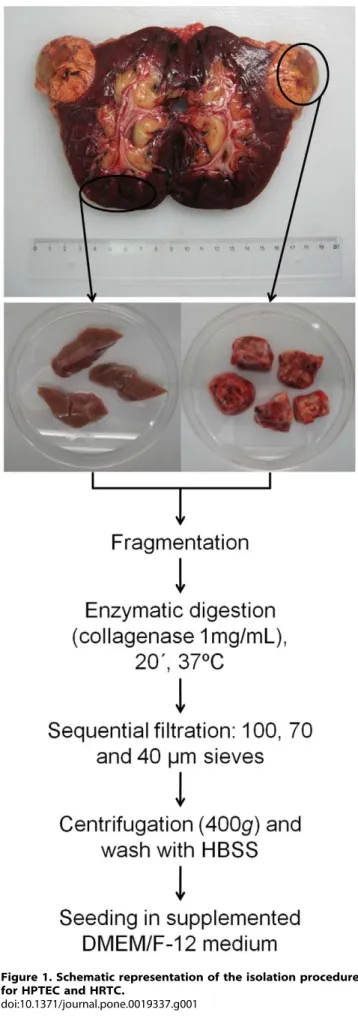
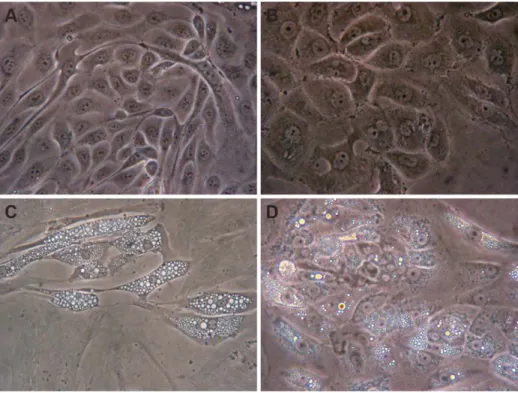
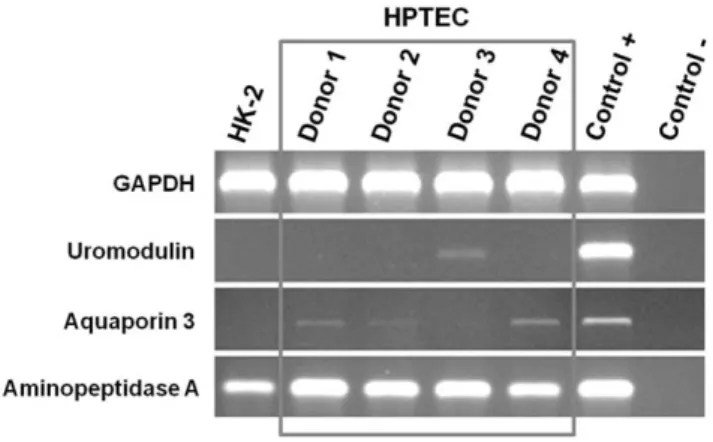
A Rapid and Simple Procedure for the Establishment of Human Normal and Cancer Renal Primary Cell Cultures from Surgical Specimens
8
0
0
Texto
Imagem

Documentos relacionados