(S
A
LIGHT
SINTESE
ANDRE ALE
T CONTR
E DE ÁCI
EXANDRE
ROLLED
DOS NU
COTTA G
SYNTH
UCLEICO
Disser Douto pela Ciênci LISBOA, 20UERRA VI
ESIS OF
OS CONT
rtação apres or em Bioquí Universidad ias e Tecnolo 010IDAL PINH
F NUCLE
TROLAD
sentada par ímica – Espe de Nova deogia
HEIRO
EIC ACID
DA POR
a obtenção ecialidade Bi Lisboa, FaDS
LUZ)
do Grau de otecnologia, aculdade de e eNº de Arquivo “copyright”
A meus pais
To my parents
ACKNOWLEDGEMENTS As we express our gratitude, we must never forget that the highest appreciation is not to utter words, but to live by them. John F. Kennedy In the course of the years passed, I had the privilege and honor of having the support of many people and institutions, whose contribution was decisive for the completion of the work here presented. Without them, none of it would have been possible. To all of you, I present my deepest appreciation, in particular to:
Aos meus orientadores Professor João Carlos Lima e Professor Pedro Viana Baptista, por me terem ensinado como um aluno e discutido ideias como um par. Pela vossa incansável disposição e dedicação à minha formação científica. Pela porta sempre aberta. Pelas palavras de apreço nas victórias, pela sensatez nos infortúnios e pelos berros na deriva. Por terem embarcado neste projecto. We are what we repeatedly do. Excellence then, is not an act, but a habit. Aristotle Ao meu amigo João Carlos, pelas infindáveis conversas acerca de tudo e mais alguma coisa, dentro e fora do laboratório. Por me ter ensinado a verdadeira essência da ciência. Por ter sido sempre o contra‐peso em qualquer situação explosiva e me ensinar a lidar com isso. Por ter sido um oásis de excelência que me ensinou a “ser arrogante o suficiente para acreditar em mim e humilde o suficiente para aprender com os melhores”. Por ter sido o contra‐peso que equilibrou a balança. I am easily satisfied with the very best. Winston Churchill Ao meu amigo Pedro, por nunca estar satisfeito, por toda a exigência e intransigências, que fizeram não mais do que me conduzir a ser mais, maior e melhor. Pelas infindáveis conversas e discussões, por vezes
acesas, acerca de política, bola ou temas menos elevados. Por ter sido o peso que desiquilibrou a balança.
À Fundação para a Ciência e Tecnologia, pelo apoio financeiro (SFRH/BD/24276/2005) que permitiu a condução dos trabalhos realizados e o patrocínio à participação em conferencias nacionais e internacionais.
Ao Departamento de Quimica e ao Departamento de Ciências da Vida, Faculdade de Ciencias e Tecnologia, Universidade Nova de Lisboa e aos seus membros pelo apoio e por disponibilizar as condições necessárias à execução dos trabalhos realizados. Em particular ao Zé Luis, Ricardo Franco, Jorge Caldeira, João Paulo Noronha, Filipe Folgosa, Luz, Rosario, Vitor, Idalina e Conceição, César, Maggie, Ana Paula, Sofia, João, Dora, Renato, Marco e Carla.
Ao Grupo de Fotoquímica e Quimica Supramolecular, por ter sido a minha escola de Química, e aos seus membros que me acompanharam desde o inicio da minha licenciatura, em particular, Carlos Pinheiro, Leticia, Laura, Carlos Lodeiro, César, Alexandre, Márcia, Avó, Yoan e Raquel. Uma palavra especial ao Professor Pina por me ter acolhido e por tudo o que me ensinou. À Ana Marta, pela sua profunda amizade, contagiante boa disposição e atenção que fazia o trabalho valer a pena. À Raquelita, por todos os cigarros e terapia, pela realidade nua e crua, pela tão querida amizade. Ao Bruno por ter sido o meu parceiro de cowboiada fora do laboratório, em tantos momentos de trabalho. Uma palavra de agracedimento em particular ao Professor Jorge Parola, ao meu orientador não oficial, por todo o apoio durante os vários projectos. Por me ter ensinado as artes obscuras da síntese orgânica. Pela sensatez durante o delírio colectivo.
Ao Centro de Investigação de Genética Molecular Humana, Pólo 1, por ter sido a minha escola de bio‐ coisas e aos seus membros, em particular, Maria, Ana, Quaresma, Red, Larguinho, Conde, Goku, Tavares, Inês, Veigas, Rita, Chang, Solange, Madalena e Carina, por todos os momentos imortais que tornaram o 315 a minha segunda casa durante os quatro anos de trabalho experimental. Um grande bem‐haja ao Revolt e Crossfire por todos os momentos de trabalho árduo que proporcionaram.
Uma palavra em particular ao Gonçalo, meu parceiro de tormentas e calmarias, por toda atenção e contribuições ao meu trabalho. Pela amizade dentro e fora do laboratório, nas aulas, na gestão do
laboratório e seus colaboradores, pelo apoio incondicional. Uma palavra em particular ao Rosinha, Man, pelas infindáveis conversas dentro e fora do laboratório e do campo de futebol. À Marta por todos os vidros partidos, pelos cigarros e msn, pela amizade que 7500 km não mudaram.
Às meninas da Conservação e Restauro, minhas colegas e amigas, por todos os momentos de diversão e por me terem concedido guarida durante tempos de escrita. Em particular à Joana, Ana, Catarina, Micaela e Ana. Ao João Pina e ao Professor Sergio Seixas de Melo, Faculdade de Ciências e Tecnologia, Universidade de Coimbra, por toda a ajuda e medição dos tempos de vida. Ao Zé Inácio e à Professora Isabel Sá Nogueira, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, pelo auxílio nas experiencias de transcrição in vitro usando nucleótidos marcados com radioactividade.
To Professor Milan Stojanovic, Columbia University, for the amazing opportunity to join the spider project and his valuable contributions regarding work and much more. To Professor Hao Yan, Biodesign Institute, for the opportunity to join his lab, learn DNA structural nanotechnology and for allowing me to continue the spider project while writing my thesis. To my Biodesign colleagues, in particular to Ashok and Chad, for their friendship that went beyond work. A special thank to Jeanette for the long AFM hours, helpful scientific discussions, mutual therapy and her precious friendship.
To my American friends, Lauren, Ashley, Twig, Doug, Kelly, Kylie, Derrick, Brian, Zach, Katie, Lellee, Amanda, Johna, Jennifer and Tiffany. A special thank to Caity for being my friend and family in a strange land, so far away from home. For taking care of me and opening the doors to her life. “It isn’t a big thing. It’s the million little things”. A todos os meus amigos, em particular, Nucha, Quico, Cardoso, Tiago, Joana, Mario, Sara, Artur, Pedro, Tiago, Nuno, Pierre, Rato, Pitcher, Ruben e Rita, por terem contribuído de forma decisiva para que esta etapa tenha sido fantástica. Uma palavra especial para a Mariana pela companhia e gostos em comum que poucos partilham. À Joaninha, pela extraordinária amizade a sensatez, que me iluminou incontáveis vezes o caminho. À Ana Diniz, pelo apoio incondicional e força que me deu durante uma parte
considerável do doutoramento. Ao Gonçalo, o meu primo acima de todos os outros, pela ajuda mutua nos tempos difíceis e comemoração nas victórias, pelo respeito e conselho que sempre procuro para decisões dificeis. Ao Terrinha von Dudster, por todos os tempos que passamos juntos, a fazer tudo ou nada, pelas nades, balázios, giros sem destio, torranço na praia e raquetada, por estar sempre presente em todos os momentos. Ao Caldas, e à sua família, pela amizade e carinho que nutriram desde o inicio da minha vida. À minha família, em particular à Tia Xanda e o meu Padrinho, às minhas irmãs, cunhado e sobrinhos, e em especial aos meus pais, pelo apoio incondicional, por terem sempre acreditado em mim, por me terem guiado, por tudo o que sou e serei. Sem eles nada disto teria sido possível. My deepest appreciation for the help and contribution to this journey that is now getting to its end. Thank you! Andre
SUMÁRIO
O principal objectivo da tese aqui apresentada foi a criação e desenvolvimento de um sistema para a síntese enzimática de ácidos nucleicos controlados por luz. A ideia baseia‐se na funcionalização de nucleótidos com grupos protectores fotolábeis (ou nucleótidos engaiolados), que não são reconhecidos como substratos pelas polimerases. Através da absorção de luz, o grupo protector fotolábil é removido e o nucleótido liberto, sendo de seguida incorporado na cadeia de ácido nucleico a ser sintetizada. A libertação específica do nucleótido desejado, de entre uma mistura de nucleótidos, é conseguida através da funcionalização de cada tipo de nucleótido com um grupo protector diferente, apresentando um espectro de absorção distincto. Utilizando radiação monocromática o nucleótido é liberto inequivocamente, levando à sua incorporação. A sequência de irradiação definiria, em ultima análise, a sequência da cadeia a ser sintetizada. De modo a ultrapassar a dependência de uma cadeia molde na síntese de ADN (ou ARN), foi utilizada uma polimerase de ADN que não necessita de cadeia molde – Terminal deoxinucleotidil Transferase.
Derivados da 4‐metilcumarina foram escolhidos como grupos protectores fotolábeis e a síntese de nucleótidos engaiolados foi alcançada com sucesso. A caraterização fotofísica e fotoquímica da [7‐dietilcumarina‐4‐il]metil fosfato (DEACM‐P) foi efectuada. Foi observada uma dependência entre o pH e a fotoquímica de libertação da DEACM‐P, e um novo modelo para a fotoquímica dos derivados da 4‐metilcumarina foi proposto. Este modelo tem em conta a concentração do ião hidóxilo na formação do fotoproduto 4‐hidroximetilcumarina. A caracterização fotofísica e fotoquímica da P3‐[7‐dietilcumarina‐4‐ il]metil adenosina trifosfato (DEACM‐ATP), P3‐[7‐dietilcumarina‐4‐il]metil guanosina trifosfato (DEACM‐ GTP), P3‐[7‐metoxycumarina‐4‐il]metil adenosina trifosfato (MCM‐ATP) e P3‐[7‐metoxycumarina‐4‐ il]metil guanosina trifosfato (MCM‐GTP) foi efectuada. Os grupos DEACM e MCM apresentam espectros de absorção em regiões distintas (λmax = 390 nm e 325 nm, respectivamente), permitindo a irradiação e
libertação selectiva desejada.
Nucleótidos engaiolados foram utilizados em reacções de trancrição in vitro. Níveis residuais de transcrição foram observados quando utilizados nucleótidos derivados com um grupo cumarínico. Após irradiação foram obtidos produtos de transcrição completos e específicos, demonstrando que a luz pode ser utilizada para a activação da síntese de ácidos nucleicos. Ambos os derivados de DEACM e MCM foram usados como grupos protectores, apresentando um comportamento semelhante. Derivados de ATP e GTP foram usados com sucesso como actuadores na síntese de ARN activada por luz, embora não foi possível obter transcritos quando DEACM‐GTP foi utilizado. A incorporação de nucleótidos numa cadeia de ácido nucleic em síntese activada por luz foi conseguida com sucesso utilizando a T7 RNA Polimerase e a Terminal deoxinucleotidil Transferase. Foi observado um efeito inibitório devido à presença do produto de fotólise 7‐dietilamino‐4‐hidroximetilcumarina (DEACM‐OH) sob a T7 RNA Polimerase. No entanto, o efeito inibitório pôde ser parcialmente suprimido através da adição de β‐ciclodextrina à reacção de transcrição in vitro.
ABSTRACT
The main objective of this thesis was the design and development of a system for the enzymatical synthesis of nucleic acids controlled by light. The overall concept is based on the functionalization of nucleotides with photoremovable protecting groups (or caged‐nucleotides), that cannot be recognized as substrates by the polymerases. Upon light absorption, the photo‐protecting group is cleaved and the nucleotide released, thus being incorporated in a growing nucleic acid chain. The specific release of the desired nucleotide, from a nucleotide mixture, is achieved functionalizing each type of nucleotide with a different caging group, presenting a distinct absorption spectrum. Through irradiation with monochromatic light, the specific nucleotide can be released unambiguously, leading to its incorporation. The irradiation sequence would, ultimately, define the sequence of the strand being formed. In order to overcome the template‐directed DNA (or RNA) synthesis, a template‐independent DNA polymerase was used – Terminal deoxynucleotidyl Transferase.
Derivatives of 4‐methylcoumarin were chosen as photoremovable protecting groups and the successful synthesis of caged‐nucleotides was achieved. The photophysical and photochemical characterization of [7‐diethylaminocoumarin‐4‐yl]methyl phosphate (DEACM‐P) was performed. A dependence of the DEACM‐P photochemistry on pH was found, and a new model for 4‐methylcoumarin derivatives photochemistry was proposed. This model accounts for the hydroxyl concentration in the 4‐hydroxymethylcoumarin photoproduct formation. The photophysics and photochemistry characterization of P3‐[7‐diethylaminocoumarin‐4‐yl]methyl adenosine triphosphate (DEACM‐ATP), P3‐ [7‐diethylaminocoumarin‐4‐yl]methyl guanosine triphosphate (DEACM‐GTP), P3‐[7‐methoxycoumarin‐4‐ yl]methyl adenosine triphosphate (MCM‐ATP) and P3‐[7‐methoxycoumarin‐4‐yl]methyl guanosine triphosphate (MCM‐ATP) was performed. The DEACM and MCM groups present absorption spectra in different regions (λmax = 390 nm and 325 nm, respectively), allowing for the desired selective irradiation
and cleavage.
Caged‐nucleotides were applied to in vitro transcription reactions. When the nucleotide was functionalized with a coumarin derivative, only residual RNA product formation could be detected. After irradiation, full size specific transcription product was obtained, showing that light can be used to activate the synthesis of nucleic acids. Both DEACM and MCM derivatives were used as caging groups, presenting similar behavior. Both ATP and GTP were successfully used as actuators for the light‐ controlled synthesis of RNA, although no transcription was attained when DEACM‐GTP was used. The light‐activated incorporation of nucleotides in a growing nucleic acid strand was successfully performed using the T7 RNA Polymerase and the Terminal deoxynucleotidyl Transferase. It was found that the 7‐diethyl‐4‐hydroxymethylcoumarin (DEACM‐OH) photoproduct presented an inhibitory effect over the T7 RNA Polymerase, but that the inhibition could be partially suppressed through the addition of
SYMBOLS AND NOTATIONS
3’‐OH – 3’‐hydroxyl Uf – Mataga solvent polarity ε ‐ molar extinction coefficient (in M‐1 cm‐1) μ ‐ dipole moment ν ‐ frequency Φf – fluorescence quantum yield Φchem – photochemical quantum yield τ ‐ lifetime A – absorbance Abs ‐ absorption ADP – adenosine diphosphate AMP – adenosine monophosphate AMPA ‐ α‐amino‐3‐hydroxy‐5‐methyl‐4‐ isoxazolepropionic acid anti‐hh – anti‐head‐to‐head anti‐ht – anti‐head‐to‐tail ATP – adenosine triphosphate BAPTA ‐ 1,2‐bis(o‐aminophenoxy)ethane‐ N,N,N’N’‐tetraacetic acid Bhc – 6‐bromo‐7‐hydroxycoumarin BSA – bovine serum albumin cAMP – cyclic adenosine monophosphate cGMP – cyclic guanosine monophosphate CM – coumarin CNS – central nervous system CTP – cytidine triphosphate dATP – deoxyadenosine triphosphate dCTP – deoxycytidine triphosphate DEACM – 7‐diethylamino‐4‐methylcoumarin DEACM‐ATP – P3‐[7‐diethylaminocoumarin‐4‐ yl]methyl adenosine 5’‐triphosphate DEACM‐GTP ‐ P3‐[7‐diethylaminocoumarin‐4‐ yl]methyl guanosine 5’‐triphosphate DEACM‐OH – 7‐diethylamino‐4‐ hydroxymethylcoumarin DEACM‐P – [7‐diethylaminocoumarin‐4‐ yl]methyl phosphate dGTP – deoxyguanosine triphosphate DMB ‐ 3’,5’‐dimethoxybenzoin group DMNB – dimethoxy‐2‐nitrobenzyl group DMNPE ‐ 1‐(4, 5‐dimethoxy‐2‐nitrophenyl)ethyl group DNA – deoxyribonucleic acid dNTP – deoxyribonucleotide DTT – dithiothreitol dTTP – deoxythymidine triphosphate E. coli – Escherichia coli EDTA – ethylenediamine tetraacetic acid EGTA – ethylene glicol tetraacetic acid FISH – fluorescence in situ hybridizationGTP – guanosine triphosphate GFP – green fluorescent protein HEPES ‐ 4‐(2‐hydroxyethyl)‐1‐ piperazineethanesulfonic acid HMPA – hexamethylphosphoramide HOMO – highest occupied molecular orbital HPLC – high performance liquid chromatography Ka – association constant kchem – photochemical rate constant kIC – internal conversion rate constant kISC – intersystem crossing rate constant kf – fluorescence rate constant knr – non‐radiative rate constant LUMO – lowest unoccupied molecular orbital MCM – 7‐methoxy‐4‐methylcoumarin MCM‐ATP – P3‐[7‐methoxy‐4‐hydroxymethyl‐4‐ yl]methyl adenosine 5’‐triphosphate MCM‐GTP – P3‐[7‐methoxy‐4‐hydroxymethyl‐4‐ yl]methyl guanosine 5’‐triphosphate MCM‐OH – 7‐methoxy‐4‐ hydroxymethylcoumarin MCM‐P – [7‐methoxy‐4‐hydroxymethyl‐4‐ yl]methyl phosphate mRNA – messenger ribonucleic acid NB – nitrobenzyl group ncPNA – negatively charged peptide nucleic acid NMDA – N‐methyl‐d‐aspartate NMR – nuclear magnetic resonance NPE – nitrophenylethyl group NSF – N‐ethylmaleimide sensitive factor NTP (or rNTP) – ribonucleotide OD – optical density qPCR – quantitative polymerase chain reaction PAGE – polyacrylamide gel electrophoresis PCR – polymerase chain reaction pHP ‐ p‐hydroxyphenacyl group PL – photocleavable linker RISC – RNA induced silencing complex RNA – ribonucleic acid rRNA – ribosomal ribonucleic acid RT‐PCR – reverse transcription polymerase chain reaction SDS – sodium dodecyl sulphate siRNA – small interference ribonucleic acid Sn – singlet excited state n syn‐hh – syn‐head‐to‐head syn‐ht – syn‐heat‐to‐tail TAE – tris acetate EDTA TBE – tris borate EDTA TdT – terminal deoxynucleotidyl transferase THF – tetrahydrofuran TICT – twisted intramolecular charge transfer TLC – thin layer chromatography
Tn – triplet excited state n Tris – tris(hydroxymethyl)aminomethane tRNA – tranfer ribonucleic acid TTP – thymidine triphosphate UTP – uridine triphosphate UV – ultra‐violet
TABLE OF CONTENTS
ACKNOWLEDGEMENTS ... VII SUMÁRIO ... XIII ABSTRACT ... XV SYMBOLS AND NOTATIONS ... XVII TABLE OF CONTENTS ... XXI FIGURE INDEX ... XXV TABLE INDEX ... XXIX CHAPTER 1. General Introduction ... 1 1.1. Light to Synthesize Nucleic Acids ...3 1.2. Nucleic Acids Synthesis ...3 1.2.1. Nucleic Acids Structure ...3 1.2.2. Synthesis of Nucleic Acids in vivo ...7 1.2.3. Nucleic Acids Polymerases ...9 1.2.4. In vitro synthesis of Nucleic Acids ... 18 1.2.5. Light Control of RNA Synthesis ‐ What component should be controlled? ... 21 1.3. Caged Molecules ... 22 1.3.1. Caged Compounds ... 22 1.3.2. Caged Compounds in Bio‐Applications ... 23 1.3.3. Photolabile Protecting Groups ... 30 1.4. Photophysics and Photochemistry of Coumarins ... 35 1.4.1. Introduction to Photochemistry ... 35 1.4.2. Coumarin Ground‐State and Photophysical Properties ... 45 1.4.3. Coumarin Photochemical Properties ... 52 1.5. Light‐controlled Nucleic Acids Typewriter – an Overview ... 59 CHAPTER 2. Materials and Methods ... 63 2.1. General Information ... 64 2.2. Synthesis of Coumarin Derivatives ... 64 2.2.1. Synthesis of 7‐diethylamino‐4‐methylhydroxycoumarin (DEACM‐OH) ... 64 2.2.2. Synthesis of [7‐diethylaminocoumarin‐4‐yl]methyl di‐tert‐butyl phosphate (DEACM‐tBut) ... 652.2.3. Synthesis of [7‐diethylaminocoumarin‐4‐yl]methyl phosphate (DEACM‐P) ... 66 2.2.4. Synthesis of P3‐[7‐diethylaminocoumarin‐4‐yl]methyl adenosine 5’‐triphosphate (DEACM‐ATP) ... 66 2.2.5. Synthesis of 7‐methoxy‐4‐methylhydroxycoumarin (MCM‐OH) ... 67 2.2.6. Synthesis of [7‐methoxycoumarin‐4‐yl]methyl di‐tert‐butyl phosphate (MCM‐tBut) ... 68 2.2.7. Synthesis of [7‐methoxycoumarin‐4‐yl]phosphate (MCM‐P) ... 68 2.2.8. Synthesis of P3‐[7‐methoxycoumarin‐4‐yl]methyl adenosine 5’‐triphosphate (MCM‐ATP) and P3‐[7‐methoxycoumarin‐4‐yl]methyl guanine 5’‐triphosphate (MCM‐GTP) ... 69 2.2.9. Synthesis of P3‐[7‐diethylaminocoumarin‐4‐yl]methyl guanine 5’‐triphosphate (DEACM‐GTP) ... 69 2.3. Photophysical and Photochemical Characterization ... 70 2.3.1. Absorption and Emission Titrations ... 70 2.3.2. Fluorescence and Photochemical Quantum Yield Determinations ... 72 2.3.3. Time‐Resolved Fluorescence Spectroscopy Measurements ... 74 2.3.4. Flash Photolysis Experiments ... 74 2.3.5. DEACM‐ATP Irradiation Profiles ... 75 2.4. Light‐controlled in vitro Synthesis of Nucleic Acids ... 75 2.4.1. Transcription Template Cloning and Purification ... 75 2.4.2. In vitro Transcription ... 76 2.4.3. Reverse Transcription (RT) and Real‐Time PCR Reaction ... 77 2.4.4. Light Activated Polymerization Using Terminal deoxynucleotidyl Transferase ... 77 2.5. DEACM‐OH Inhibition Experiments ... 78 2.5.1. DEACM‐OH Inhibition Effect ... 78 2.5.2. DEACM‐OH Inhibition Suppression: β‐lactoglobulin ... 79 2.5.3. DEACM‐OH/β‐Cyclodextrin Association Constant Determinations ... 79 2.5.4. DEACM‐OH Inhibition Suppression: β‐cyclodextrin ... 79 CHAPTER 3. Photophysical and Photochemical Characterization of DEACM Derivatives ... 81 3.1. Synthesis of P3‐[7‐diethylaminocoumarin‐4‐yl]methyl adenosine 5’‐triphosphate (DEACM‐ATP) ... 82 3.2. Ground State Properties of DEACM‐OH and DEACM‐P ... 86 3.3. Dependence of DEACM‐OH and DEACM‐P Photophysics and Photochemistry on pH ... 89 3.4. Flash Photolysis Studies of DEACM‐H, DEACM‐OH and DEACM‐P ... 101
3.5. Characterization of DEACM‐ATP Caged Nucleotide ... 108 CHAPTER 4. Light Activated in vitro Transcription Reactions ... 117 4.1. Using Light to Control the Synthesis of RNA ... 118 4.1.1. DEACM‐ATP Irradiation Profiles ... 119 4.1.2. Light Activated in vitro Transcription ... 120 4.1.3. Inhibition Effect of DEACM‐OH ... 123 4.2. Suppression of DEACM‐OH Inhibition in Light‐controlled in vitro Transcription Reactions ... 126 4.2.1. β‐Lactoglobulin ... 127 4.2.2. Cyclodextrins ... 129 4.3. Light Activated Polymerization Using Terminal deoxynucleotidyl Transferase ... 136 CHAPTER 5. Light Controlled Synthesis of Nucleic Acids Using Multi‐Wavelength Excitation .... 143 5.1. Synthesis of 7‐methoxy‐4‐methylcoumarin Derivatives ... 144 5.2. Photochemical Characterization of DEACM‐GTP, MCM‐ATP and MCM‐GTP ... 150 5.3. Light‐activated in vitro Transcription Reactions Using DEACM‐ and MCM‐Caged Nucleotides .... 153 5.4. Light‐Input, RNA‐Output Logic Gates ... 155 CHAPTER 6. Conclusions and Future Perspectives ... 161 REFERENCES ... 169
FIGURE INDEX
Figure 1.1 – Chemical structure of nucleotides ...4 Figure 1.2 – Natural occurring nitrogen bases in DNA and RNA ...5 Figure 1.3 – The DNA double helix ...7 Figure 1.4 – Central dogma in genetics ...8 Figure 1.5 – Schematic representation of the polymerization of a DNA strand ... 10 Figure 1.6 –Crystal structure of Taq DNA polymerase I ... 11 Figure 1.7 – Crystal structure of the RNA Polymerase/DNA template complex ... 14 Figure 1.8 – Schematization of the transcription bubble complex in RNA Polymerase ... 15 Figure 1.9 – Proposed structure of the Terminal deoxynucleotidyl Transferase ... 17 Figure 1.10 – Polymerase Chain Reaction ... 19 Figure 1.11 – The first photo‐activated biomolecule to be called “caged”: Caged‐ATP with a nitrobenzyl group ... 23 Figure 1.12 – Photocleavage mechanism of nitrophenil protecting group. ... 31 Figure 1.13 – Photocleavage mechanism of benzoin protecting group ... 32 Figure 1.14 – Photocleavage mechanism of p‐hydroxyphenancil protecting group ... 33 Figure 1.15 – Franck‐Condon principle and vertical transitions ... 38 Figure 1.16 – The Jablonski diagram ... 39 Figure 1.17 – The solvatochromic effect ... 41 Figure 1.18 – Photophysical and photochemical processes ... 44 Figure 1.19 – Coumarin moiety and some common derivatives ... 45 Figure 1.20 – Absorption and emission spectra of the coumarin derivatives shown in Figure 1.19 ... 46 Figure 1.21 – Absorption and emission spectra of ClMMC ... 48 Figure 1.22 – Photophysical deactivating processes in 7‐aminocoumarin derivatives ... 51 Figure 1.23 – Effect of the solvent polarity on the non‐radiative rate constant (knr) of DEACM ... 52 Figure 1.24 – Structures of coumarin dimers ... 53 Figure 1.25 – Mechanism of (coumarin‐4‐yl)methyl photochemistry proposed by Schade and co‐workers ... 56 Figure 3.1 – Synthesis of 7‐diethylamino‐4‐hydroxymethylcoumarin (DEACM‐OH) ... 83 Figure 3.2 – Synthesis of [7‐diethylaminocoumarin‐4‐yl]methyl di‐tert‐butyl phosphate (DEACM‐tBut) .. 84 Figure 3.3 – Synthesis of [7‐diethylaminocoumarin‐4‐yl]phosphate (DEACM‐P) ... 84Figure 3.4 – Activation of ADP precursor ... 85 Figure 3.5 – Synthesis of P3‐[7‐diethylaminocoumarin‐4‐yl]methyl adenosine triphosphates (DEACM‐ATP) ... 86 Figure 3.6 – A) DEACM‐OH and DEACM‐P structure ... 87 Figure 3.7 – Spectrophotometric titration of DEACM‐OH and DEACM‐P ... 88 Figure 3.8 – Fluorimetric titration of DEACM‐OH and DEACM‐P ... 89 Figure 3.9 – DEACM‐OH and DEACM‐P acid‐base equilibria ... 90 Figure 3.10 – Photochemical and fluorescence quantum yields of DEACM‐P as function of pH ... 91 Figure 3.11 – Time resolved fluorescence measurements of DEACM‐OH and DEACM‐P as function of pH ... 92 Figure 3.12 – Overall deactivating rate constant of DEACM‐HPO4– species as function of hydroxyl anion concentration. ... 95 Figure 3.13 – Kinetic model for the photochemistry of DEACM‐P involving a pH dependent equilibrium affecting 1[CP] ... 95 Figure 3.14 ‐ Overall deactivating rate constant of DEACM‐HPO4– based on the kinetic model presented in Figure 3.13. ... 97 Figure 3.15 – Kinetic model for the photochemistry of DEACM‐P involving buffer quenching of 1[CP] ... 97 Figure 3.16 ‐ Overall deactivating rate constant of DEACM‐HPO4– based on the kinetic model presented in Figure 3.15 ... 98 Figure 3.17 ‐ Kinetic model for the photochemistry of DEACM‐P involving a nucleophilic attack to an intermediary formed from 1[CP] state. ... 99 Figure 3.18 – Flash Photolysis transient spectroscopy of DEACM‐P ... 101 Figure 3.19 – Effect of the pH on the DEACM‐P transient intermediary species ... 102 Figure 3.20 – Transient spectra of Coumarin 1 and DEACM‐OH ... 103 Figure 3.21 – Effect of oxygen on the transient spectra of Coumarin 1, DEACM‐OH and DEACM‐P ... 106 Figure 3.22 – Transient absorption spectra of 6,7‐dimethoxycoumarin ... 107 Figure 3.23 – Absorption and emission spectra of DEACM‐OH, DEACM‐P and DEACM‐ATP solutions ... 108 Figure 3.24 – DEACM‐ATP spectrophotometic titration in 10 mM Tris‐phosphate buffer ... 109 Figure 3.25 – Acid‐base equilibria of adenosine triphosphate ... 110 Figure 3.26 – DEACM‐ATP fluorimetric titration in 10 mM Tris‐phosphate buffer ... 111 Figure 3.27 – Photochemical quantum yield of DEACM‐ATP disappearance and DEACM‐OH production as function of pH ... 114
Figure 3.28 – Photochemical quantum yields of DEACM‐ATP disappearance, and DEACM‐OH and ATP appearance ... 115 Figure 4.1 – Photolysis of DEACM‐ATP and ATP release ... 118 Figure 4.2 – Irradiation profiles of four different concentrations of DEACM‐ATP in water, pH 7.0 ... 120 Figure 4.3 – In vitro transcription using DEACM‐ATP ... 121 Figure 4.4 – Relative quantification of full‐length transcription products as function of ATP released after DEACM‐ATP irradiation ... 122 Figure 4.5 – Effect of DEACM‐OH in transcription reactions ... 123 Figure 4.6 – Effect of DEACM‐OH presence in transcription reactions... 124 Figure 4.7 – Molecular structure of Clorobiocin, Novobiocin and Coumermycin A1 ... 125 Figure 4.8 – Crystal structure of β‐lactoglobulin complexed with a cholesterol molecule ... 128 Figure 4.9 ‐ Effect of the presence of β‐lactoglobulin in transcription reactions ... 129 Figure 4.10 – DEACM derivatives involved in a light controlled in vitro transcription reaction (DEACM‐OH and DEACM‐ATP) and β‐cyclodextrin ... 130 Figure 4.11 – Effect of the presence of β‐cyclodextrin in transcription reactions ... 131 Figure 4.12 – Formation of DEACM‐OH/β‐cyclodextrin complex ... 132 Figure 4.13 – DEACM‐OH fluorescence emission intensity at 570 nm as function of β‐cyclodextrin concentration ... 133 Figure 4.14 – β‐Cyclodextrin and reduction of inhibition ... 135 Figure 4.15 – Incorporation of deoxyribonucleotides to a 20‐mer oligonucleotide by the Terminal deoxynucleotidyl Transferase ... 138 Figure 4.16 – Incorporation of ribonucleotides to a 20‐mer oligonucleotide by the Terminal deoxynucleotidyl Transferase ... 140 Figure 4.17 – Effect of the presence of DEACM‐OH in a template independent TdT‐catalyzed ATP addition to a single stranded 20‐mer oligonucleotide ... 141 Figure 4.18 – Light activated template independent TdT‐catalyzed ATP addition to a single stranded 20‐ mer oligonucleotide ... 142 Figure 5.1 – Structure of coumarins with strong electro‐donating groups in the 7‐ position and/or electro‐ withdrawing groups in the 3‐ position ... 145 Figure 5.2 – Synthesis of the (7‐methoxycoumarin‐4‐yl)methyl acetate ... 146 Figure 5.3 – Synthesis of the 7‐methoxy‐4‐hydroxymethylcoumarin (MCM‐OH) ... 147 Figure 5.4 – Synthesis of MCM‐tBut ... 147
Figure 5.5 – Synthesis of (7‐methoxycoumarin‐4‐yl)methyl phosphate (MCM‐P) ... 148 Figure 5.6 – Synthesis of MCM‐ATP and MCM‐GTP ... 149 Figure 5.7 – Synthesis of DEACM‐GTP ... 149 Figure 5.8 – Absorption and emission spectra of DEACM‐ATP and DEACM‐GTP ... 150 Figure 5.9 – Absorption and emission spectra of MCM‐ATP and MCM‐GTP ... 151 Figure 5.10 – Selective excitation and photochemistry of DEACM and MCM caging groups ... 152 Figure 5.11 – Light‐activated in vitro transcription reaction using DEACM‐ATP, DEACM‐GTP, MCM‐ATP or MCM‐GTP ... 154 Figure 5.12 – Truth tables for AND, OR and NOT logic gates ... 157 Figure 5.13 – Design of a light‐input/RNA‐output OR logic gate ... 157 Figure 5.14 ‐ Design of a light‐input/RNA‐output AND logic gate ... 158 Figure 5.15 – Design of a light‐input/RNA‐output NOT logic gate ... 159
TABLE INDEX
Table 1.1 – Photophysics of common coumarins as function of solvent polarity ... 47 Table 1.2 – Photophysics of ClMMC as function of solvent polarity ... 49 Table 1.3 – Effect of acid moiety in the photophysical and photochemical properties of 7‐methoxy‐4‐methylcoumarin ester derivatives ... 57 Table 1.4 – Photophysical and photochemical properties of cyclic adenosine monophosphates esters of differently substituted (coumarin‐4‐yl)methyl alcohols ... 58 Table 2.1 – Intensity of irradiation setup used for determination of photochemical quantum yields and nucleotide release as function of the excitation wavelength and slit width ... 72 Table 3.1 – Photophysical and photochemical properties of DEACM‐OH and DEACM‐P ... 93 Table 3.2 – Decay times of DEACM‐ATP as function of pH ... 112CHAPTER 1. General Introduction
1.1. Light to Synthesize Nucleic Acids
In 2004, my supervisors and I discussed for the first time a system in which light would be used to control the enzymatic synthesis of nucleic acids. The idea was born under the context of supramolecular chemistry, the field of chemistry that makes use of the known properties of molecules to produce a desired molecular event. We were aware that the process of in vitro DNA and RNA polymerization depends on a DNA template that determines the sequence of the forming strands, and that customized DNA synthesis is, thus far, only possible via chemical synthesis. A system that could put together the efficiency and simplicity of enzymatic polymerization of nucleic acids and a means to control the sequence would be of great relevance to molecular biology and supramolecular chemistry. Based on our photochemistry expertise, we immediately thought of using light to achieve this control. Light as a “reagent” would present several advantages, namely to eliminate the addition of mass to the system in each cycle avoiding, theoretically, the necessity of purification steps.
The first step was to perform a careful analysis of natural and in vitro polymerization processes of nucleic acids to identify its key elements, evaluate the parameters upon which we would need to actuate in order to obtain sequence control. Also, the essential and common components of RNA and DNA synthesis should be analyzed to assess the possibility of extending this system to custom RNA synthesis. 1.2. Nucleic Acids Synthesis 1.2.1. Nucleic Acids Structure
The genome of all living organisms is constituted by deoxyribonucleic acid (DNA), where the genetic information is stored. DNA may then be transcribed into ribonucleic acid (RNA) that, if suitable, can then be translated into protein.[1] Cellular processes of nucleic acids “management” (processing, DNA/RNA/protein interaction and elimination) are complex, stimulating and of crucial importance to understand life and Nature. However, they are also beyond the scope of this work.
DNA and RNA are chemically very similar ‐ both present a linear primary structure composed of monomers called nucleosides. All nucleosides present a common structure: a pentose linked to a nitrogen base at the 1’ position (Figure 1.1). In RNA, the pentose is ribose; in DNA, it is deoxyribose. A nucleotide is formed when a phosphate group is linked to the 5’ position of the pentose.
Figure 1.1 – Chemical structure of nucleotides. The presence or absence of a –OH group at the 2’ position in a
ribose sugar ring distinguishes between ribose (upper right) or 2’‐deoxyribose (down right). When a nitrogen base is attached to the 1’ position in the (deoxy)ribose a nucleoside is formed (left). When a phosphate group is present at the 5’ position of the (deoxy)nucleoside a nucleotide is attained (left). Figure adapted from [2].
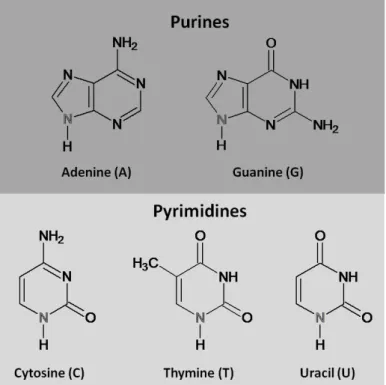
The bases adenine, guanine, and cytosine are found in both DNA and RNA; thymine is only found in DNA, and uracil is only found in RNA. Adenine and guanine are purines, which contain a fused pyrimidine and imidazole rings; cytosine, thymine, and uracil are pyrimidines, containing a single ring (Figure 1.2).
Nucleotides can have one, two, or three phosphate groups esterified at the 5‐hydroxyl forming nucleotide monophosphates, diphosphates or triphosphates, respectively. The nucleotide triphosphates are used in the synthesis of nucleic acids.[2]
Figure 1.2 – Natural occurring nitrogen bases in DNA and RNA. The adenine and guanine nitrogen bases are
purines, comprising a fused pyrimidine and imidazole rings (up). The cytosine, thymine and uracil are composed by a single pyrimidine ring (down). In gray are represented the nitrogen atom from which the coupling to the (deoxy)ribose sugar is made to form a nucleoside. The way these nucleotides are organized into the DNA double helix ‐ the secondary structures ‐ was first described in 1953 by J. Watson and F. Crick.[3] I have to me this description as one of the most important papers in Life Sciences and, therefore, deserves to be remembered in its original version:
“We wish to put forward a radically different structure for the salt of deoxyribose acid. This structure has two helical chains each coiled round the same axis. We have made the usual chemical assumption, namely, that each chain consists of phosphate diester groups joining β‐D‐deoxyribofuranose residues with 3’,5’ linkages. The two chains (but not their bases) are related by a dyad perpendicular to the fibre axis. Both chains follow right‐handed helices, but owing to the dyad the sequences of the atoms in the two chains run in opposite directions. Each chain loosely resembles Furberg’s model No. 1; that is, the bases are on the inside of the helix and the phosphates on the outside. The configuration of the sugar and the atoms near it is close to Furberg’s standard configuration, the sugar being roughly perpendicular to the attached base.
There is a residue on each chain every 3.4 Å in the z‐direction. We have assumed an angle of 36° between adjacent residues in the same chain, so that the structure repeats after 10 residues on each chain, that is, after 34 Å. The distance of a phosphorus atom from the fibre axis is 10 Å. As the phosphates are on the outside, cations have easy access to them. (…) The novel feature of the structure is the manner in which the two chains are held together by the purine and pyrimidines bases. The planes of the bases are perpendicular to the fibre axis. They are joined together in pairs, a single base from the other chain being hydrogen‐bonded to a single base from the other chain, so that the two lie side by side with identical z‐co‐ordinates. One of the pair must be a purine and the other a pyrimidines for bonding to occur. (…) If it is assumed that the base only occur in the structure in the most plausible tautomeric forms (that is, with the keto rather than the enol configurations) it is found that only specific pairs of bases can bond together. These pairs are: adenine (purine) with thymine (pyrimidines), and guanine (purine) with (pyrimidines). In other words, if an adenine forms one member of a pair, on either chain, then on these assumptions the other member must be thymine; similarly for guanine and cytosine. The sequence of bases on a single chain does not appear to be restricted in any way. (…) It has been found experimentally that the ratios of the amounts of adenine to thymine, and the ratio of guanine to cytosine, are always very close to unity for deoxyribose nucleic acid”.
As a small additional note regarding DNA double helix base pairing, adenine forms two hydrogen bonds with thymine, and guanine three hydrogen bonds with cytosine (see Figure 1.3). Regarding RNA structure, and depending on the type of RNA, the structure can vary significantly between linear single stranded chains (mRNA), small single‐stranded chains with secondary structures (tRNA) or complex secondary structures associated with proteins (rRNA).
Figure polari nitrog 1.2.2 1.2.2 The c essen inform but congl term begin Then e 1.3 – The ization of the gen base pairs . Synthesis o .1. Replicati cellular proc ntial that its mation as th as a gener lomerate of ination. Initi ns, the paren synthesis o DNA double two strands l s responsible f of Nucleic Ac on cess through s genome is he parental o ral consider f enzyme ac ation involve ntal strands of daughter
e helix. A) S
leads to the fo for the forma cids in vivo h which DNA duplicated one. The rep ation, replic ctivities. The es recognitio must be sep strands can Schematic rep ormation of a tion of the do
A is synthes so each of plication pro
cation of d ere are thre on of an orig parated and be initiated presentation a major and m ouble helix. Fig
sized is calle the daught cess is differ duplex DNA e stages in gin by a com
(transiently d. Elongation
of a DNA d minor groove. gure B adapte
ed replicatio ter cells can rent in proka
is a comp replication: mplex of prot ) stabilized i n is underta double helix. B) Hydrogen ed from [2].
n. For a cel n contain th aryotic and e plex endeav initiation, e teins. Before n the single ken by anot
The anti‐par bonding betw
ll to divide e same gen eukaryotic c vor involvin elongation, e DNA synth ‐stranded st ther complex rallel ween it is netic ells, g a and hesis tate. x of
proteins tha the end of taken when involved, su stress. Any chemical co systems tha replication a 1.2.2.2. Tra As referred of RNA are Figure 1.4 – genome. In t is then proce ribosomes oc at moves alo the replicat n the cell n uch as DNA r event that onstitution o at recognize apparatus its nscription above, the i produced du Central dogm the presence essed and tra ccur. Figure a ong DNA, un tion site, join needs to div
repair mecha introduces a of its nitrog e and correc
self, which in information uring the tra ma in genetic of a Transcrip ansported to t dapted from nwinding par ning and/or vide, but are
anisms. Dam a deviation
en bases is ct these dam ndicates the stored in DN nscription pr cs. The DNA r ption complex the cytoplasm [4]. rental strand termination e also other mage in DNA from the us a threat to mages. The ir importanc NA is passed rocess. replication du x comprising t m (in eukaryo d, while dau n reactions a r processes can occur w sual double‐ o the cell. In
repair syste ce for surviva
on to RNA (
uring cell divis the RNA Polym tes), where t ghter strand are necessar in which D hen a cell is helical struc njuries in DN ems might b al.[4] (Figure 1.4) a sion yields tw merase, RNA he Translatio ds are synthe ry. Replicatio NA polymer subjected to cture of DNA NA are mini be as comple
and all differ
wo copies of t is synthesized n into protein
esized. At on is only rization is o external A and the imized by ex as the
rent types
he original d. The RNA ns through
Transcription is also the first stage in gene expression, and the main step at which it is controlled. Regulatory proteins determine whether a particular gene is available to be transcribed by the RNA polymerase (enzyme that connects the nucleotides to form a strand). The initial (and often the only) step in regulation is the decision of whether to transcribe a gene or not. Transcription initiation requires the binding of several proteins which are called transcription factors.[4,5] The presence of cellular signals to these protein complexes determine the existence of transcription (as an on/off state), but also its transcription rate. This modulation of gene expression is crucial for cell function since it regulates which proteins are being synthesized at what rate, and defines how the cell interacts with external medium. In prokaryotic cells, which have no nuclei, translation of an mRNA into protein can begin from the 5’ end of the mRNA even while the 3’ end is still being synthesized. In eukaryotic cells not only is the nucleus separated from the cytoplasm where translation occurs, but also the primary transcripts of protein‐ coding genes are precursor mRNAs (pre‐mRNAs) that must undergo several modifications to yield a functional mRNA, termed RNA processing. This mRNA must then be exported to the cytoplasm before it can be translated into protein.[2] 1.2.3. Nucleic Acids Polymerases The control of replication and transcription processes in the cell requires many protein complexes with different functions. Although the proteins present in each process vary from each organism, there is a type of enzyme that is not only the core of both DNA and RNA synthesis processes, but also present in all living organisms: the polymerases. 1.2.3.1. DNA Polymerases
DNA polymerases are enzymes that catalyze the synthesis of deoxyribonucleic acid in a template‐ dependent process that results in a faithful copy of the original DNA molecule. Based on their functions, DNA polymerases can be broadly classified into two groups: replicative DNA polymerases (DNA replicases), that are responsible primarily for duplicating genomic DNA and repair polymerases, that primarily fix damaged DNA strands. Replicative DNA polymerases must synthesize extended lengths of DNA with high speed and accuracy to ensure that each daughter cell receives a true copy of the genome upon cell division. In general, these DNA replicases are complex assemblies of several proteins that
function together for efficient DNA replication. Polymerases dedicated to DNA repair generally have a simpler architecture and appear designed for DNA synthesis localized to areas of DNA damage.[6] Figure 1.5 – Schematic representation of the polymerization of a DNA strand. A) The polymerization requires the presence of a template (long chain) and a primer (short chain) with a free 3’‐OH group. B) In the presence of a free nucleotide that forms a Watson‐Crick base pair with the template’s nucleotide, the addition catalysis might occur.
C) Upon catalysis, the formation of a phosphodiester bond between the n nucleotide and the n+1 incoming
nucleotide occurs, leading to the release of a pyrophosphate.
Polymerases are unusual enzyme catalysts since DNA is not only a substrate (template) but also the product. In order to make a copy of DNA, these enzymes are designed to:
Chem the n pyrop Figure is in c Closed the ac [6]. 1) Recogn template 2) Recogn to form a 3) Catalyz on the pr 4) Reposi mically, polym nucleotide t phosphate is e 1.6 –Crystal contact with d Conformati ctive site. The
nize and bin for synthesi nize and bind base pair ze nucleotidy rimer and the tion the new merases cata o be added s released (Fi
l structure of
bulk solution on (right) cla e closed confo nd a partially is d a deoxynu yl transfer in e incoming d wly extended alyze the nu d to the gro igure 1.5).
Taq DNA pol
n, allowing fo mps the tem ormation is ach
y single stra
cleoside 5'‐t n which a cov dNTP d DNA polym ucleophilic at owing chain. ymerase I. In r the diffusio plate, growin hieved after a
nded (or pri
triphosphate
valent link is
mer for the ne
ttack of a pr . A n+1 olig
the Open Co n of free nuc ng strand and a rotation of t imed) DNA s e (dNTP) and s formed bet ext cycle of n rimer 3’‐OH gonucleotide onformation (l cleotides in a incoming nu he fingers sub strand, whic d match it wi tween the 3' nucleotidyl tr
end to the e chain is ge
left) the polym nd out of the cleotide, seal b‐domain. Fig ch serves as th the temp ‐hydroxyl gr ransfer α‐phosphate enerated an merase active
e active site. ling the acces ure adapted f the plate roup e of nd a e site The ss of from
From a structural point of view, the shape of the polymerase is by far the most prominent feature common to all the polymerase structures determined to date. As described first for the Klenow fragment from E. coli DNA Pol I, the polymerase resembles a half‐open right hand with the "palm" sub‐domain forming a cleft that is flanked by the "fingers" and "thumb" sub‐domains.[6] Together, the three sub‐ domains hold the primer‐template DNA and position the incoming dNTP for incorporation into DNA. The palm sub‐domain contains the catalytic site where nucleotidyl transfer takes place. The fingers sub‐ domain interacts with and positions the template DNA strand and the incoming dNTP. The thumb sub‐ domain primarily binds the duplex DNA in a sequence‐independent manner along the minor groove (Figure 1.6).[6,7]
The active site contains several acidic and polar amino acid residues as well as two metal cations (usually Mg2+) that are essential for catalysis. In particular, two aspartate residues are absolutely conserved between the polymerase families, and these provide the carboxylate oxygens that coordinate the metal ions. Ion A is located near the 3'‐ hydroxyl group of the DNA primer and the α‐phosphate of the incoming dNTP. In this location, ion A is ideally positioned to lower the pKa of the hydroxyl group and facilitate the formation of a hydroxide anion, which can initiate nucleophilic attack on the α‐phosphate of incoming dNTP. Metal ion B co‐ordinates oxygen in all three phosphate groups of the dNTP, likely helping align the triphosphate moiety for attack by the 3'‐hydroxyl, as well as stabilizing the charge on the transition state. Other polar residues in the active site, and possibly ion B, help stabilize the charged pyrophosphate group as it dissociates from the polymerase after nucleotidyl transfer is complete.[7‐9]
The ability of a DNA polymerase to faithfully complement a DNA template depends on how well it selects for correct pairing between the template and the incoming nucleotide. The dNTP‐binding site is located in a narrow junction between the fingers and thumb domains. The 3’‐end of the primer lies right next to the dNTP‐binding site, and together with residues from the fingers domain forms a highly constrained binding pocket for the new base pair .[8,10‐12] DNA polymerases can select for correct Watson‐Crick base pairs and reject distorted mismatched base pairs when the incoming dNTP initially fits into the binding pocket. The dissociation constant for interaction between polymerase and the correct nucleotide is 20 μM versus 4 to 8 mM for nucleotides that do not match the DNA template.[9,10] The nucleotide binding site in DNA polymerases can also discriminate between dNTPs and rNTPs (ribonucleoside triphosphates) such that the polymerase synthesizes DNA, not RNA. This selection is possible due to the presence of
phenylalanine or tyrosine residues that work as a steric gate. The longer –OH group in NTPs present then much lower association constants in template‐polymerase complex.[9]
When the correct dNTP is inside the catalytic site, the polymerase can change from an open to a close conformation, where the nucleotidyl transfer can occur (Figure 1.6). This conformational change is the rate limiting step in the nucleotide addition reaction. A 10‐fold increase in the association constant of the correct dNTP‐template‐polymerase complex is observed, as a consequence of the clamping down of the catalytic site, due to the conformational change. Also, the closed conformation permits the correct alignment of the incoming dNTP triphosphate moiety for nucleophilic attack. After catalysis, an open conformation is taken, from which the pyrophosphate product is released. Then, dissociation between the polymerase and the template (now with n+1 nucleotides) may happen, although it is usually prevented either through interaction with other proteins, or in the case of processive DNA polymerases, by the thumb domain forming a clamp‐like structure that wraps the double stranded DNA. This way, it is more likely the polymerase to slide to the new incorporation position than releasing the template strand.[7‐9]
1.2.3.2. RNA Polymerases
DNA‐dependent RNA polymerases are responsible for the vital process of synthesis of RNA from a double‐stranded DNA template. Although nuclear transcription within eukaryotes is performed by a complicated multi‐enzyme RNA polymerase machine, most mitochondria, chloroplast and bacteriophage genes are transcribed by a homologous family of smaller nucleus‐encoded RNA polymerase.[4] While larger cellular RNA polymerases present multiple subunits related with regulation and proof‐reading functions, single‐subunit RNA polymerases share many of the biochemical characteristics, including catalytic specifications. These single‐unit RNA polymerases (Figure 1.7) resemble DNA polymerases in structure and catalysis mechanism, but the process of RNA synthesis is conceptually more complex.[13‐16]
Figure 1.7 – C As a major d RNA polym the recogni double stra polymerase a template sequence w Crystal struct distinction, b erases do no ition of a sp
nd is perfor e recognizes for the RN with the othe ture of the RN besides the o ot require a pecific seque med by the the specific NA synthesis er DNA strand NA Polymeras obvious diffe primed tem ence – prom enzyme so t promoter se s. The RNA d, which is c e/DNA templ erence in stru plate to init moter – to st that one of t equence in o is thus com alled the cod late complex. ucture of the iate polymer tart transcrip the chains ca one of the do mplementary ding strand ( . Figure adapt e nucleotide rization.[17,18 ption. The u an be used a ouble helix s y to this stra
Figure 1.8).[1 ted from [14]. s (rNTP vs. d ] However, i nwinding of as template. strands that and, and id 17] dNTP), the t involves f the DNA The RNA is used as entical in
Figure 1.8 – Schematization of the transcription bubble complex. The unwinding of the double helix is essential for the hybridization of the incoming ribonucleotides and the DNA template strand, forming a bubble. Incorporation of the successive ribonucleotides leads to the formation of a RNA chain that becomes single‐stranded upon rewinding of the duplex DNA strands.
Transcription can be divided into four stages, in which a bubble is created, RNA synthesis begins, the bubble moves along the DNA and finally is terminated:[4,15,18‐21]
1) Template recognition begins with the binding of RNA polymerase to the double‐stranded DNA at the promoter to form a "closed complex". Then the DNA strands are separated to form the "open complex" that makes the template strand available for base pairing with ribonucleotides.
2) Initiation describes the synthesis of the first nucleotide bonds in RNA. The enzyme remains at the promoter site while it synthesizes the first ~9 nucleotide bonds. The initiation phase is often protracted by the occurrence of abortive events, in which the enzyme makes short transcripts, releases them, and then starts synthesis of RNA again. The initiation phase ends when the enzyme succeeds in extending the chain downstream of the promoter region.
3) During elongation the enzyme moves along the DNA and extends the growing RNA chain. As the enzyme moves, it unwinds the DNA helix to expose a new segment of the template in single‐stranded condition. Nucleotides are covalently added to the 3' end of the growing RNA chain, forming an RNA‐DNA hybrid in the unwound region. Behind the unwound region, the DNA template strand pairs with its original partner to reform the double helix. The RNA emerges as a free single strand. 4) Termination involves recognition of the point at which no further bases should be added to the chain. When the last base is added to the RNA chain, the transcription bubble collapses as the RNA‐DNA hybrid is disrupted, the DNA reforms in duplex state, and the enzyme and RNA are both released. The sequence of DNA required for these reactions defines the terminator.
The catalysis of nucleotidyl transfers is very similar to the one observed in DNA polymerases involving two divalent cations (usually Mg2+).[22] Recognition of ribonucleotides as substrate and differentiation from their deoxy‐ analogues is believed to be due to the interaction of a tyrosine residue which interacts specifically with the 2’‐OH from the nucleotide’s ribose sugar, as opposed to the bulky residue present in DNA polymerases. The differentiation of the correct nitrogen base is achieved by the same “close‐fit” model as in DNA polymerases.[23] The nature of the changes from initiation to elongation complex that allow translocation of the enzyme along the template, away from the promoter, is not known. It has been suggested that the non‐template strand may play an important role in the elongation complex, perhaps in RNA displacement.[14] 1.2.3.3. Terminal deoxynucleotidyl Transferase Deoxynucleotidyl transferases belong to the only family of DNA polymerases that elongates DNA strands in a template independent manner. Unlike any other polymerase, Terminal deoxynucleotidyl Tranferase (TdT) has only minor preference for the incorporation of deoxyribo‐ over ribonucleotides on DNA strands in vitro. However, incorporation of ribonucleotides by TdT leads to premature termination of chain elongation.[24] All known nucleotidyl tranferases contain a catalytic domain which is topologically different from the structures of other DNA polymerases. Despite these topological differences, the local structure of the catalytic site is made of three carboxylate side chains and two divalent cations in the
same palm bindi Figure The p absen incor group Altho the m for Td addit gene adapt e spatial arra domain con ng of the DN e 1.9 – Propos presence of a nce of activ
porated is t ps of nucleot
ough TdT wa most poorly u
dT remained tion of nuc
rating subtle tation of the ngement. Fu ntaining the NA primer an sed structure a lasso‐like 1 ity over 3’‐O thought to d tide’s ribose s one of the understood e d elusive for leotides to e randomiza e vertebrate urthermore, catalytic car nd nucleotide of the Termi 16 amino aci OH recessed derive from group.[25] e first DNA po enzymes tha several deca single‐stran tion of this immune syst the hand m rboxylate tri e (Figure 1.9 nal deoxynuc d loop impe d DNA chain the lack of
olymerase a at catalyzes D ades. It is now nded DNA genetic mat tem (for a re etaphor use iad and a fin ). cleotidyl Trans edes the pres ns. The lack interactions ctivities iden DNA synthes w recognized during V(D) terial, TdT p eview see [29 d in DNA po nger and thu sferase. Figur sence of a te of specificit s between t ntified in ma is. Indeed, th d that TdT is )J recombin plays a crucia
9] and refere olymerases h umb domain re adapted fro emplate stra ty towards t he protein a ammals,[26] it he specific p responsible nation.[27,28]
al role in the ences therein olds true wi involved in om [25]. nd, showing the sugar be and 2’ or 3’ t remains on hysiological for the rand By delibera e evolution n). th a the the eing ‐OH e of role dom ately and
![Figure 1.8). [1 ted from [14]. s (rNTP vs. d] However, inwinding ofas template.strands that and, and id17] dNTP), the t involves f the DNA The RNA is used as entical in](https://thumb-eu.123doks.com/thumbv2/123dok_br/15239942.1022849/44.892.274.582.160.539/figure-from-however-inwinding-template-strands-involves-entical.webp)
![Figure 1.20 – Absorption (A) and emission (B) spectra of the coumarin derivatives shown in Figure 1.19. Figure adapted from [145].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15239942.1022849/76.892.129.733.362.608/figure-absorption-emission-spectra-coumarin-derivatives-figure-figure.webp)
![Figure 1.21 – Absorption and emission spectra of ClMMC in cyclohexane (full line) and dioxane:water mixture, 1:4 (dashed lines). Figure taken from [147].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15239942.1022849/78.892.242.609.160.437/figure-absorption-emission-spectra-cyclohexane-dioxane-mixture-figure.webp)

![Figure 1.23 – Effect of the solvent polarity on the non‐radiative rate constant (k nr ) of DEACM. A significant increase in k nr is observed for high polarity solvents. Figure adapted from [157].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15239942.1022849/82.892.257.604.152.412/polarity-radiative-constant-significant-increase-observed-polarity-solvents.webp)

![Table 1.4 – Photophysical and photochemical properties of cyclic adenosine monophosphates esters of differently substituted (coumarin‐4‐yl)methyl alcohols. In CH 3 OH/H 2 O‐HEPES buffer (pH = 7.2). Data taken from [172]. Caged Comp](https://thumb-eu.123doks.com/thumbv2/123dok_br/15239942.1022849/88.892.111.746.223.513/photophysical-photochemical-properties-adenosine-monophosphates-differently-substituted-coumarin.webp)
