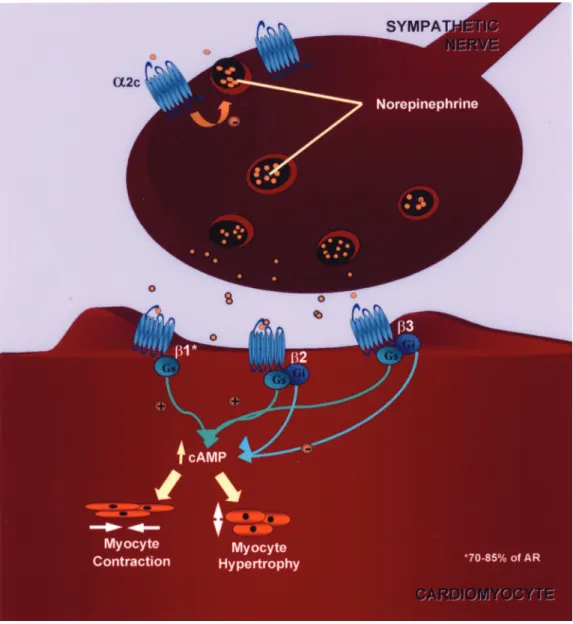
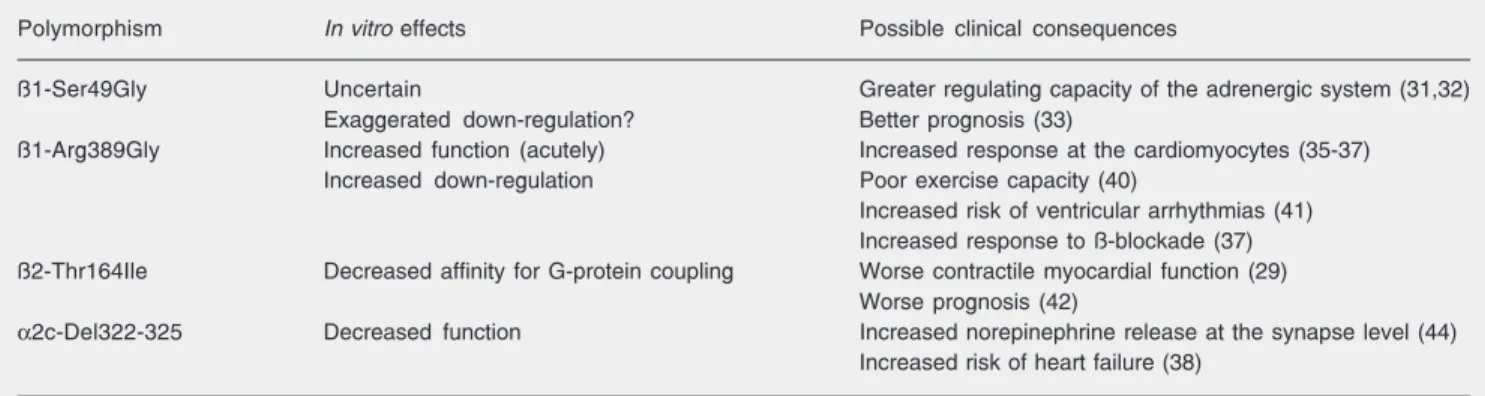
The role of adrenergic receptor polymorphisms in heart failure
Texto
Imagem

Documentos relacionados
Objective: To correlate serum functional circulating antibodies with beta adrenergic (Ab- β ), muscarinic (Ab-M) or muscarinic and beta adrenergic (Ab-M β ) activity, the autonomic
Association between beta-1 and beta-2 adrenergic receptor gene polymorphisms and the response to beta-blockade in patients with stable congestive heart failure. Small KM, Wagoner
Objective: This study assessed the association between the two functional polymorphisms of the beta1-adrenergic receptor gene (ADBR1), Ser49Gly and Arg389Gly, and the presence
lower in postinfarction myocytes (Figure 4) although the relative increases induced by 1 µM isoproterenol were the same for sham and myocardial infarction rats. The functional
Myocardial signaling defects and impaired cardiac function of a human beta 2-adrenergic receptor polymorphism expressed in transgenic mice. Alpha 1-adre- nergic receptor responses
IL-1 beta, IL-1 receptor antagonist and soluble type II IL-1 receptor in synovial fluid of patients with temporomandibular disorders. Arch
1 – Expressions of transforming growth factor-beta (TGF-β 1 ) and transforming growth factor-beta receptor I (TβRI) in the aortic tissues of aortic dissection (AD) and
Evaluation of the Influence of the Codon 16 Polymorphism of the Beta-2 Adrenergic Receptor Gene on the Incidence of Arterial Hypotension and Ephedrine Use in Pregnant Patients
