinamide adenine dinucleotide phosphate-oxidase
NK cell Natural killer cell
NTF2 Nuclear transport factor 2
PAR2 Protease activated receptor 2
PDA Potato Dextrose Agar
PMSF Phenylmethylsulfonyl fluoride
Q Quaternary ammonium
S Methyl sulfonate
SDS-PAGE Sodium dodecyl sulfate and polyacrylamide gel electrophoresis
SP Sulphopropyl
SPT Skin prick test
TCTP Translationally controlled tumor protein
TH1 cell Type 1 T helper cells TH2 cell Type 2 T helper cells
U Activity units
UBI University of Beira Interior
Vmax Maximal rate
1
1. Introduction
2
Introduction
Allergic diseases affect approximately one-third of the general population [1]. Allergens are antigens that induce the immune system to produce IgE antibodies and which trigger symptoms in a sensitized individual [2]. Several hundred allergenic proteins, glycoproteins and lipoproteins, with different biological sources, including weed, grass, tree pollens and animal dander, molds, house dust mites, parasites, insect venoms, occupational allergens such as natural rubber latex, drugs, and foods, elicit IgE antibody production when introduced into an immunocompetent and genetically predisposed host. Allergens are classification into two groups: Indoor and outdoor allergens. The main sources of indoor allergens are the mites, cockroaches, pets like dogs and cats and fungi, whereas the main sources of outdoor allergens are the grasses, grains, herbs, trees and fungi [3].
The fungi are a major cause of allergies and are also associated with various other complications. Epidemiologic studies have associated mold sensitivity, with the development, persistence, and severity of asthma. Currently, the allergic diseases have been increasing in a significant way in the western population increasing in this way all the problems associated with them, making necessary to investigate the main sources of allergens such as molds and thus understand and design the best strategies to combat these diseases [4].
1.1. Hypersensitivity reactions
Hypersensitivity defines an undesirable reaction produced by the normal immune system that may in some cases be a fatal reaction. According to various authors, hypersensitivity refers to a pre-sensitized state of an individual being abnormally sensitive to the foreign substances causing inflammation and cellular damage [5].
Hypersensitivity reactions can be classified into four types: Type I or Anaphylactic reactions, type II or Cytotoxic reactions, type III or Immune Complex reactions and type IV or Cell-Mediated reactions (Figure 1) [5].
3
Figure 1. Types of hypersensitivity reactions (adapted from [6]).Type I or anaphylactic reaction is characterized by occuring within minutes after exposure to antigen. In this type of reaction the IgE binds to high affinity receptors (FcεRI) on mast cells and basophils. When a relevant allergen binds to IgE bound to FcεRI on mast cells and basophils, these cells degranulate and release several mediators such as histamine, prostaglandins and leukotrienes. Type II or cytotoxic reactions include activation of complement by IgG or IgM binding to an antigen on a cell. These antigens are expressed by lysed cell and cell with altered function. Drug allergies, chronic urticarial, thyroiditis and myastenia gravis were examples of hypersensitivity type II reactions [7]. Type III or immune complex reactions involve reactions against soluble antigens circulating in serum. Usually involve IgG antibodies with formation of antigen-antibody immune complexes that are deposited in organs, activate complement, and cause inflammatory damage. This type of reaction occurs when slightly high antigen-antibody ratio is present.
Type IV or cell-mediated reactions involve reactions against cell-associated or circulating antigens. These reactions are only confered by T lymphocytes and may involve CD4+ Th1 T cells, CD4+ Th2 T cells, or cytolytic CD8+ T cells. Macrophages and epithelioid cells may also be involved in some cases. These reactions develop over a period of 2-4 days [5, 6].
1.1.1. Allergic reactions
Concept of allergy was defined by von Pirquet in 1906 as an altered reactivity to antigen stimulation which may lead to protective immunity or hypersensitivity. In 2001 the European Academy of Allergology and Clinical Immunoloy defined allergic reaction as a hypersensitivity reaction mediated by immunological mechanisms. Allergy involves signs or symptoms that are
4
objectively reproducible, and which are triggered by exposure to known stimuli at a dose which is tolerated by normal individuals. These reactions occur in certain people, minutes after contact with the antigen, which is called allergen, in the case of this form of hypersensitivity type I or anaphylactic reactions. These reactions usually result in exacerbated IgE production specific for a given allergen, but may also be mediated by IgG. In certain cases the exaggerated IgE production is a personal or familial tendency as in the case of atopy [8].The development of atopic allergic disease (Figure 2) is divided into two phases: the first is the sensitization and the second phase is the secondary contact [9].
Figure 2. Immunopathology of allergic reactions (adapted from [6]).
In sensitization phase the allergens (normally proteins) which come into contact with the respiratory or oral mucosal will be endocytosed by antigen-presenting cells (APCs) such as monocytes, macrophages, and dendritic cells. The antigen is broken down to reveal the epitope. Once processed, the antigen is bound to the MHC class II molecules on the surface of these cells and the complex is presented to the CD4+ T cell receptor. The CD4+ T cell will be activated, deliver and initiate the production of cytokines such as interleukin-4 (IL-4), interleukin-5 (IL-5) and interleukin-13 (IL-13). The IL-4 and IL-13 induce isotype switching to IgE in B cell, with the same specificity as the activated CD4 + T cell, allowing the synthesis IgE specific for that allergen. This IgE will bind to low-affinity IgE receptor (FcεRI) present in mast cells, monocytes, macrophages and dendritic cells. Mast cells are usually located in the mucous, a privileged place to come into contact with allergens. In a situation in which the levels of IgE specific for a given allergen are increased, it increases the proportion of specific IgE on the surface of mast cells that in turn increases the probability of an IgE bound to FcεRI
5
recognize the allergen. The Increase in IgE, IL-4 and IL-13 contributes to the transendothelial migration of eosinophils and T cells while increase IL-5 is the major factor of eosinophil activation. The secondary contact phase occurs in a similar procedure to the sensitization phase. However, given the high levels of IgE bound to mast cells the recognition of allergen by these cells is much faster leading to an elevated mast cell degranulation. This degranulation release cytokines and various mediators such as histamine, tryptase and derivatives of arachidonic acid [8].In the industrialized countries, however, IgE responses to innocuous antigens predominate and allergy is an important cause of disease. Almost half the populations of North America and Europe have allergies to one or more common environmental antigens and, although rarely life-threatening, these cause much distress and lost time from school and work. Because of the medical importance of allergy in industrialized societies, much more is known about the pathophysiology of IgE-mediated responses than about the normal physiological role of IgE [6]. It is estimated that about 50% of children and young people have one or more allergy within the first 18 years of life [10].
Fungal allergens represent a major cause of atopic disorders. Most fungi possess multiple and diverse allergens. Some are metabolic products secreted outside the organism; other are cytoplasmic and structural components released on lysis or autolysis of the fungal cell [11]. Fungal allergens are usually associated with allergic lower respiratory tract diseases. Epidemiologic studies have associated mold sensitivity, especially to A. alternata and
Cladosporium herbarum, with the development, persistence, and severity of asthma. In
addition, sensitivity to Aspergillus fumigatus has been associated with severe persistent asthma in adults. A. fumigatus causes Allergic bronchopulmonary aspergillosis (ABPA) a disease characterized by exacerbations of asthma, recurrent transient chest radiographic infiltrations, coughing up thick mucus plugs, peripheral and pulmonary eosinophilia, and increased total serum IgE and fungus-specific IgE levels, especially during exacerbation. [4].
A. alternata is one of the principal species associated with allergic disease. Exposure and
sensitisation to this fungi have been recognised as risk factor for the development and persistence of allergic respiratory diseases [12].
1.2. Alternaria alternata
A. alternata is a fungus which has been responsible for causing leaf spot and other diseases
on several species of plant. It is an opportunistic pathogen on numerous hosts causing leaf spots, rots and blights on many plant parts. It can also cause upper respiratory tract infections and asthma in humans with compromised immunity.
6
A. alternata was first studied in 1817 by Christian von Esenbeck having been given the name Alternaria tenuis (Figure 3), thus established the genus Alternaria [13]. However, in 1912 Karlvon Keissler, who continued the study of A. tenuis, changed the name for A. alternata. In recognition of his research his name was added to fungus, which came to be named A.
alternata Keissl.
Figure 3. A. tenuis description by Christian von Esenbeck (1817) (adapted of [13])
A. alternata is taxonomically classified as follows: Kingdom: Fungi; Phylum: Ascomycota;
Class: Dothideomycetes (Euascomycetes); Order: Pleosporales; Family: Pleosporaceae, Genus: Alternaria; Species: Alternaria alternata.
Alternaria is usually found in fresh fruit and vegetables, however it also occurs in preserves,
juices, or sauces prepared from damaged products [14, 15]. Alternaria sp. species produce many metabolites toxic to plants, animals and humans. In humans those toxic metabolites are normally associated to allergic reactions such as allergic bronchopulmonary mycosis, allergic sinusitis, hypersensitivity pneumonitis, and atopic dermatitis, and allergic asthma. The allergens from A. alternata are presented in the following section.
1.2.1. Allergens from Alternaria alternata
The prevalence of Alternaria allergies was studied by Amato et al. using Skin prick tests (SPT). It was observated that 3% of individuals in Portugal and 20% of individuals in Spain showed a positive test. It was also demonstrated that A. alternata is an important factor in the onset of childhood allergic asthma [16].
To date, 17 allergens of A. alternata have been isolated. The table 1 presented the allergens from A. alternata listed by WHO/IUIS website, Allergome (platform for allergen Knowledge website).
7
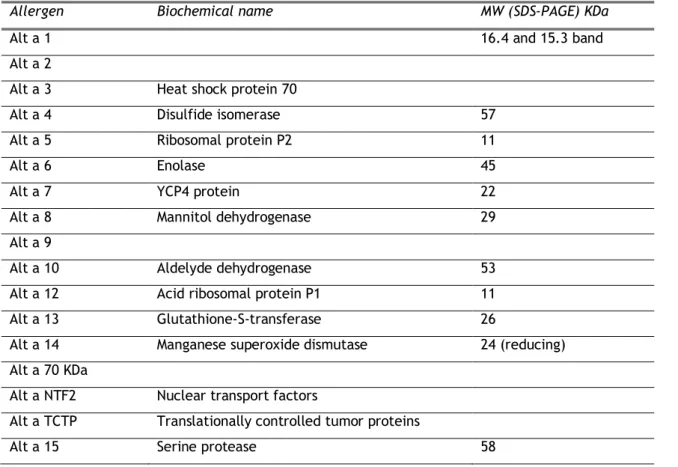
Table 1. The allergens of A. alternataAllergen Biochemical name MW (SDS-PAGE) KDa
Alt a 1 16.4 and 15.3 band
Alt a 2
Alt a 3 Heat shock protein 70
Alt a 4 Disulfide isomerase 57
Alt a 5 Ribosomal protein P2 11
Alt a 6 Enolase 45
Alt a 7 YCP4 protein 22
Alt a 8 Mannitol dehydrogenase 29
Alt a 9
Alt a 10 Aldelyde dehydrogenase 53
Alt a 12 Acid ribosomal protein P1 11 Alt a 13 Glutathione-S-transferase 26
Alt a 14 Manganese superoxide dismutase 24 (reducing) Alt a 70 KDa
Alt a NTF2 Nuclear transport factors
Alt a TCTP Translationally controlled tumor proteins
Alt a 15 Serine protease 58
Alt a 1 is a major allergen of A. alternata. This protein is a dimer with 29-KDa that migrates as two separate bands of 16.4 and 15.3 KDa in SDS-PAGE conditions [17]. Recombinant Alt a 1 and native Alt a 1 reacted with IgE from patients sensitive to Alternaria. Vailes et al. analyzed specific IgE and IgG responses to recombinand Alt a 1 in asthmatic and atopic dermatitis patients concluding that 93 % and 47 %, respectively, had antibodies to recombinand Alt a 1 [18, 19].
Alt a 2 allergen was identified first time by Bush et al. as a major allergen of A. alternata. In this study the recombinant Alt a 2 bound to IgE from 61% of patients sensitive to Alternaria [20]. However, Asturias et al. verified that this allergen only bound to IgE from 21% of patients sensitive to Alternaria, concluding that Alt a 2 was in fact a minor allergen from A.
alternata [21].
Alt a 3 allergen was identified and cloned by Vouge et al. The cloned fragments were identical to regions of heat shock protein 70 KDa from C. herbarum. In this study, Alt a 3 was identified on immunoblot in 5% of the sera of patients sensitive to A. alternaria [18].
8
Alt a 4 was cloned in 1990s by Achatz et al. This allergen is a protein disulfide isomerase of 57 KDa and contains at least one thioredoxin domain. Almost 42% of Alternaria-sensitive individuals feature hypersensitivity this allergen [22]–[24].Alt a 5 and Alt a 12 are ribosomal proteins. Alt a 5 is a 60s acidic ribosomal protein P2 with 11-KDa. 8-14% of Alternaria-sensitive patients feature IgE that binds to Alt a 5 in sera. Alt a 12 is a 60s acidic ribosomal protein P1 with 11-KDa, similarly to Alt a 5 [24].
Alt a 6 is an enolase, also called phosphopyruvate hydratase or 2-phospho-D-glycerate hydrolyase. This allergen was cloned and it was found that 22% of the A. alternata-sensitive individuals specifically reacted to recombined Alt a 6 [22], [24]. Kustrzeba et al. purified this enolase using chromatographic methods from crude extracts of A. alternata. Alt a 6 purified in this study presented a molecular weight of 47 KDa, pH optimum of 6.8 [25].
Alt a 7 is an analogous yeast protein YCP4 of 22 KDa with a sequence of 204-amino acid. Almost 7% of Alternaria-sensitive individuals present IgE that binds to this protein in sera [22, 24].
Schneider et al. cloned Alt a 8 allergen. This is an enzyme-NADP-dependent mannitol 2-dehydrogenase (MtDH) of 29-kDa with a complete sequence of 266-amino acid. Using IgE-ELISA and immunoblots, recombinant Alt a 8 was recognized by 41% of A. alternata-allergic individuals. The KM was determined using D-fructose and NADPH as substrats showing a KM =
474 mM and KM = 18.7 µM, respectively [26].
Alt a 9 is a protein with molecular weight of 43 KDa, however its structure and biochemical function are unknown. 5% of Alternaria-sensitive individuals feature IgE that binds to this protein in sera [22], [27].
Alt a 10 is a NAD-dependent aldehyde dehydrogenase (ALDH) of 53-KDa with 497-amino acids. 2% of patiens are hypersensitive to this allergen [22].
Alt a 13 is a glutathione-S-transferase (GST) of 26-KDa with 231-amino acids. 82% of
Alternaria-sensitive individuals feature IgE that binds to Alt a 13 in sera. Studies with
recombinant Alt a 13 and native alt a 13 have demonstrated similar allergenicity and enzymatic activity [28].
Alt a 70 KDa is a protein with 70 KDa, however its structure and biochemical function are unknown. Studies carried out by Portnoy et al. showed positive skin test in 87% of Alternaria-sensitive individuals, in a study using 16 subjects [29].
Alt a 14 is a mitochondrial enzyme, Mn-dependent Superoxide dismutase (MnSOD). It was shown that 6.6% of patiens are hypersensitive to this allergen [30, 31]. In other study,
9
Gaabriel et al. concluded that Alt a 14 is a minor allergen of A. alternata that can trigger a cross-reactive IgE response to Asp f 6, a MnSOD from A. fumigatus [12].Alt a NTF2 is a small homodimeric nuclear transport factor 2 of 28-KDa with 124 and 125 amino acids. The recombinant proteins, obtained by Weichel et al., were able to bind and cross-inhibit IgE binding and to elicit skin reactions in fungi-sensitized individuals [32], [33]. Alt a TCTP is a translationally controlled tumor protein. This protein has similar cross-reactive IgE epitopes as its C. herbarum homologue and show about 4 % prevalence of IgE reactivity in
Alternaria-sensitive individuals [34], [35].
Alt a 15 is a subtilisin-like serine protease of 53 KDa with 507 amino acids. Gabriel et al. cloned this allergen and performed a study where 3 of 53 mold-sensitized subjects had IgE+ binding in reducing immunoblot (GenBank: AHZ97469.1). Boitane et al. in another study concluded that A. alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2 using stratum of this fungi [36]. However structure and kinetic parameters are unknown, since the native protein was not already purified.
Postigo et al applied the concept of molecular diagnostics to study a large group of patients labeled as "allergic to Alternaria" and demonstrated that 6.6% of these patients were not sensitized to the major allergen, Alt a 1 or to enolase Alt a 6. Moreover, the sensitivity of these patients to a MnSOD, orthologous to Aspergillus allergen, Asp f 6, could justify the initial labeling [30]. On the other hand, a main difference to other sources is that fungi may colonize the human body, and they may damage airways by the production of toxins, proteases, enzymes and volatile organic compounds. For some of the allergens (e.g. serine proteases), extensive cross-reactivity was demonstrated, making these proteins fungal pan-allergens [24]. Therefore, the pan-allergens included in serine proteases family, associated to date with Alternaria provide one of the targets in the study of allergic phenomena caused by fungal products. Moreover, verification of the sensitizing nature of fungal species including
Curvularia, which do not express orthologous of Alt a 1 but, have a significant correlation
with Alternaria sensitization data, suggests that some other allergens such as serine protease described as a major allergen of Curvularia [37], could have their orthologous in Alternaria. In a recent study, Gabriel et al. concluded that A. alternata vacuolar serine protease Alt a 15 behaves as a cross-reactive minor allergen in an A. alternata-sensitized population. Alt a 15 (or the highly homologous protein Cur l 4) may also be considered a marker for Curvularia
lunata sensitiza-tion in the absence of Alt a 1 sensitization, which may underlie the initial
10
Purification and characterization of the serine protease of A. alternata will allow to obtain relevant data about this protein and its cross-reactivity, which represents a significant added value to clinical diagnosis of allergic patients.1.3.
Purification of fungal serine protease
In recent years, several studies have been carried out using different purification methods in order to study and characterize the fungal serine proteases. Chromatography is the principal method used in separation of these enzymes because it presents high efficiency in the fractionation of proteins in complex mixtures maintaining its physicochemical properties. The purification of proteins using chromatographic methods is usually divided into three steps: capture, purification and polishing. The capture step aims to collect and concentrate the protein matrix. The purification step is an enlarged separation of the proteins forming the protein matrix, which is the most important step in the purification process. In this step the protein was obtained with a high degree of purity. The polishing step is a final step aiming to increase the purity of the protein matrix by removing the proteins which resemble the protein of interest. Each of these steps may be composed of one or more chromatographic methods [39]. During the purification steps different physicochemical properties of the protein of interest can be exploited using different chromatographic methods (Table2) [40].
Table 2. Physicochemical properties of the proteins exploited by different chromatographic methods
Chromatographic method Basis of separation
Ion exchange Charge, charge distribution Interaction hydrophobic Hydrophobicity
Affinity chromatography Molecular recognition Gel filtration Size, shape
Most of chromatographic processes that have been used in the purification of fungal serine proteases are based on ion exchange chromatography, or both on ion exchange and interaction hydrophobic chromatograph. Additionally, affinity chromatography and gel filtration have also been used. Each of these methods will be presented and discussed in the next sections.
11
1.3.1 Purification based on ion exchange chromatography
The ion exchange chromatography (IEX) has been the most frequently used method in the purification of fungal serine proteases. IEX separates biomolecules according to differences in their net surface charge (Figure 4) [41].
Figure 4. Separation principles in ion exchange chromatography (adapted from [41])
The principal functional groups utilized in IEX were: Quaternary ammonium (Q) and Diethylaminoethyl (DEAE) used for anion exchange chromatography (AEX) and Methyl sulfonate (S), Sulphopropyl (SP) and Carboxymethyl (CM) for cation exchange chromatography (CEX). In Table 3 are summarized different methods used in the recent years for purification of fungal serien proteases by IEX.
12
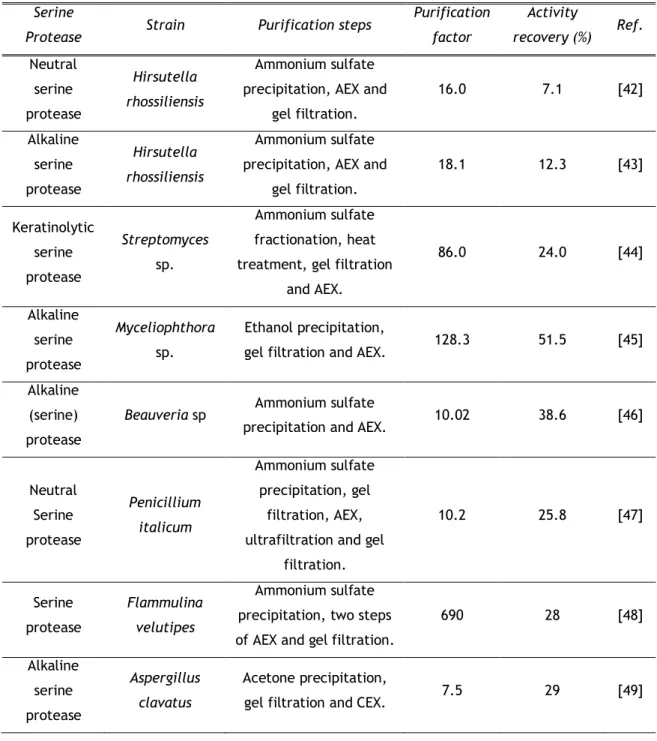
Table 3. Purification of fungal serine protease using IEXSerine
Protease Strain Purification steps
Purification factor Activity recovery (%) Ref. Neutral serine protease Hirsutella rhossiliensis Ammonium sulfate precipitation, AEX and
gel filtration. 16.0 7.1 [42] Alkaline serine protease Hirsutella rhossiliensis Ammonium sulfate precipitation, AEX and
gel filtration. 18.1 12.3 [43] Keratinolytic serine protease Streptomyces sp. Ammonium sulfate fractionation, heat treatment, gel filtration
and AEX. 86.0 24.0 [44] Alkaline serine protease Myceliophthora sp. Ethanol precipitation,
gel filtration and AEX. 128.3 51.5 [45] Alkaline
(serine) protease
Beauveria sp Ammonium sulfate
precipitation and AEX. 10.02 38.6 [46]
Neutral Serine protease Penicillium italicum Ammonium sulfate precipitation, gel filtration, AEX, ultrafiltration and gel
filtration. 10.2 25.8 [47] Serine protease Flammulina velutipes Ammonium sulfate precipitation, two steps of AEX and gel filtration.
690 28 [48] Alkaline serine protease Aspergillus clavatus Acetone precipitation,
gel filtration and CEX. 7.5 29 [49]
Bin Wang and co-workers purified both the neutral and alkaline serine proteases from mycelial extract of Hirsutella rhossiliensis by ammonium sulfate precipitation, AEX using a Q-Sepharose column with a linear NaCl gradient and finally a gel filtration step. The neutral serine protease was obtained with a 16-fold increase in specific activity with a recovery of 7.1% [42] whereas the purification of the alkaline serine protease results in an 18.1-fold increase in specific activity with an activity recovery of 12.3% [43]. A similar approach was applied for purification of a keratinolytic alkaline serine protease (KERAB) from Streptomyces sp. using ammonium sulfate fractionation, heat treatment, gel filtration and AEX with a Q-Sepharose column. The KERAB was isolated with an 86-fold increase in specific activity with
13
24% of the total activity of the crude [44]. Another purification strategy was developed by Zanphorlin et al. in 2011 being purified a alkaline serine protease from Myceliophthora sp [45]. The purification protocol consisted of ethanol precipitation, gel filtration and AEX using a Source 15-Q column equilibrated with 20mM Tris buffer at pH 8.5 and eluted with a linear gradient of NaCl. The alkaline serine protease was purified 128.3 fold with a final recovery of 51.5% [45]. This purification strategy shows a greater increase in specific activity and recovery rate, being the best purification scheme when a single step of AEX is used with quaternary ammonium as principal functional group.DEAE is another functional group used in AEX. The alkaline serine protease from Beauveria sp. was purified with two chromatographic steps. The first was an ammonium sulfate precipitation and the second step involved AEX using a DEAE-cellulose column equilibrated and eluted with 50mM phosphate buffer pH 7. Serine protease was purified 10.02-fold with 38.6 % of yield [47]. Recently, a neutral serine protease from Penicillium italicum was purified by Abidi and co-workers using three steps. The two initial consisted of an ammonium sulfate precipitation and gel filtration. The last step included AEX with a DEAE-Sepharose column equilibrated and washed with 20 mM Tris-HCl buffer. The bound proteins were eluted with a linear gradient of sodium chloride [47]. The serine protease was purified 10.2-fold with a recovery of 25.8% [47], being a value slightly lower when compared to the protocol used in the purification of a alkaline serine protease from Beauveria sp., showing a modest increase in the degree of purity. Another approach using DEAE was developed by Iketani et al. to purify a serine protease with caspase- and legumain-like activities from Flammulina velutipes [48]. The purification process started with an ammonium sulfate precipitation followed by two steps of anion exchange chromatography using two different types of DEAE columns equilibrated with 20mM Tris-HCl buffer pH 8 and eluted with a linear gradient of NaCl in the same buffer. After a gel filtration final step the serine protease showed a 690-fold increase in specific activity as estimated by hydrolysis of Ac-YVAD-MAC, with an overall yield of 28% [48]. However, this result was obtained with a non-specific extract contrarily to the previous protocol that used a highly specific substratum.
Alkaline serine protease from Aspergillus clavatus was purified by acetone precipitation, gel filtration and a cation exchange chromatography using a CM-sepharose column equilibrated with 25mM Tris-HCl buffer pH 8 and eluted with a linear gradient of NaCl in the range of 0-0.5 M in the buffer. This was the only protocol that used CEX obtaining a 7.5-fold with a recovery 29% and a specific activity of 37600U/mg of protein [49].
14
1.3.2 Purification based on ion exchange and hydrophobic
interaction chromatograph
Hydrophobic interaction chromatography (HIC) is based on the interaction of biomolecules to a weakly hydrophobic surface at high salt concentrations, followed by elution with a descending salt gradient (Figure 5) [50].
Figure 5. Separation principles in hydrophobic interaction chromatography (adapted for [51] ) HIC can be considered a general chromatographic technique being used in the purification of fungi serine proteases together with ion exchange chromatography. In table 4 are summarized different methods used in the recent years for purification of fungal serine proteases by IEX and HIC.
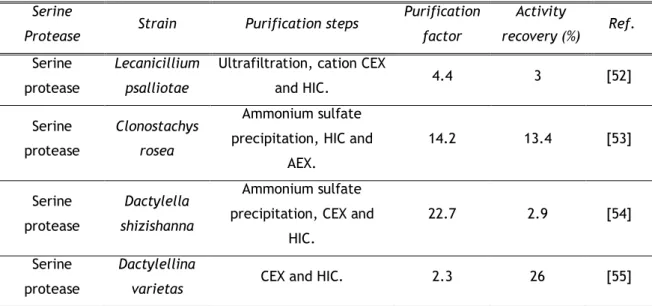
Table 4. Purification of fungal serine proteases using IEX and HIC Serine
Protease Strain Purification steps
Purification factor Activity recovery (%) Ref. Serine protease Lecanicillium psalliotae
Ultrafiltration, cation CEX
and HIC. 4.4 3 [52] Serine protease Clonostachys rosea Ammonium sulfate precipitation, HIC and
AEX. 14.2 13.4 [53] Serine protease Dactylella shizishanna Ammonium sulfate precipitation, CEX and
HIC.
22.7 2.9 [54]
Serine protease
Dactylellina
15
Ke-Qin Zhang and co-workers purified four fungi serine proteases using different methods based on IEX and HIC. Extracellular serine protease produced by Lecanicillium psalliotae was purified using ultrafiltration and two chromatographic steps. A first CEX step was applied with a SP Sepharose column equilibrated with 10 mM sodium phosphate buffer pH 6.0 and with 10 mM sodium phosphate buffer pH 6.0 containing 0.5 M NaCl as eluent. The HIC step was performed with a Phenyl Sepharose column equilibrated with 50 mM sodium phosphate buffer pH 7.0 and 1 M ammonium sulfate. The elution was performed with 50 mM sodium phosphate buffer pH 7.0 and a 4-fold with a recovery of 3% were achieved [52].The second serine protease purified by this working group was the extracellular serine protease from Clonostachys rosea (PrC) using a different protocol that results in an increased purification factor and activity recovery. PrC was purified by a precipitation step with ammonium sulfate followed by a HIC step with Phenyl Sepharose column equilibrated with 20 mM Tris-HCl pH9.0, containing 1M ammonium sulfate and eluted with a linear salt gradient of 1 to 0 M in 20 mM Tris-HCl buffer pH 9.0. Finally an AEX step using a SOURCE 15Q column equilibrated with 20 mM Tris-HCl pH 9.0 and eluted with continuous NaCl gradient (0-0.5 M) was applied. This strategy resulted in 13.4% PrC recovery and a 14.2-fold purification with a specific activity of 241 U mg-1 [53].
In 2005, Ke-Qin Zhang and co-workers purified an extracellular serine protease from
Dactylella shizishanna (Ds1) using ammonium sulfate precipitation, CEX and HIC. The first
chromatographic step was carried out using a SP Sepharose column with a three stage isocratic elution of 6%, 30% and 100% of 10 mM sodium phosphate buffer pH 6.5 with 1 mMNaCl. The last step was HIC using a Phenyl Sepharose column eluted with a three stage isocratic elution of 30%, 50% and 100% of the buffer without salt. D s 1 was purified 22.7-fold with 2.9% recovery and 240.3 U mg-1 specific activity [54].
The last serine protease purified by IEX and HIC was the extracellular serine protease from
Dactylella varietas (Dv1). This protocol consisted of a CEX step with SP Sepharose
equilibrated with 10 mM sodium phosphate pH 6.0 and eluted with the same buffer containing 0.5 M NaCl. The second and last step consisted of HIC using Phenyl Sepharose equilibrated with 50 mM sodium phosphate pH 7.0 containing 1 M ammonium sulfate, then eluted with same buffer without salt. Dv1 was purified 2.3-fold with 26% recovery and 27.5 U mg-1 of
16
1.3.3 Purification based on gel filtration chromatography
Gel filtration chromatography (GF), also known as size exclusion chromatography, is used to separate molecules of different sizes (Figure 6) [56].
Figure 6. Separation principle in gel filtration chromatography (adapted from [56])
Usually this technique is used in the polishing step, after other chromatographic steps, however sometimes it can also be applied as unique step. In Table 5 are summarized the different methods used in the recent years for purification of fungal serine proteases by gel filtration.
Table 5. Purification of fungal serine proteases using gel filtration Serine
Protease Strain Purification steps
Purification factor Activity recovery (%) Ref. Serine Protease Graphium putredinis Ethanol precipitation
and gel filtration. 8.263 36.49 [57] Serine
protease
Trichoderma haszianum
Ethanol precipitation
and gel filtration. 11.50 29.39 [57] Serine
protease
Penicillium waksmanii
Ethanol precipitation
and gel filtration. 53.5 80.3 [58]
S. Savitha and co-workers purified two extracellular serine proteases from Graphium
putredinis and Trichoderma harzianum using the same protocol with two chromatographic
steps. The first was ethanol precipitation and the second step involved gel filtration using a Sephadex G 100 column equilibrated with 50 mM sodium phosphate buffer, pH 7.0 and eluted with the same buffer containing 0.5 M NaCl. G. putredinis extracellular serine protease was purified 8.63-fold with 36.49 % of yield and a specific activity of 14.85 IU/mg. On the other
17
hand, T. harzianum serine protease was obtained with a specific activity of 14.50 IU/mg, showing a better purification fator (11.50-fold) but a slight lower yied (29.39 %) [57].The extracellular serine protease secreted by Penicillium waksmanii was purified by ethanol precipitation and gel filtration using a Sephadex G-75 column equilibrated and eluted with 50 mM acetate buffer, pH 5.5 with 50 mM NaCl. A 53.5- fold with a recovery 80.3% and a specific activity of 27.1U/mg were obtained [58].
Although both studies are based on the same purification steps it was observed that in the second study, the authors obtained better results both in specific activity as well as in purification factor and recovery.
1.3.4 Purification based on affinity chromatography
Affinity chromatography separates proteins on the basis of a reversible interaction between a protein and a specific ligand coupled to a chromatographic matrix (Figure 7). The technique offers high selectivity, high resolution, and usually high capacity for the protein of interest [59].
Figure 7. Separation principles in affinity chromatography (adapted from [41])
Concanavalin A (Con A) is a functional group widely used in affinity chromatography for separation of fungi serine proteases. This functional group was applied to purify and identify a serine protease by C. lunata (Cur l 1) usually associated to human allergies. The column was washed with 10 mM Tris-HCl pH 7.2, 5 mM CaCl2 and 5 mM MgCl2 buffer (buffer A) and eluted
with 0.2 M methyl α-D mannopyranoside in buffer A. The Con A bound fraction was then purified by gel filtration on a Superdex G-75 column. The Cur l 1 was identified by ELISA and immunoblotting with Curvularia allergic patient’s sera. The proteolytic activity was exhibited with gelatin, skimmed milk and casein. Serine protease specific inhibition experiments were also performed showing a significant inhibition of proteolytic activity that was observed with
18
phenylmethylsulfonyl fluoride (PMSF) (80%), pefabloc (72%), leupeptin (67%) and aprotinin (67%) confirming it is a serine protease [37].Pseudogymnoascus destructans serine protease (PdSP1) is an extracellular serine protease
responsible for high mortalities in North American cave bat populations. This protease was isolated by preparative isoelectric focusing and lectin affinity chromatography, using a Con A Sepharose. The column was washed with 20 mM Tris, 0.5M NaCl, pH 8 buffer and eluted with 20 mM α-D-methylglucoside, 0.5M NaCl, 20 mM Tris, pH 4 buffer. The presence of a serine protease was confirmed by the complete inhibition of the protease activity in the pooled fraction, using PMSF. Structural characterization of PdSP1 by MALDI-TOF MS, Orbitrap MS/MS, and Edman amino-terminal peptide sequencing matched it directly to a supposed protein accession from the sequenced P. destructans genome that was further identified as a MEROPS family S8A subtilisin-like serine peptidase [60].
19
1.4.
Aim of the present study
The global aim of the present work was to develop new chromatographic strategies to purify the native serine protease from an A. alternata extract and to carry out its biochemical characterization.
Thus the work had the following specific objectives:
a) Purification of the native serine protease using two chromatographic methods: Affinity chromatography with a Con A Sepharose column specific for glicoproteins such as serine proteases and IEX chromatography with DEAE Sepharose fast flow.
b) Biochemical characterization of the purified serine protease by studying the effect of pH, temperature, metal ions and inhibitors in the enzyme activity, as well as the kinetic parameters KM and Vmax.
20
2. Materials and Methods
21
2.1. Materials
2.1.1. Chemicals
All reagents were of analytical grade and were supplied as indicated in Table 6. Protein determination was performed using the Pierce™ BCA Protein Assay Kit.
Table 6. Reagents used in the experimental work
Reagent Purity
grade
Provider
Enzyme activity determination
Casein from bovine milk Trichloroacetic acid Folin & Ciocalteu’s phenol reagent
Sodium carbonate Sodium acetate trihydrate
L-Tyrosine ---- 99% ---- 99.5% 99% 98% Sigma Sigma Sigma Sigma Sigma Sigma Protein purification Hydrochloric acid Tris base Calcium chloride Magnesium chloride methyl α-D mannopyranoside Sodium chloride 37% 99% 90% --- 99% 99% Fisher Sigma Applichem Vaz Pereira Sigma Fisher Effect of inhibitors on purified enzyme activity phenylmethylsulfonyl fluorid ethylenediamine tetraacetic acid ethylene glycol-bis
(β-aminoethylether)-tetraacetic acid 98.5% 98.5% Sigma VWR BDH prolab Sigma
22
Molecular weight
Acrilamide Sodium dodecyl sulphate molecular weights from 11 to 245 kDa
Tris-HCl Bromophenol blue Glycerol Β-mercaptoethanol 40% --- Applichem Vaz Pereira NZYColour Protein Marker II by Nzytech gene &
enzymes Effect of pH on purified enzyme activity Acetic acid Sodium acetate Monosodium phosphate Disodium phosphate Glycine Sodium hydroxide 99% 99% 99% 99% 99% Pure Chem-lab Merck Acros organics Panreac Fisher Vencilab
Effect of metal ions on purified enzyme activity Magnesium chloride Calcium chloride Manganese Chloride Zinc Chloride Iron sulfate Copper sulfate Cobalt chloride 90% 97% 98% 99% 99% 98% Vaz Pereira Applichem Acros organics Sigma Sigma Sigma Sigma
23
2.1.2. Materials and Equipments
In Table 7 are specified the different materials and equipments used in each experiment of the work.
Table 7. Materials and equipments
Materials and equipments characteristics
Meter pH Termo scientific
Balance
Sartorius CP225D Max 220g; d= 0,01 mg
Kern plj 510-3M Max 510g; d= 0,001g
Spectrophotometers Pharmacia Biotech
Ultrospec 3000
Centrifugal Beckman Coulter
Allegra® 25R
Electrophoresis System
SE260 Mighty Small II Deluxe Mini Vertical Electrophoresis
Power supply
Hoefer®
Biorad POWER PAC Basic
Concentrators Vivacell 250 Sartorius
stedium
“Fast Protein Liquid Chromatography” (FPLC®)
LCC-501 Plus controller Pumps P-500
Conductivity detector UV-1 optical drive Recorder REC 112
Collector FRAC-100 fractions
Amersham Pharmacia Biotech
24
1)2.2. Methods
2.2.1. Strains and fungal extracts (microorganism and culture
conditions)
A. alternata strain CBS 104.26 (Centraalbureau von Schimmelcultures, Utrecht, The
Netherlands) was grown amid Czapek Dox Broth for 15 days at a constant temperature of 25 ° C, with occasional agitation. After 15 days growth, the mycelia were separated from the medium by vacuum filtration using Whatman culture filter paper 22 µm. The mycelium was then placed at -40º C overnight and the next day pulverized using a mortar round.
Culture filtration extracts were obtained according to the method described by Martinez et al [61]. Briefly, culture filtrations were obtained by successive filtration using Whatman No. 1, AP, and sterilizing filters. The filtration was dialyzed by tangential ultrafiltration with a 5 KDa cutoff point.
2.2.2. Enzyme-activity determination
Protease activity was determined using a modified method from R.B. Wang et al [54]. 100 µL protease solution was added to 300 µL of 1% (w/v) of casein previously dissolved in 50mM phosphate buffer (pH 7.5) and pre-incubated at 40ºC. The mixture was incubated at 40ºC for 15min and the reaction was quenched using 600µL of 110 mM trichloroacetic acid (TCA). The mixture was centrifuged at 15,000 × g for 15 min at 4º C. Then 250µL of the clear supernatant was mixed with 625 µL 500 mM sodium carbonate buffer and 125 µl Folin-phenol agent, followed by incubation at 37º C for 30 min. The absorbance value of the resulting supernatant was measured at 680 nm against a blank control. The protease activity was determined using the Tyrosine Standard Curve with the following L-tyrosine concentrations: 0.0055 µmoles, 0.011 µmoles, 0.0275 µmoles, and 0.55 µmoles. A standard curve should be generated each time the assay is performed. The units of protease activity per ml of protease sample were determined using the following equation (1):
µmoles of tyr = µmole tyrosine equivalents released; reaction volume = Total volume (in milliliters) of assay; reaction time= Time of assay (in minutes) as per the Unit definition; Sample vol. = Volume of Enzyme (in milliliters) of used enzyme; Vol. assayed = Volume (in milliliters) used in Colorimetric Determination
One unit (U) of protease activity was defined as the amount of enzyme that hydrolysed the substrate and produced 1µg tyrosine in 1 minute under the assay conditions.
25
2.2.3. Protein determination
The protein concentration was determinate by bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit) using bovine serum albumin (BSA) as a standard [62]. This method combines the known reduction of Cu+2 to Cu+1 by protein in an alkaline medium with the highly sensitive and
selective colorimetric detection of the cuprous cation using a unique reagent containing bicinchoninic acid. The purple-colored reaction product of this assay is formed by the chelation of two molecules of BCA with one cuprous ion. This water-soluble complex exhibits a strong absorbance at 562nm that is nearly linear with increasing protein concentrations. Initially, 25 µL of each BSA standard (were prepared used Table 8) and sample were pipetted into a microplate well. Thereafter were added 200µL of the WR, 50 parts of BCA Reagent A with 1 part of BCA Reagent B, to each well and mix plate thoroughly on a plate shaker for 30 seconds and was incubated at 37°C for 30 minutes. The absorbance was measure at or near 562 nm on a plate reader. The protein content during the purification stages was calculated from a standard curve.
Table 8. Guide to prepare of BSA standards
Vial Volume of Diluent (µl) Volume and Source of BSA (µl) Final BSA Concentration (µg/ml)
A 0 300 µl of Stock 2000 B 125 375 µl of Stock 1500 C 325 325 µl of Stock 1000 D 175 175 µl of vial B dilution 750 E 325 325 µl of vial C dilution 500 F 325 325 µl of vial E dilution 250 G 325 325 µl of vial F dilution 125 H 400 100 µl of vial G dilution 25 I 400 0 0
2.2.4. Protease purification
The sample was loaded onto a Con A Sepharose (GE Healthcare) affinity column washed with 10 mM Tris-HCl pH 7.2 buffer with 5 mM CaCl2 and 5 mM MgCl2 (buffer A). Con A bound proteins were eluted with 0.4 M methyl α-D mannopyranoside (buffer B) and were collected fractions of 3 ml at a flow rate of 0.5 ml min-1.
The Con A bound proteins were resuspended in the 20 mM Tris-HCl buffer (buffer C) and loaded onto a DEAE Sepharose fast flow (Sigma) anion exchange column. The column was washed with buffer C and the bound proteins were eluted by stepwise elution using 40%, 80% and 100% 20 mM Tris-HCl, pH 8, 0.5 M NaCl (buffer D). Fractions of 1.5 ml were collected at a
26
flow rate of 0.75 ml min-1. All the purification steps were conducted at room temperatures.All chromatography steps were carried out on an FPLC system.
2.2.5. Biochemical characterization of purified enzyme
2.2.5.1. Electrophoretic analysis
Two-dimensional (2D) gel electrophoresis analysis was performed according to the method described by Liu et al. [63]. Proteins were separated by 2-DE analysis using the Ettan IPGphor 3 System (GE Healthcare) for the first dimension isoelectric focusing (IEF) and the Ettan DALTSix System (GE Healthcare) to perform SDS- PAGE in the second dimension. The IEF was performed with ImmobilineTM DryStrip of 24 cm (pH 3-10), which were rehydrated overnight
at room temperature (20°C) with 450 µL of the rehydration buffer (7 M Urea, 2 M thiourea, 20 mM DTT, 1% IPG buffer and bromophenol blue). The isoeletric focusing was performed at 20°C, Step 1: 500V for 1 h, Step 2: 1000V for 1 h, Step 3: Grad 10000V for 4 h and Step 4: Step 10000V for 2 h. After completion of the Ettan IPGphor 3 program, the strips were stored at −80°C. The individual strips were then equilibrated to resolubilize proteins and reduce disulfide bonds. The second-dimension electrophoresis was performed using a 12.5% SDS-polyacrylamide gel. The gels were run at 100 V for around 4.5 h, until the dye front reached the bottom of the gel. The protein spots on analytical and preparative 2-DE gels were visualized by Blue silver colloidal Coomassie G-250, prepared according to the method described by [64].
Purity and molecular weight of the enzyme were determination using Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) described by Laemmli et al. [65]. The same treated by 5µl of a reduction buffer (500mM Tris-HCl (pH 6.8), 10% (w/v) SDS, 0.02% bromophenol blue (w/v), 0.2% glycerol (w/v), 0.02% β-mercaptoethanol (v/v) to the 20 µl of concentrated samples and denatured at 100º C during 10 minutes and electrophoretic assay were executed on 5% (w/v) stacking gel and a 15% (w/v) separating gel with a running buffer (25mM Tris, 192mM glycine and 0.1% (w/v) SDS at 120V for 120 minutes. The molecular mass of the enzyme was estimated using a ready-to-use mixture of 12 highly purified pre-stained proteins, covering a wide range of molecular weights from 11 to 245 kDa (NZYColour Protein Marker II by Nzytech gene & enzymes).
2.2.5.2. Effect of pH on purified enzyme activity
The effect of pH on the enzyme activity was measured at different pH between 3 to 12 using the following buffers: 0.1 M of acetate buffer (pH 5), sodium buffer (pH 6 and 7), Tris-HCl buffer (pH 8 and 9) and glycine-NaOH (pH 10 to 13). To assay relative activity, the enzyme was dissolved in each buffer with different pH and the mixture was incubated for 15 minutes at room temperature. Protease activity was determined using a method described in Section 2.2.2. The activity at beginning of the experiment was considered as control (100%).
27
2.2.5.3. Effect of temperature on purified enzyme activity
The effect of temperature on the enzyme activity was determined by incubating the enzyme reaction at temperatures between 30 °C to 70 °C for 15 minutes using 20 mM Tris-HCl buffer pH 8. Protease activity was determined using a method described in Section 2.2.2. The non-heated enzyme was considered as control (100%).
2.2.5.4. Effect of protease inhibitors on purified enzyme activity
For the determination of protease type, purified protease was pre-incubated for 1h with following inhibitors: 1mM phenylmethylsulfonyl fluorid (PMSF), 1mM ethylenediamine tetraacetic acid (EDTA) and 1 mM ethylene glycol-bis (β-aminoethylether)-tetraacetic acid (EGTA). Protease activity was determined using a method described in Section 2.2.2. The residual activity was measured at 50ºC, pH 8 using casein as a subtrate. Control without inhibitor was taken as 100%.
2.2.5.5. Effect of metal ions on purified enzyme activity
The effect of metal ions on the protease activity was determined in the presence of the following ions: Mg2+, Ca2+, Mn2+, Zn2+, Fe2+, Cu2+ and Co2+ at a final concentration of 5 mM. The
enzyme was previously incubated for 30 minute at room temperature. Protease activity was determined using a method described in Section 2.2.2. The residual activity was measured at 50ºC, pH 8 using casein as a subtrate. Control without inhibitor was taken as 100%.
2.2.6. Determination of kinetic parameters
The kinetic constants Km and Vm of the purified enzyme were calculated using a constant amount of the enzyme incubated in the presence of increasing amount (0.2 mg/ml to 20 mg/ml) of 2% (w/v) of casein. Protease activity was determined using a method described in Section 2.2.2. The residual activity was measured at 50ºC, pH 8. Maximum velocity of the reaction (Vmax) and the Michaelis constant (KM) were calculated based on Michaelis–Menten
28
3. Results and Discussion
29
3.1. Protease purification
As previously mentionated, it was found that some patients labeled as "allergic to Alternaria" were not sensitized to the major allergen, Alt a 1 or to enolase Alt a 6. Moreover, these patients were sensitized to a serine protease described as a major allergen of Curvularia, which do not express orthologous of Alt a 1, but have a significant correlation with Alternaria sensitization data. This suggests that some other allergens such as Curvularia serine protease [37], could have their orthologous in Alternaria. In a preceding work a serine protease from mRNA from A. alternaria was cloned [38]. Using Clustal Omega, a multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments between three or more sequences, the proteins sequences of serine protease from C. lunata (UniProt: ACF19598.1) and A. alternata (UniProt: AHZ97469.1) were compared. It was verified 89.92% identity between the two sequences (Figure 8).
Figure 8. Multiple Sequence Alignment used Clustal Omega 1.2.1. Input Parameters: Output guide tree: false, Output distance matrix: false, Dealign input sequences: false, mBed-like clustering guide tree: true, mBed-like clustering iteration: true, Number of iterations: 0, Maximum guide tree iterations: -1, Maximum HMM iterations: -1, Output alignment format: clustal, Output order: aligned, Sequence Type: protein. ACF19598.1 subtilisin-like serine protease from C. lunata and AHZ97469.1 subtilisin-like serine protease, partial from A. alternata.
![Figure 1. Types of hypersensitivity reactions (adapted from [6]).](https://thumb-eu.123doks.com/thumbv2/123dok_br/18901423.935335/17.892.154.786.105.475/figure-types-hypersensitivity-reactions-adapted.webp)
![Figure 2. Immunopathology of allergic reactions (adapted from [6]).](https://thumb-eu.123doks.com/thumbv2/123dok_br/18901423.935335/18.892.172.765.389.745/figure-immunopathology-allergic-reactions-adapted.webp)
![Figure 3. A. tenuis description by Christian von Esenbeck (1817) (adapted of [13])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18901423.935335/20.892.275.661.258.518/figure-tenuis-description-christian-von-esenbeck-adapted.webp)
![Figure 4. Separation principles in ion exchange chromatography (adapted from [41])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18901423.935335/25.892.406.527.254.536/figure-separation-principles-ion-exchange-chromatography-adapted.webp)

![Figure 7. Separation principles in affinity chromatography (adapted from [41])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18901423.935335/31.892.417.521.617.853/figure-separation-principles-affinity-chromatography-adapted.webp)