Research paper
Hybrid compounds with two redox centres: Modular synthesis of
chalcogen-containing lapachones and studies on their antitumor
activity
Andr
e A. Vieira
a,f,1, Igor R. Brand
~
ao
a, Wagner O. Valença
b,1, Carlos A. de Simone
c,
Bruno C. Cavalcanti
d, Claudia Pessoa
d,e, Teiliane R. Carneiro
d, Antonio L. Braga
a,**,
Eufr
^
anio N. da Silva Júnior
b,*aDepartamento de Química, UFSC, 88040-900, Florianopolis, SC, Brazil
bInstituto de Ci^encias Exatas, Departamento de Química, UFMG, 31270-901, Belo Horizonte, MG, Brazil cDepartamento de Física e Informatica, Instituto de Física, USP, 13560-160, Sao Carlos, SP, Brazil~ dDepartamento de Fisiologia e Farmacologia, UFC, 60430-270, Fortaleza, CE, Brazil
eFiocruz
eCeara, 60180-900, Fortaleza, CE, Brazil
fInstituto de Química, Departamento de Química Org^anica, UFBA, 40170-290, Salvador, BA, Brazil
a r t i c l e
i n f o
Article history:
Received 11 February 2015 Received in revised form 20 June 2015
Accepted 22 June 2015 Available online 26 June 2015
Keywords: b-lapachone Quinone Selenium Sulfur Antitumor Selenide Anticancer
a b s t r a c t
Chalcogen-containingb-lapachone derivatives were synthesized using a straightforward methodology and evaluated against several cancer cell lines (leukaemia, human colon carcinoma, prostate, human metastatic prostate, ovarian, central nervous system and breast), showing, in some cases, IC50values
below 1mM. The cytotoxic potential of the lapachones evaluated was also assayed using non-tumor cells: human peripheral blood mononuclear cells, two murinefibroblast lines (L929 and V79 cells) and MDCK (canine kidney epithelial cells). These compounds could provide promising new lead derivatives for anticancer drug development. This manuscript reports important findings since few authors have described C-3 substitutedb-lapachone with potent antitumor activity. The methodology employed allowed the preparation of the compounds from lapachol within a few minutes in a green approach.
©2015 Elsevier Masson SAS. All rights reserved.
1. Introduction
In medicinal chemistry, within a broad range of privileged structures, naphthoquinones (NQs) could be considered as a special class due to their notable biological activity against various dis-eases, including cancer [1], and unique properties, for instance, their characteristics as oxidants and electrophiles[2]. Within the context of cancer, the cytotoxicity of quinones is mainly related to the generation of reactive oxygen species (ROS) and the alkylation of crucial proteins and nucleic acids which provoke cell damage[3].
The naturally occurring naphthoquinone lapachol (1) was iso-lated for thefirst time by Arnaudon in 1858[4]and synthesized in 1927 by Fieser [5]. It is the most abundant naphthoquinoidal compound isolated from the core of trees of the Bignoniaceae family, popularly known in Brazil asip^e. This natural product has been extensively studied due to its important biological activities, for instance, trypanocidal[6], leishmanicidal[7]and antitumoral [8e10]. From lapachol (1),
b
-lapachone (b
-lap) can be easily ob-tained by simple acid cyclization.b
-lap (also known as ARQ 501) has been extensively studied in recent years[11,12]and is currently in multiple phase II clinical trials as a monotherapy and applied in combination with other cytotoxic drugs[13,14].Besides the biological activity presented by
b
-lap, this com-pound can be used as a prototype for preparing new bioactive substances[6]. Recently, through the strategy of the redox centre modification ofb
-lap, imidazoles with trypanocidal and*Corresponding author.
**Corresponding author.
E-mail addresses:[email protected](A.L. Braga),[email protected](E.N. da Silva).
1 These authors contributed equally to this work.
Contents lists available atScienceDirect
European Journal of Medicinal Chemistry
j o u r n a l h o m e p a g e : h t t p : / / w w w . e l s e v i e r . c o m / l o c a t e / e j m e c h
http://dx.doi.org/10.1016/j.ejmech.2015.06.044
0223-5234/©2015 Elsevier Masson SAS. All rights reserved.
antimycobacterial activities have been described[15,16]. Di Chenna and coworkers[17]reported the synthesis and antitumor activity of imine derivatives obtained from
b
-lap (Scheme 1). Methodologies for the synthesis of lapachones have also recently emerged, for instance, the use of asymmetric organocatalysis[18,19]. da Silva Júnior and Namboothiri et al. showed the potential of chiral squaramide-catalyzed asymmetric synthesis via cascade reactions of 1,3-dicarbonyls with Morita-Baylis-Hillman acetates of nitro-alkenes for the preparation of asymmetric lapachones[18]. Rueping et al. described the synthesis of chiral lapachones from the reaction of lawsone witha
,b
-unsaturated aldehydes in the presence of an organocatalyst to generatea
-lapachone derivatives, and after isomerization C-ring-substitutedb
-lapachones were obtained[19]. Jimenez-Alonso and collaborators described other examples of C-ring-modified lapachones derived from the Knoevenagel conden-sation of lawsone with unsaturated aldehydes (Scheme 1). Authors have discussed the action of these compounds asin vitroinhibitors of human topoisomerase II[20].In addition,
b
-lapachone-based 1,2,3-triazoles with activity against cancer cells, with IC50 values below 2m
M, have been described by our group[21]. We gained some insights related to the generation of reactive oxygen species (ROS) and carried out some preliminary studies on the mechanism of action in tumor cells. Investigations on the formation of thiobarbituric acid reactive substances (TBARS) and oxidative DNA damage after treatment, detected by the comet assay with the bacterial enzymes for-mamidopyrimidine DNA-glycosylase and endonuclease III, were also conducted[21].Organoselenium compounds have been widely studied due to their recognized biological activity against several major diseases,
including diabetes, Alzheimer's disease and strokes, and they play an important role in cancer prevention and treatment[22]. Several mechanisms have been proposed regarding the anticancer activity of these compounds, such as antioxidant protection by sele-noenzymes, specific inhibition of tumor cell growth by Se metab-olites, modulation of the cell cycle and apoptosis, and an effect on DNA repair, but the specific mode of action is still not fully understood.
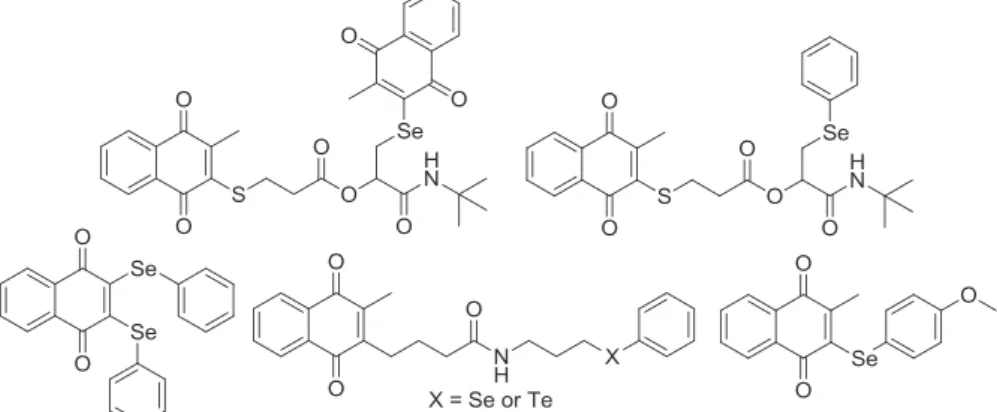
Several kinds of organoselenium compounds are able to mimic the activity of some selenoenzymes, and antioxidant properties are recognized features of these compounds[23]. In the past few years, Jacob and co-workers have conducted studies aimed at the syn-thesis and antitumor evaluation of chalcogen-containing quinones able to disturb ROS in cancer cell lines to deadly levels[24]. The strategy employed was based on a combination of ROS generators, for instance, a quinoidal system, and ROS users, exemplified by organochalcogens. In general terms, ROS in cancer cells are elevated to levels close to the critical redox, resulting in the induction of apoptosis[24,25]. ROS-generating quinones efficiently enhance the formation of, for example, H2O2, O2 andOH radicals, but inter-esting effects can be observed by diminishing the capacity of the cell to eliminate these species (antioxidant defense). Pre-existing ROS can be used by organochalcogen compounds, accelerating re-actions with redox-sensitive proteins and enzymes, and this pro-cess can cause malfunction of the cell andfinally cell death[26]. Following this approach, 1,4-naphthoquinone-based chalcogens and peptidomimetic compounds with the presence of chalcogen and quinone redox centres and notable antitumor activity have been reported[24e27](Fig. 1).
Based on recent findings reported by our group and other
Scheme 1.Structural modifications of the antitumorb-lapachone (ARQ 501).
authors, we envisioned a straightforward synthesis of chalcogen-containing
b
-lapachone. The compounds were evaluated against ten types of cancer cell lines, HL-60 (leukaemia), MOLT-4 (leukaemia), HCT-116 (human colon carcinoma), HCT-8 (colon), PC3 (prostate), PC3M (human metastatic prostate), OVCAR-3 (ovarian), OVCAR-8 (ovarian), SF295 (central nervous system) and MDA-MB-435 (breast), and normal cell lines, exemplified by pe-ripheral blood mononuclear cells (PBMC), V79 and L929. The preparation of this class ofb
-lapachones by exploiting the elec-trophilic reactivity of different dichalcogenides and studies on their antitumor properties are as yet unreported.2. Results and discussion
2.1. Chemistry
The chemical reactivity of lapachol (1) is mainly related to the isoprenyl lateral chain which is susceptible to reaction with different electrophiles. As previously reported, the reactions with sulfuric acid [28], bromine[29], iodine[30]andmeta -chloroper-oxybenzoic acid [31], to name a few, can provide the respective substituted bioactive
b
-lapachone derivatives [32] (Scheme 1). Bearing in mind the nucleophilic characteristic of lapachol (1), the selenium-containingb
-lapachone derivatives 2e8 and the sulfur analogous 9e13 were synthesized using a recently described methodology[33]through reaction with electrophilic species RYI (Y¼S or Se). The chalcogenylation of lapachol (1) using molecular iodine as a catalyst, DMSO as a stoichiometric oxidant and different nucleophiles under microwave irradiation afforded the selenium-and sulfur-containingb
-lapachone derivatives (Schemes 2 and 3).This methodology represents a rapid, green and efficient method to prepare chalcogen-containing
b
-lapachone, with a reaction time of only 10 min.A proposed reaction pathway for the chalcogen functionaliza-tion of lapachol (1), based on the informafunctionaliza-tion reported above, is illustrated inScheme 4. Initially, the electrophilic specie RYI (Y¼S or Se) is formed, probably through the reaction of diorganyl dichalcogenide with the catalyst. The electrophile generated then undergoes nucleophilic attack of the double bond of the isoprenyl lateral chain of the lapachol (1) leading to the formation of the intermediate chalcogeniranium ion. In the next step, a cyclization and the elimination of Hþ lead to the formation of chalcogen-containing
b
-lapachone (Scheme 4). Only the formation of the ortho-quinone was observed in the reaction. The other possible product, para-quinone (a
-lapachone derivative), was not evidenced.The chemical shift of the hydrogens of the pyranic ring of
b
-lap derivatives appears in the same region (d
2.5e3.5). In general, the signals of the hydrogens of the two methyl groups appear atd
1.6 and 1.5. The other signals corresponding to the substitution pattern observed for each substance are totally in accordance with those expected for the prepared compounds. For all compounds, the signals consistent with the proposed structures can be observed on the13C NMR spectra.The unpublished compounds3e8and9e13were obtained as orange and red solids, respectively, in moderate to high yields, and their structures were determined by IR,1H and13C NMR. Electro-spray ionization mass spectra were also obtained. Suitable crystals of compound5were obtained, as an example, and the structure was confirmed by crystallographic methods.
Fig. 1.Organochalcogen compounds described by Jacob and co-workers.
Scheme 2.Synthesis of selenium-containingb-lapachone2e8.
2.2. X-ray analysis
The bond lengths and angles are in good agreement with the expected values, as reported in the literature[34]. The atoms of the rings (C1eC10) are coplanar and the largest deviation [0.072(2) Å] from the least-squares plane is exhibited by atom C9. Atoms O1, O2 and O3 are in the mean least-squares plane of the rings with de-viations of 0.112(2), 0.199(2) and 0.132(3) Å, respectively. Regarding the pyran ring, the atoms C12 and C13 are out of the leastsquares plane, giving this ring the conformation of a half chair. The puckering parameters calculated for this conformation were: q2¼0.178(6) Å, q3¼0.459(6) Å, Q¼0.492(3) Å,
q
¼21.2(7)and4¼132.1(3)[35]. The bond angles between atoms are 96.6for C16eSeeC12 and 109.9for C11eC12eSe. The dihedral angle be-tween the leastesquares plane calculated through the atoms [C1eC10] and [C16eC21] is 141.37(1) (Fig. 2). All H atoms were located by geometric considerations placed (CeH¼0.93e0.97 Å) and refined using a riding model with Uiso(H)¼1.5Ueq(C-methyl) or 1.2Ueq(other). An Ortep-3 diagram of the molecule is shown in
Fig. 2andTable S1shows the main crystallographic data.
2.3. Biological activity
All of the substances described (Schemes 2 and 3) were evalu-atedin vitrousing the MTT assay against ten cancer cell lines: HL-60, MOLT-4, HCT-116, HCT-8, PC3, PC3M, OVCAR-3, OVCAR-8,
SF295 and MDA-MB-435.
b
-Lapachone and doxorubicin were used as positive controls (Table 1). Normal cells, human peripheral blood mononuclear cells (PBMC), murinefibroblasts cell lines (L929 and V79) and canine kidney epithelial cells (MDCK) were used to evaluate the selectivity of the compounds. As previously described [36], the compounds were classified according to their activity as highly active (IC50 < 2m
M), moderately active (2m
M<IC50<10m
M), or inactive (IC50>10m
M).Chalcogen-containing
b
-lapachone (compounds 2, 4e10 and 12e13)were considered moderately active, with IC50values in the range of 2.03e10.0m
M. However, high activity was observed in some lineages evaluated. For HL-60, compounds6(IC50¼0.94m
M) and7(IC50¼0.53m
M) were considered to be highly active. Com-pounds 4, 9e10, 12e13 with IC50 values in the range of 1.22e1.92m
M were also promising against HL-60 (Table 1). Another lineage sensitive to the compounds described herein was MOLT-4 (leukaemia) with four active compounds 6e7 and 12e13 (IC50 values between 0.73 and 1.89m
M). Recently, we described the synthesis of nor-b
-lapachone-based 2,1,3-benzothiadiazole-linked-triazole with potent activity against MOLT-4 [37], showing the importance of this class of compounds against leukaemia cancer cell lines.Into the new compounds prepared, we inserted phenylselenyl and phenylthio substituents with the presence of electron-withdrawing (chlorine, bromine and trifluoromethyl) and electron-donating (methyl and methoxy) groups. Phenylselenyl Scheme 3.Synthesis of sulfur-containingb-lapachone9e13.
Scheme 4. Proposed mechanism for the formation of chalcogen-containingb-lapachone.
(unsubstituted), butylselanyl, benzylthio, phenylthio and ethylthio substituents were also considered. In general terms, the choice of the groups was based on two important characteristics: diselenides and disulfides able to generate electrophilic species in the reaction medium and the presence of groups capable of modifying the electronic nature of the molecules studied.
Regarding 3-phenylseleno-
b
-lapachone (2), potent antitumor activity was observed against PC3, OVCAR-3 and OVCAR-8 (IC50¼ 0.55, 1.93 and 1.76m
M, respectively). The insertion of a bromine group (electron-withdrawing group in thepara-position) intensified the activity and compound 6 was active against all cancer cell lines evaluated, with IC50 values in the range of 0.94e1.72m
M. However, this compound presented cytotoxicity against the four normal cell lines used in the study, and the selectivity index was low (Table 2). We considered compound7, with the presence of a trifluoromethyl group, one of the most promising of this series. This compound was very active against HL-60 and MOLT-4, as discussed previously, but when evaluated against the normal cell line PBMC the IC50value was 6.36m
M. The selectivity index for7 was 12.0. A comparison with doxorubicin (selectivity index¼10.0) indicates that7should be considered for further studies.Compounds12and13, with the presence of a sulfur atom, were active against seven cancer lineages with IC50values<2
m
M. For these substances moderate cytotoxicity was observed against the normal cell line PBMC, with IC50values between 3.40 and 3.94m
M. Lapachones represent an important family of compounds withrelevant antitumor activity [38]. Since Pink and co-workers re-ported the use of
b
-lapachone against tumors that overexpress NAD(P)H:quinone oxidoreductase (NQO1), this ubiquitous fl avo-protein remains an important intracellular target ofb
-lap in tumor cells[39].b
-Lap is able to kill different cancer cells that overexpress endogenous NQO1 [40], for instance, solid tumors, and their mechanism of action is related to NQO1-dependent futile redox cycling, which consumes O2 and generates ROS, as recently described by Bey and co-workers[41]. As discussed by the authors, in a redox cycle ofb
-lap in the presence of NQO1, the hydroquinone form ofb
-lap is unstable. Through 2 one-electron oxidations, probably using O2, at equilibrium the hydroquinone can be bio-oxidized tob
-lap. In this type of redox cycle superoxide is formed [41].The presence of two individual redox centres in tellurium-containing naphthoquinoidal compounds was confirmed by elec-trochemical studies carried out by Jacob et al.[42]. Their results indicated the presence of multifunctional properties related to the generation of ROS (quinoidal moiety) and GPx-like peroxidation catalysis by the chalcogenium atom, which provokes cell damage. Recently, Braga and co-workers shed light on the mechanism of the GPx-like activity of selenides and selenoxides [43]and demon-strated the participation of hydroxy perhydroxy selenane, as a stronger oxidizing agent in the oxidation of PhSH with H2O2 cata-lyzed by selenoxides.
In view of the above, the possible mechanism of action involving chalcogen-containing
b
-lapachone is shown inScheme 5. In com-pounds with two redox centres, that is, a quinoidal moiety able to generate ROS and selenium or sulfur redox centres for GPx-like peroxidation catalysis, both being biologically important in cellular redox balance, either centre can be responsible for the cell damage[44]. In general, naphthoquinones are able to generate ROS and the catalytically active organochalcogens are usually capable to use ROS. With the chalcogen-containingb
-lapachones herein described, the aim was explore simultaneously these different properties presented by the quinoidal system and chalcogens in order to obtain more active compounds, which are able to act as redox modulator derivatives, since lethal cocktails of reactive spe-cies can push cancer cell lines over a critical redox threshold and finally kill them through apoptosis as recently proposed by Jacob et al.[24].Studies on the mechanism of action of selected compounds are currently underway in our laboratories and will be reported in due course. The aim is to describe, in a concise and complete manner, the proposed mode of action of
b
-lap derivatives. Initially, to determine the involvement of ROS in the antitumor activity, we performed experiments to measurement the protein oxidation and lipid peroxidation through protein carbonylation and TBARS assays, respectively, using selected compounds6,12and13and ovarian OVCAR-3 cancer cells. Compounds6,12and13presented IC50in the range of 1.22e2.81m
M for the cancer cell lines evaluated and are among the most active substances of this class. Besides the bio-logical aspects, these compounds represent three distinct organo-chalcogen groups, exemplified by phenylselenyl, phenylthio and ethylthio substituents.Proteins are major targets for ROS, and ROS-induced protein modifications can result in the unfolding, or other alteration, of the protein structure. The carbonylation of proteins is an irreversible type of oxidative damage, which is considered to be a widespread indicator of severe oxidative damage, often leading to a loss of protein function[45,46]. Also, lipid peroxidation has been shown to be involved in oxidative stress. Thus, the extent of compound-induced lipid peroxidation was determined by the reaction of thi-obarbituric acids (TBARS) which are formed as a byproduct of lipid peroxidation (i.e. as fat degradation products). As shown inFig. 3A Fig. 2.(a) Ortep-3 projection of5showing the atom labeling and 50% probability
displacement ellipsoids. (b) Projection down direction of the quinonoid ring of5.
Table 1
Cytotoxic activity expressed as IC50mM (95% CI) for compounds2e8and9e13against cancer and normal cell lines after 72 h exposure, obtained by nonlinear regression for all cell lines from three independent experiments.
Compd IC50mM (95% CI)
HL-60 MOLT-4 HCT-116 HCT-8 PC3 PC3M OVCAR-3 OVCAR-8 SF295 MDA-MB-435 PBMC V79 L929 MDCK
2 5.18 4.75e5.71
4.50 3.72e5.43
2.41 2.13e2.76
3.29 2.81e3.75
0.55 0.30e0.90
10.49 9.63e11.47
1.93 1.73e2.11
1.76 1.63e2.06
2.11 1.71e2.66
2.26 1.93e2.84
3.04 2.64e3.69
2.54 2.34e2.99
2.49 1.91e2.89
2.08 1.61e2.31
3 3.64 2.72e4.88
5.73 5.22e6.78
19.27 16.87e20.93
7.87 7.31e9.16
5.17 4.32e5.76
18.13 15.33e19.20
13.15 12.08e14.31
15.48 14.41e16.14
9.52 9.16e10.33
9.31 8.48e9.96
11.13 10.76e11.71
10.11 9.48e10.23
10.47 10.13e13.34
9.60 9.04e9.99
4 1.80 1.45e2.22
2.29 1.56e2.64
4.68 4.08e5.40
2.90 2.48e3.36
3.06 2.45e3.83
3.79 3.22e4.46
3.86 3.15e4.49
4.75 4.35e4.93
5.05 4.51e5.52
4.04 3.55e4.46
2.69 2.26e2.87
2.22 1.66e2.52
1.89 1.47e2.38
3.20 2.71e3.55
5 2.73 2.22e3.35
4.95 4.51e5.62
7.68 6.43e9.26
7.99 7.34e8.70
3.28 2.68e4.02
6.04 5.74e6.80
4.00 3.58e4.46
3.49 3.40e3.70
2.64 2.33e2.87
3.45 3.12e3.72
6.55 6.13e7.04
5.25 4.58e5.58
5.81 5.51e6.39
4.95 4.37e5.44
6 0.94 0.77e1.15
1.15 0.86e1.53
1.19 0.90e1.40
1.32 1.19e1.44
0.96 0.77e1.28
1.72 1.30e2.26
1.25 1.17e1.32
1.55 1.42e1.70
0.98 0.86e1.15
1.28 1.02e1.51
2.03 1.49e2.45
2.26 1.72e2.60
1.53 1.09e1.80
1.07 0.77e1.46
7 0.53 0.23e1.16
0.73 0.38e1.11
3.78 3.11e4.57
3.37 2.68e4.25
1.69 1.26e2.29
3.33 2.64e4.21
2.04 1.78e2.34
2.55 2.04e2.81
2.19 1.86e2.44
2.38 1.93e2.72
6.36 5.65e6.81
3.11 2.75e3.50
3.69 3.20e4.19
3.00 2.62e3.39
8 2.80 2.51e3.15
4.34 3.89e4.77
5.96 5.51e6.43
7.89 7.18e8.69
3.68 2.67e5.03
2.51 1.61e2.83
3.65 2.70e4.16
2.78 2.19e3.02
3.33 3.04e4.31
3.63 2.78e4.45
4.79 4.37e5.22
5.37 4.84e5.77
4.69 4.02e5.69
4.34 3.76e4.92
9 1.75 1.42e2.14
2.27 1.78e2.90
2.82 2.55e3.10
3.42 2.85e3.84
2.93 2.49e3.07
3.75 3.48e3.92
2.24 1.94e2.60
2.82 2.44e3.42
3.89 3.59e4.49
3.45 2.99e3.67
4.25 3.78e4.74
4.66 4.33e5.02
3.48 3.15e3.86
3.67 3.23e4.36
10 1.19 0.68e2.05
2.19 1.56e2.73
3.22 2.36e3.45
2.99 2.39e3.22
3.90 3.16e4.33
3.05 2.59e3.50
2.68 2.51e2.99
2.65 2.05e2.91
3.31 2.76e3.48
3.56 3.22e3.73
2.76 2.14e3.19
2.39 2.19e2.62
3.22 2.59e3.62
2.73 2.31e2.93
11 2.27 1.72e3.01
3.51 2.35e3.86
10.31 9.71e11.03
4.33 3.64e5.15
8.67 7.79e9.19
9.16 8.69e9.98
7.60 7.10e8.45
6.80 5.92e7.49
6.03 5.46e6.88
5.92 5.02e6.61
5.07 4.55e5.51
5.67 5.04e6.17
4.69 4.33e5.32
3.81 3.45e4.30
12 1.22 1.03e1.53
1.89 1.58e2.23
1.48 1.24e1.76
2.05 1.55e2.36
1.32 0.85e1.76
2.00 1.53e2.70
1.63 1.35e2.00
2.23 1.92e2.67
1.11 0.80e1.48
2.07 1.94e2.28
3.94 3.29e4.78
2.54 1.81e2.98
2.96 2.70e3.16
1.87 1.32e2.41
13 1.48 1.28e1.71
1.22 0.66e1.62
1.58 1.42e1.75
1.85 1.71e1.98
2.38 1.71e3.30
2.81 2.57e3.04
1.81 1.65e2.07
1.58 1.09e2.01
1.88 1.65e2.18
2.34 2.14e2.91
3.40 3.07e4.03
2.21 1.45e2.71
1.91 1.28e2.34
2.64 2.01e3.07
b-lap 1.57 1.11e1.69
0.78 0.49e0.87
0.87 0.74e0.95
0.95 0.78e1.07
1.65 1.40e1.94
1.82 1.57e2.02
1.90 1.69e2.23
1.16 0.97e1.25
0.95 0.70e1.03
0.25 0.16e0.33
>20.63 e e 13.50
12.92e12.08
DOXO 0.06 0.02e0.09
0.02 0.02e0.04
0.15 0.09e0.21
0.19 0.15e0.23
0.02 0.02e0.04
0.04 0.02e0.04
0.32 0.26e0.43
0.41 0.36e0.49
0.51 0.43e0.58
0.96 0.87e1.10
0.55 0.41e0.58
0.28 0.21e0.36
0.23 0.15e0.30
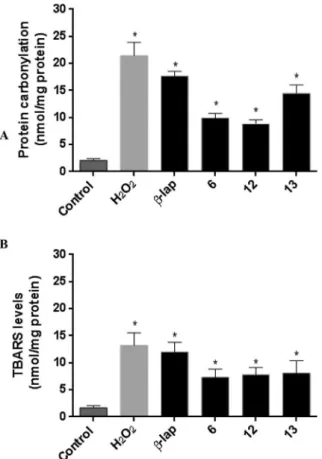
0.43 0.32e0.53
and B, after 12 h of exposure, OVCAR-3 cells treated with com-pounds6,12and13resulted in a significant (p<0.05) increase in protein carbonylation and TBARS levels at 5
m
M.The increases in the protein carbonylation and TBARS levels are related to the generation of reactive oxygen species and this is the first evidence associated with the mechanism of action of these compounds in cancer cell lines and these data corroborate our proposal for ROS generated by the chalcogen-containing
b
-lapachone.3. Conclusions
We have described herein a rapid, green and efficient method to prepare a high diversity of selenium- and sulfur-containing
b
-lapachone compounds, in moderate to high yields. For the first time, two redox centres (ortho-quinoidal and chalcogens) inb
-lap derivatives have been described. In general, all of the compounds were moderately and/or highly active against cancer cell lines andsome of the substances have presented a higher selectivity index than doxorubicin, a standard drug used clinically against several types of cancer.
4. Experimental section
4.1. Chemistry
1H and13C NMR spectra were recorded at room temperature using a Bruker AC 200 and Varian Unity AS 400 in CDCl3at 400 MHz and 200 MHz or at 100 MHz and 50 MHz, respectively. Chemical shifts (
d
) are reported (in ppm) relative to the TMS (1H NMR) and the solvent (13C NMR). APPI-Q-TOF MS measurements were per-formed with a micrOTOF Q-II (Bruker Daltonics) mass spectrometer equipped with an automatic syringe pump (KD Scientific) for sample injection. The APPI-QTOF mass spectrometer was run at 4.5 kV with a desolvation temperature of 180C. The mass spec-trometer was operated in the positive ion mode. A standardScheme 5.Possible mechanism of action for chalcogen-containingb-lapachone based on previously-described data[38e42]. Table 2
Selectivity index for the most active compounds (only IC50values<2mM for cancer cell lines were considered) [Selectivity index, represented by the ratio of cytotoxicities between normal cells and different lines of cancer cells].
Compd PBMC, V79, L929 and MDCKvs. HL-60
PBMC, V79, L929 and MDCKvs. MOLT-4
PBMC, V79, L929 and MDCKvs. HCT-116
PBMC, V79, L929 and MDCKvs. HCT-8
PBMC, V79, L929 and MDCKvs. PC3
PBMC, V79, L929 and MDCKvs. PC3M
PBMC, V79, L929 and MDCKvs. OVCAR-3
PBMC, V79, L929 and MDCKvs. OVCAR-8
PBMC, V79, L929 and MDCKvs. SF295
PBMC, V79, L929 and MDCKvs. MDA-MB-435
2 e e e e 5.5, 4.6, 4.5
and 3.7
e 1.5, 1.3, 1.2 and
1.0
1.7, 1.4, 1.4 and 1.1
e e
6 2.1, 2.4, 1.6 and 1.1
1.7, 1.9, 1.3 and 0.9
1.7, 1.8, 1.2 and 0.8
1.5, 1.7, 1.1 and 0.8
2.1, 2.3, 1.5 and 1.1
1.1, 1.3, 0.8 and 0.6
1.6, 1.8, 1.2 and 0.8
1.3, 1.4, 0.9 and 0.6
2.0, 2.3, 1.5 and 1.0
1.5, 1.7, 1.1 and 0.8
7 12.0, 5.8, 6.9 and 5.6
8.7, 4.2, 5.0 and 4.1
e e e e e e e e
12 3.2, 2.0, 2.4 and 1.5
2.0, 1.3, 1.5 and 0.9
2.6, 1.7, 2.0 and 1.2
e 2.9, 1.9, 2.2
and 1.4
1.9, 1.2, 1.4 and 0.9
2.4, 1.5, 1.8 and 1.1
e 3.5, 2.2, 2.6
and 1.6
e
13 2.2, 1.4, 1.2 and 1.7
2.7, 1.8, 1.5 and 2.1
2.1, 1.3, 1.2 and 1.6
1.8, 1.1, 1.0 and 1.4
e e 1.8, 1.2, 1.0 and
1.4
2.1, 1.3, 1.2 and 1.6
1.8, 1.1, 1.0 and 1.4
e
Doxorubicin 10.0, 5.4, 4.4 and 8.4
53.0, 27.0, 22.0 and 42.0
3.7, 1.9, 1.5 and 3.0
2.9, 1.5, 1.2 and 2.3
53.0, 27.0, 22.0 and 42.0
17.0, 9.0, 7.3 and 14.0
1.7, 0.8, 0.7 and 1.3
1.3, 0.6, 0.5 and 1.0
1.0, 0.5, 0.4 and 0.8
0.5, 0.2, 0.2 and 0.4
atmospheric pressure photoionization (APPI) source was used to generate the ions. The sample was injected using a constantflow (3
m
L/min). The solvent was an acetonitrile/methanol mixture. The APPI-Q-TOF MS instrument was calibrated in the mass range of 50e3000 m/z using an internal calibration standard (low concen-tration tuning mix solution) supplied by Agilent Technologies. Data were processed employing Bruker Data Analysis software version 4.0. Column chromatography was performed using Merck Silica Gel (230e400 mesh). Thin layer chromatography (TLC) was performed using Merck Silica Gel GF254(0.25 mm thickness). For visualization, the TLC plates were placed under ultraviolet light in acidic vanillin. The product yields included in all tables refer to isolated yields. All reactions were performed in 10 mL sealed tubes in a commercially available monomode reactor (CEM Discover) with IR monitoring and a non-invasive pressure transducer. All reactions under mi-crowave irradiation were performed in 10 mL sealed tubes in a commercially available monomode reactor (CEM Discover) with IR monitoring and a non-invasive pressure transducer. Lapachol (1) was extracted from the heartwood of Tabebuia sp. (Tecoma). Initially, a saturated aqueous sodium carbonate solution was added to the sawdust of ipe. After the formation of the lapachol sodium salt, hydrochloric acid was added, allowing the precipitation of lapachol. Afterfiltration, a yellow solid was obtained. This solid was purified by a series of recrystallizations with appropriate solvents [47]. Compound2has been previously described[33].4.2. General procedure for the synthesis of the selenium-containing
b
-lapachone derivatives under microwave irradiationThe lapachol (1) (0.5 mmol), the diaryl diselenides (0.25 mmol), iodine (20 mol %) and DMSO (1 equiv.) were placed in a dry mi-crowave tube. The tube was sealed and placed into a CEM Discover microwave. Initially, an irradiation power of 100 W was applied. After the temperature reached 50 C the instrument was auto-matically adjusted to maintain a constant temperature. After 10 min the reaction was quenched with a solution of 10% sodium thiosulfate and the aqueous layer was extracted with ethyl acetate. The organic phase was dried over MgSO4,filtered, and the solvent was completely removed under vacuum to give the crude product. Purification was carried out by flash chromatography with a mixture of hexane/ethyl acetate (96:4), affording the desired compounds2e13.
4.2.1. 2,2-Dimethyl-3-(p-tolylselanyl)-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (3)
Yield: 71%; mp 183e184C;1H NMR (CDCl
3, 200 MHz)
d
: 8.05 (d, 1H,J¼8.0 Hz), 7.78 (d, 1H,J¼8.0 Hz), 7.64 (t, 1H,J¼4.0 Hz), 7.53e7.48 (m, 3H), 7.07 (d, 2H,J¼4.0 Hz), 3.34 (dd, 1H,J¼4.0 and 10.0 Hz), 3.07 (dd, 1H,J¼8.0 and 18.0 Hz), 2.72 (dd, 1H,J¼8.0 and 18.0 Hz), 2.32 (s, 3H), 1.68 (s, 3H), 1.54 (s, 3H);13C NMR (CDCl3, 50 MHz)
d
: 179.5, 177.9, 161.5, 154.0, 138.6, 135.5, 134.8, 132.1, 130.8, 130.1, 130.0, 128.6, 124.1, 112.5, 82.9, 45.6, 27.8, 25.2, 21.2; IR (film): 3442, 2979, 2932, 2920, 1694, 1648, 1635, 1607, 1590, 1572, 1486, 1450, 1382, 1366, 1307, 1284, 1240, 1219, 1178, 1164, 1113, 1089, 930, 889, 809, 768, 726, 658 cm 1; HRMS (APPIþ) m/z: calculated for C22H21O3Se [MþH]þ: 413.0656; found: 413.0654.4.2.2. 3-((4-methoxyphenyl)selanyl)-2,2-dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (4)
Yield: 62%; mp 139e140C;1H NMR (CDCl
3, 400 MHz)
d
: 8.04 (d, 1H,J¼8.0 Hz), 7.77 (d, 1H,J¼8.0 Hz), 7.64 (t, 1H,J¼8.0 Hz), 7.54e7.49 (m, 3H), 6.80 (d, 2H,J¼8.0 Hz), 3.78 (s, 3H), 3.30e3.26 (m, 1H), 3.04 (dd, 1H,J¼8.0 and 18.0 Hz), 2.69 (dd, 1H,J¼8.0 and 18.0 Hz), 1.67 (s, 3H), 1.53 (s, 3H);13C NMR (CDCl3, 100 MHz)d
: 179.5, 177.9, 161.5, 160.1, 137.6, 134.8, 132.1, 130.8, 130.0, 128.6, 124.1, 118.2, 114.9, 112.4, 82.9, 55.3, 45.6, 27.8, 25.0, 23.4; IR (film): 3442, 3281, 3065, 2996, 2973, 2932, 2837, 1694, 1650, 1635, 1609, 1588, 1572, 1490, 1452, 1382, 1366, 1309, 1287, 1246, 1176, 1162, 1115, 1089, 1032, 991, 928, 768 cm 1; HRMS (APPIþ) m/z: calculated for C22H21O4Se [MþH]þ: 429.0605; found: 429.0604.Fig. 3.Determination of protein oxidation (A) and lipid peroxidation (B) levels of ovarian OVCAR-3 cancer cells after treatment with compounds (6,12and13) at 5mM for 12 h. The negative control was treated with the vehicle (DMSO) used for diluting the test substances, and H2O2(150mM) was used as the positive control. *p<0.05 compared to control applying ANOVA followed by Tukey's test. Data are presented as mean values±S.E.M. from three independent experiments in triplicate.
4.2.3. 3-((4-chlorophenyl)selanyl)-2,2-dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (5)
Yield: 50%; mp 190e191C;1H NMR (CDCl3, 400 MHz)
d
: 8.05 (d, 1H,J¼8.0 Hz), 7.78 (d, 1H,J¼8.0 Hz), 7.65 (t, 1H,J¼8.0 Hz), 7.54e7.50 (m, 3H), 7.23 (d, 2H,J¼8.0 Hz), 3.39 (dd, 1H,J¼4.0 and 10.0 Hz), 3.09 (dd, 1H,J¼8.0 and 18.0 Hz), 2.72 (dd, 1H,J¼8.0 and 18.0 Hz), 1.67 (s, 3H), 1.54 (s, 3H); 13C NMR (CDCl3, 100 MHz)
d
: 179.3, 177.9, 161.4, 136.5, 134.8, 134.8, 131.9, 130.9, 130.0, 129.5, 128.7, 126.5, 124.1, 112.2, 82.7, 45.9, 27.7, 25.2, 23.3; IR (film): 3442, 2992, 2977, 2935, 1694, 1648, 1635, 1607, 1590, 1472, 1450, 1382, 1366, 1307, 1284, 1240, 1219, 1178, 1164, 1115, 1089, 1011, 930, 889, 817, 770, 726 cm 1; HRMS (APPIþ) m/z: calculated for C21H18ClO3Se [MþH]þ: 433.0109; found: 433.0106.4.2.4. 3-((4-bromophenyl)selanyl)-2,2-dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (6)
Yield: 99%; mp 189e190C;1H NMR (CDCl3, 400 MHz)
d
: 8.07 (d, 1H,J¼8.0 Hz), 7.79 (d, 1H,J¼8.0 Hz), 7.67 (t, 1H,J¼8.0 Hz), 7.54 (t, 1H,J¼8.0 Hz), 7.48 (d, 2H,J¼8.0 Hz), 7.41 (d, 2H,J¼8.0 Hz), 3.41 (dd, 1H,J¼4.0 and 8.0 Hz), 3.10 (dd, 1H,J¼4.0 and 16.0 Hz), 2.74 (dd, 1H, J¼8.0 and 16.0 Hz), 1.68 (s, 3H), 1.55 (s, 3H);13C NMR (CDCl3, 100 MHz)d
: 179.4, 178.0, 161.4, 136.7, 134.8, 132.5, 131.9, 130.9, 130.07, 128.8, 127.2, 124.1, 122.9, 112.2, 82.7, 45.9, 27.7, 25.3, 23.4; IR (film): 3440, 2992, 2977, 2932, 1694, 1650, 1635, 1607, 1588, 1572, 1466, 1382, 1366, 1307, 1284, 1240, 1178, 1164, 1115, 1091, 1074, 1007, 770 cm 1; HRMS (APPIþ) m/z: calculated for C21H18BrO3Se [MþH]þ: 476.9599; found: 476.9595.4.2.5. 2,2-Dimethyl-3-((3-(trifl uoromethyl)phenyl)selanyl)-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (7)
Yield: 84%; mp not determined, orange oil; 1H NMR (CDCl 3, 400 MHz)
d
: 8.02 (dd, 1H,J¼8.0 and 4.0 Hz), 7.83 (s, 1H), 7.76 (d, 2H,J¼8.0 Hz), 7.63 (t, 1H,J¼8.0 Hz), 7.54e7.48 (m, 2H), 7.40 (t, 1H,
J¼8.0 Hz), 3.46 (dd, 1H,J¼4.0 and 10.0 Hz), 3.09 (dd, 1H,J¼4.0 and 18.0 Hz), 2.74 (dd, 1H,J¼8.0 and 18.0 Hz), 1.66 (s, 3H), 1.54 (s, 3H); 13C NMR (CDCl
3, 100 MHz)
d
: 179.2, 177.9, 161.4, 137.9, 134.8, 131.8, 131.2, 131.1, 130.9, 129.9, 129.7, 129.6, 128.7, 125.0, 124.9, 124.1, 112.1, 82.6, 45.9, 27.6, 25.3, 23.4; IR (film): 2981, 2985, 1699, 1652, 1637, 1607, 1590, 1574, 1486, 1454, 1417, 1384, 1321, 1303, 1270, 1256, 1229, 1166, 1127, 1091, 1068, 989, 928, 795, 775 cm 1; HRMS (APPIþ) m/z: calculated for C22H18F3O3Se [MþH]þ: 467.0373; found: 467.0365.4.2.6. 3-(butylselanyl)-2,2-dimethyl-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (8)
Yield: 94%; mp 116e117C;1H NMR (CDCl3, 400 MHz)
d
: 8.05 (d, 1H,J¼8.0 Hz), 7.78 (d, 1H,J¼8.0 Hz), 7.64 (t, 1H,J¼8.0 Hz), 7.50 (t, 1H,J¼8.0 Hz), 3.13e3.02 (m, 2H), 2.72 (t, 2H,J¼2.7 Hz), 2.63 (dd, 1H,J¼8.0 and 18.0 Hz), 1.71 (s, 3H), 1.68e1.62 (m, 2H), 1.44 (s, 3H), 1.41e1.38 (m, 2H), 0.91 (t, 3H, J ¼ 0.9 Hz); 13C NMR (CDCl3, 100 MHz)d
: 179.5, 177.9, 161.5, 134.8, 132.1, 130.8, 130.0, 128.6, 124.1, 112.7, 83.4, 40.3, 32.5, 27.9, 25.6, 25.2, 22.9, 22.4, 13.5; IR (film): 3442, 2955, 2928, 2869, 2859, 1694, 1643, 1633, 1601, 1570, 1454, 1431, 1386, 1370, 1305, 1287, 1258, 1227, 1150, 1111, 1091, 991, 768, 722 cm 1; HRMS (APPIþ) m/z: calculated for C19H23O3Se [MþH]þ: 379.0812; found: 379.0809.4.2.7. 3-(benzylthio)-2,2-dimethyl-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (9)
Yield: 46%; mp 117e118C;1H NMR (CDCl
3, 400 MHz)
d
: 8.05 (d, 1H,J¼4.0 Hz), 7.75 (d, 1H,J¼8.0 Hz), 7.64 (t, 1H,J¼7.6 Hz), 7.51 (t, 1H,J¼7.6 Hz), 7.36e7.28 (m, 5H), 3.86 (d, 2H,J¼4.0 Hz), 2.98 (dd, 1H,J¼4.0 and 18.0 Hz), 2.73 (dd, 1H,J¼4.0 and 10.0 Hz), 2.54 (dd, 1H,J¼8.0 and 18.0 Hz), 1.58 (s, 3H), 1.40 (s, 3H);13C NMR (CDCl3, 100 MHz)d
: 179.3, 178.0, 161.2, 137.2, 134.7, 131.8, 130.7, 129.9, 128.7, 128.5, 128.5, 127.2, 124.0, 112.1, 82.5, 44.7, 36.4, 27.1, 24.4, 21.8; IR (film): 3440, 2977, 2926, 2851, 1696, 1645, 1635, 1601, 1570, 1486, 1452, 1386, 1372, 1323, 1291, 1266, 1227, 1195, 1182, 1148, 1115, 1091, 1072, 991, 775, 722 cm 1; HRMS (APPIþ) m/z: calculated for C22H21O3S [MþH]þ: 365.1211; found: 365.1203.4.2.8. 2,2-Dimethyl-3-(phenylthio)-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (10)
Yield: 52%; mp 106e107C;1H NMR (CDCl3, 400 MHz)
d
: 8.03 (d, 1H,J¼8.0 Hz), 7.78 (d, 1H,J¼8.0 Hz), 7.75 (m, 1H), 7.52e7.44 (m, 3H), 7.31e7.25 (m, 3H), 3.34 (dd, 1H,J¼4.0 and 8.0 Hz), 3.0 (dd, 1H,J¼8.0 and 16.0 Hz), 2.60 (dd, 1H,J¼8.0 and 18.0 Hz), 1.66 (s, 3H), 1.51 (s, 3H);13C NMR (CDCl
3, 100 MHz)
d
: 179.4, 178.1, 161.4, 134.9, 133.9, 132.6, 131.9, 130.9, 130.0, 129.3, 128.7, 127.9, 124.2, 112.1, 82.5, 50.4, 29.7, 27.5, 24.7, 22.3; IR (film): 3071, 3057, 2981, 2926, 2853, 1699, 1652, 1607, 1590, 1574, 1484, 1454, 1439, 1384, 1293, 1260, 1229, 1180, 1150, 1113, 1091, 1025, 991, 928, 775, 746, 740 cm 1; HRMS (APPIþ) m/z: calculated for C21H19O3S [MþH]þ: 351.1055; found: 351.1051.4.2.9. 2,2-Dimethyl-3-(p-tolylthio)-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (11)
Yield: 56%; mp 198e199C;1H NMR (CDCl
3, 400 MHz)
d
: 8.05 (d, 1H,J¼8.0 Hz), 7.78 (d, 1H,J¼8.0 Hz), 7.64 (t, 1H,J¼8.0 Hz), 7.51 (t,1H,J¼8.0 Hz), 7.34 (d, 2H,J¼8.0 Hz), 7.11 (d, 2H,J¼8.0 Hz), 3.27 (dd, 1H,J¼8.0 and 12.0 Hz), 2.97 (dd, 1H,J¼4.0 and 16.0 Hz), 2.59 (dd, 1H,J¼8.0 and 18.0 Hz), 2.32 (s, 3H), 1.66 (s, 3H), 1.51 (s, 3H);13C NMR (CDCl3, 100 MHz)d
: 190.5, 179.5, 178.2, 161.3, 138.3, 134.8, 133.3, 132.0, 130.9, 130.0, 128.7, 124.2, 112.2, 82.5, 50.7, 27.5, 24.6, 22.3, 21.1; IR (film): 3442, 2994, 2981, 2932, 2920, 1694, 1648, 1637, 1607, 1590, 1572, 1452, 1384, 1368, 1309, 1287, 1238, 1189, 1121, 1091, 815, 768 cm 1; HRMS (APPIþ) m/z: calculated for C22H21O3S [MþH]þ: 365.1211; found: 365.1208.4.2.10. 3-((3-chlorophenyl)thio)-2,2-dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (12)
Yield: 53%; mp 185e186C;1H NMR (CDCl
3, 400 MHz)
d
: 8.08(m, 1H), 7.81 (m, 1H), 7.71e7.62 (m, 1H), 7.59e7.49 (m, 1H), 7.44 (s, 1H), 7.34e7.23 (m, 3H), 4.36 (dd, 1H,J¼6.0 and 5.0 Hz), 3.44e2.98 (m, 2H), 2.63 (dd, 1H,J¼8.0 and 18.0 Hz), 1.70 (s, 3H), 1.53 (s, 3H);
13C NMR (CDCl
3, 100 MHz)
d
: 179.1, 178.1, 177.6, 161.3, 136.1, 134.9, 131.8, 131.1, 130.3, 130.2, 130.1, 128.9, 127.9, 124.1, 112.1, 50.2, 30.4, 27.8, 26.7, 24.7, 22.4; IR (film): 2981, 2932, 1699, 1652, 1637, 1607, 1590, 1574, 1486, 1454, 1433, 1384, 1307, 1291, 1260, 1240, 1229, 1178, 1150, 1111, 1091, 1040, 989, 889, 819, 726, 701 cm 1; HRMS (APPIþ) m/z: calculated for C21H18ClO3S [MþH]þ: 385.0665; found: 385.0664.4.2.11. 3-(ethylthio)-2,2-dimethyl-3,4-dihydro-2H-benzo[h] chromene-5,6-dione (13)
Yield: 68%; mp 132e133C;1H NMR (CDCl
3, 400 MHz)
d
: 8.06 (d, 1H,J¼8.0 Hz), 7.79 (d, 1H,J¼8.0 Hz), 7.65 (t, 1H,J¼8.0 Hz), 7.51 (t, 1H,J¼8.0 Hz), 3.05 (dd, 1H,J¼8.0 and 18.0 Hz), 2.90 (dd, 1H,J¼4.0 and 8.0 Hz), 2.69 (m, 2H), 2.51 (dd, 1H,J¼8.0 and 16.0 Hz), 1.69 (s, 3H), 1.41 (s, 3H), 1.29 (t, 3H,J¼1.2 Hz);13C NMR (CDCl3, 100 MHz)
d
: 179.5, 178.2, 161.4, 134.8, 132.0, 130.9, 130.1, 128.7, 124.2, 112.4, 82.9, 45.9, 29.7, 27.5, 26.6, 24.8, 21.7, 14.8; IR (film): 3442, 2959, 2924, 2869, 2853, 1694, 1641, 1633, 1601, 1590, 1570, 1486, 1454, 1391, 1368, 1307, 1293, 1260, 1229, 1189, 1117, 1093, 769 cm 1; HRMS (APPIþ) m/z: calculated for C17H19O3S [MþH]þ: 303.1055; found: 303.1040.4.3. Antitumor activity
Compounds were tested for cytotoxic activity against several human cancer cell lines obtained from the National Cancer Insti-tute, NCI (Bethesda, MD, US). The L929 cells (mousefibroblast L cells NCTC clone 929) were obtained from the American Type Culture Collection (Manassas, VA, US), MDCK cells were purchased from the Rio de Janeiro Cell Bank (Rio de Janeiro, Brazil), and the Chinese hamster lungfibroblasts (V79 cells) were kindly provided by Dr. JAP Henriques (UFRGS, Porto Alegre, RS, Brazil). Peripheral blood mononuclear cells (PBMC) were isolated from the heparin-ized blood of healthy, non-smoker donors who had not taken any medication at least 15 days prior to sampling, using a standard method of density-gradient centrifugation on Histopaque-1077 (Sigma Aldrich Co. - St. Louis, MO/USA). All cancer cell lines and PBMC were maintained in RPMI 1640 medium. The L929, MDCK and V79 cells were cultivated under standard conditions in MEM with Earle's salts. All culture media were supplemented with 20% (PBMC) or 10% (cancer, L929, MDCK and V79 cells) fetal bovine serum, 2 mM L-glutamine, 100 IU/mL penicillin and 100
m
g/mL streptomycin at 37C with 5% CODoxorubicin (0.001e1.10
m
M) was used as the positive control, and negative control groups received the same amount of vehicle (DMSO). The cell viability was determined through the reduction of the yellow dye 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H -tetra-zolium bromide (MTT) to a blue formazan product, as described by Mosmann[48]. At the end of the incubation time (72 h), the plates were centrifuged and the medium was replaced with fresh medium (200m
L) containing 0.5 mg/mL MTT. Three hours later, the MTT formazan product was dissolved in DMSO (150m
L) and the absor-bance was measured using a multiplate reader (Spectra Count, Packard, Ontario, Canada). The drug effect was quantified as the percentage of control absorbance of the reduced dye at 550 nm. All cell treatments were carried out with three replicates.4.4. Measurement of protein oxidation
The intracellular oxidation of proteins in ovarian OVCAR-3 cancer cells could be determined by measuring the carbonyl groups generated in some amino acid side chains using the dini-trophenylhydrazine (DNPH) derivatization method[49]. The level of protein carbonyls in test quinonoid compounds (5
m
M) or 150m
M H2O2-treated cells was measured for 12 h. At the end of the treatments, cells were collected and the cell lysates were prepared by ultrasonication. The protein present in the cell lysates was quantified using Lowry's method [50]. Protein samples (900m
L; 5 mg/mL) were incubated with or without DNPH solution for 60 min at 37C in the dark. The reaction mixture was added to 1 mL of 10% trichloroacetic acid (4C) for 25 min. The suspension was then centrifuged at 5.000 rpm for 30 min (4C) and the superna-tant fraction was discarded. The pellet was washed three times with ethanol/ethyl acetate (1:1, v/v), resuspended in 45 mM TriseHCl (pH 7.4) buffer and incubated for 10 min at 37C. The solubilized protein in the buffer was normalized to the protein content[50]and the absorbance was measured at 375 nm using a spectrophotometer (Spectra Count, Packard, Ontario, Canada). Total protein carbonylation content was determined as nmol/mg protein.4.5. Lipid peroxidationeTBARS assay
The extent to which the test compounds induced lipid peroxi-dation was determined by the reaction of thiobarbituric acid (TBA) with malondialdehyde (MDA), a product formed by lipid peroxi-dation. The assays were performed according to Salgo and Pryor (1996), with minor modifications [51]. Ovarian OVCAR-3 cancer cells were incubated with the test compounds (5
m
M) or 150 mM H2O2for 12 h and then lysed with 15 mM TriseHCl for 1 h. Two milliliters of trichloroacetic acid (0.4 mg/mL) and HCl (0.25 M) were added to the lysate, which was then incubated with 6.7 mg/ mL TBA for 15 min at 100 C. The mixture was centrifuged at 2500 rpm for 15 min. Since TBA reacts with other products of lipid peroxidation in addition to MDA, the results are expressed in terms of thiobarbituric reactive species (TBARS), which are determined by absorbance at 532 nm. Hydrolyzed 1,1,3,3-tetramethoxypropane was used as the standard. The results were normalized to the protein content[50].4.6. X-ray analysis
X-ray diffraction data was obtained on an Enraf-Nonius Kappa-CCD diffractometer (95 mm Kappa-CCD camera on
k
-goniostat) using graphite monochromated MoKa
radiation (0.71073 Å), at room temperature. Data collection was carried out using the COLLECT software[52]at up to 50(2q
). Thefinal unit cell parameters were based on 15,293 reflections. Integration and scaling of the re-flections, correction for Lorentz and polarization effects were
performed with the HKL DENZO-SCALEPACK system of programs [53]. The structure of the compound was solved by direct methods with SHELXS-97[54]. The model was refined by full-matrix least squares on F2using SHELXL-97[55]. The program ORTEP-3[56]was used for the graphic representations and the program WinGX[57] was used to prepare the material for publication. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre asSupplementary material(No. CCDC 1043158). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 2EZ, UK (Fax: (þ44) 1223 336033; e-mail:[email protected]).
Acknowledgments
This research was funded by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), numbers: 480719/2012-8, PVE 401193/2014-4, 449348/2014-8, 474797/2013-9 and 454171/2014-5. We would also like to thank FAPEMIG, FUNCAP, FAPESP, FAPESC, CAPES, PROCAD and INCT-catalise and also the Physics Institute of USP (S~ao Carlos) for kindly allowing the use of the KappaCCD diffractometer.
Appendix A. Supplementary data
Supplementary data related to this article can be found athttp:// dx.doi.org/10.1016/j.ejmech.2015.06.044.
References
[1] (a) L. Constantino, D. Barlocco, Curr. Med. Chem. 13 (2006) 65;
(b) A.A.S. Naujorks, A.O. da Silva, R.S. Lopes, S. Albuquerque, A. Beatriz, M.R. Marques, D.P. de Lima, Org. Biomol. Chem. 13 (2015) 428;
(c) T.A.P. de Moraes, M.J.S. Filha, C.A. Camara, T.M.S. Silva, B.M. Soares, I.S. Bomfim, C. Pessoa, G.C. Ximenes, V.A. Silva Jr., Molecules 19 (2014) 13188;
(d) M.A. Colucci, C.J. Moody, G.D. Couch, Org. Biomol. Chem. 6 (2008) 637. [2] (a) A.V. Pinto, S.L. de Castro, Molecules 14 (2009) 4570;
(b) E.A. Hillard, F.C. de Abreu, D.C.M. Ferreira, G. Jaouen, M.O.F. Goulart, C. Amatore, Chem. Commun. (2008) 2612;
(c) T.J. Monks, D.C. Jones, Curr. Drug Metab. 3 (2002) 425.
[3] (a) J.L. Bolton, M.A. Trush, T.M. Penning, G. Dryhurst, T.J. Monks, T.J. Chem, Res. Toxicol. 13 (2000) 135;
(b) A. Brunmark, E. Cadenas, Free Radic. Biol. Med. 7 (1989) 435;
(c) T.J. Monks, R.P. Hanslik, G.M. Cohen, D. Ross, D.G. Graham, Toxicol. Appl. Pharmacol. 112 (1992) 2;
(d) M.O.F. Goulart, P. Falkowski, T. Ossowski, A. Liwo, Bioelectrochemistry 59 (2003) 85;
(e) S. Vilamil-Fernandez, A.O. Stoppani, M. Dubin, Methods Enzymol. 378 (2004) 67.
[4] M. Arnaudon, Comptes Rendus Hebdomadares. Des Sianes‘L‘Acord, Des Sci. 46 (1858) 1152.
[5] L.F. Fieser, J. Am. Chem. Soc. 49 (1927) 857.
[6] A.V. Pinto, C. Neves Pinto, M.C.F.R. Pinto, R.M. Santa Rita, C. Pezzella, S.L. de Castro, Arzneim Forsch 47 (1997) 74.
[7] N.M.F. Lima, C.S. Correia, L.L. Leon, G.M.C. Machado, M.F. Madeira, A.E.G. Santana, M.O.F. Goulart, Mem. Inst. Oswaldo Cruz 99 (2004) 757. [8] K.V. Rao, T.J. McBride, J.J. Oleson, Cancer Res. 28 (1968) 1952.
[9] S. Fiorito, F. Epifano, C. Bruyere, V. Mathieu, R. Kiss, S. Genovese, Bioorg. Med. Chem. Lett. 24 (2014) 454.
[10] S.N. Sunassee, C.G.L. Veale, N. Shunmoogam-Gounden, O. Osoniyi, D.T. Hendricks, M.R. Caira, J. la Mare, A.L. Edkins, A.V. Pinto, E.N. da Silva Jr., M.T. Davies-Coleman, Eur. J. Med. Chem. 62 (2013) 98.
[11] C.J. Li, Y.Z. Li, A.V. Pinto, A.B. Pardee, Proc. Natl. Acad. Sci. U S A 96 (1999) 13369.
[12] J.S. Lee, A.H. Park, S.H. Lee, S.H. Lee, J.H. Kim, S.J. Yang, Y.I. Yeom, T.H. Kwak, D. Lee, S.J. Lee, C.H. Lee, J.M. Kim, D. Kim, PLoS One 7 (2012) e47122. [13] X.S. Miao, C. Zhong, Y. Wang, R.E. Savage, R.Y. Yang, D. Kizer, E. Volckova,
M.A. Ashwell, T.C. Chan, Rapid Commun. Mass Spectrom. 23 (2009) 12. [14] E.N. da Silva Jr., C.F. de Deus, B.C. Cavalcanti, C. Pessoa, L.V. Costa-Lotufo,
R.C. Montenegro, M.O. de Moraes, M.C.F.R. Pinto, C.A. de Simone, V.F. Ferreira, M.O.F. Goulart, C.K.Z. Andrade, A.V. Pinto, J. Med. Chem. 53 (2010) 504. [15] K.C.G. de Moura, K. Salom~ao, R.F.S. Menna-Barreto, F.S. Emery, M.C.F.R. Pinto,
A.V. Pinto, S.L. de Castro, Eur. J. Med. Chem. 39 (2004) 639.
[16] K.C.G. Moura, P.F. Carneiro, M.C.F.R. Pinto, J.A. da Silva, V.R.S. Malta, C.A. de Simone, G.G. Dias, G.A.M. Jardim, J. Cantos, T.S. Coelho, P.E.A. da Silva, E.N. da
Silva Jr., Bioorg. Med. Chem. 20 (2012) 6482.
[17] P.H. Di Chenna, V. Benedetti-Doctorovich, R.F. Baggio, M.T. Garland, G. Burton, J. Med. Chem. 44 (2001) 2486.
[18] D.K. Nair, R.F.S. Menna-Barreto, E.N. da Silva Jr., S.M. Mobin, I.N.N. Namboothiri, Chem. Commun. 50 (2014) 6973.
[19] M. Rueping, E. Sugiono, E. Merino, Angew. Chem. Int. Ed. 47 (2008) 3046. [20] S. Jimenez-Alonso, H.C. Orellana, A. Estevez-Braun, A.G. Ravelo, E. Perez-Sacau,
F. Machín, J. Med. Chem. 51 (2008) 6761.
[21] E.N. da Silva Jr., B.C. Cavalcanti, T.T. Guimar~aes, M.C.F.R. Pinto, I.O. Cabral, C. Pessoa, L.V. Costa-Lotufo, M.O. de Moraes, C.K.Z. de Andrade, M.R. dos Santos, C.A. de Simone, M.O.F. Goulart, A.V. Pinto, Eur. J. Med. Chem. 46 (2011) 399.
[22] (a) D.L. Klayman, H.H. Günter, Organoselenium Compounds: Their Chemistry and Biology, Wiley, New York, 1973;
(b) C.W. Nogueira, G. Zeni, J.B.T. Rocha, Chem. Rev. 104 (2004) 6255;
(c) A.L. Braga, J. Rafique, Synthesis of biologically relevant small molecules containing selenium. Part B. Anti-infective and anticancer compounds, in: Z. Rappoport (Ed.), The Chemistry of Organic Selenium and Tellurium Com-pounds, vol. 4, John Wiley&Sons, Ltd, Chichester, UK, 2013, pp. 1053e1117. [23] (a) K.B. Sarma, G. Mugesh, J. Am. Chem. Soc. 127 (2005) 11477;
(b) G. Roy, M. Nethaji, G. Mugesh, J. Am. Chem. Soc. 126 (2004) 2712;
(c) L. Flohe, E.A. Günzler, H.H. Schock, FEBS Lett. 32 (1979) 132;
(d) R.J. Shamberger, Biochemistry of Selenium, Plenum Press, New York, 1983;
(e) G. Mugesh, W.W. Du Mont, H. Sies, Chem. Rev. 101 (2001) 2125;
(f) A.L. Braga, J. Rafique, Synthesis of biologically relevant small molecules containing selenium. Part A. Antioxidant compounds, in: Z. Rappoport (Ed.), The Chemistry of Organic Selenium and Tellurium Compounds, vol. 4, John Wiley&Sons, Ltd, Chichester, UK, 2013, pp. 989e1052.
[24] V. Jamier, L.A. Ba, C. Jacob, Chem. Eur. J. 16 (2010) 10920.
[25] S. Shaaban, R. Diestel, B. Hinkelmann, Y. Muthukumar, R.P. Verma, F. Sasse, C. Jacob, Eur. J. Med. Chem. 58 (2012) 192.
[26] (a) N.M. Giles, N.J. Gutowski, G.I. Giles, C. Jacob, FEBS Lett. 535 (2003) 179;
(b) S. Mecklenburg, S. Shaaban, L.A. Ba, T. Burkholz, T. Schneider, B. Diesel, A.K. Kiemer, A. R€oseler, K. Becker, J. Reichrath, A. Stark, W. Tilgen, M. Abbas, L.A. Wessjohann, F. Sasse, C. Jacob, Org. Biomol. Chem. 7 (2009) 4753. [27] M. Doering, L.A. Ba, N. Lilienthal, C. Nicco, C. Scherer, M. Abbas, A.A.P. Zada,
R. Coriat, T. Burkholz, L. Wessjohann, M. Diederich, F. Batteux, M. Herling, C. Jacob, J. Med. Chem. 53 (2010) 6954.
[28] A.V. Pinto, M.C.F.R. Pinto, C.G.T. de Oliveira, An. Acad. Bras. Cienc 54 (1982) 108.
[29] S.C. Hooker, J. Chem. Soc. Trans. 61 (1892) 611.
[30] A.V. Pinto, M.C.R. Pinto, B. Gilbert, J. Pellegrino, R.T. Mello, Trans. R. Soc. Trop. Med. Hyg. 71 (1977) 133.
[31] J.S. Sun, A.H. Geiser, B. Frydman, Tetrahedron Lett. 39 (1998) 8221. [32] S.L. de Castro, F.S. Emery, E.N. da Silva Jr., Eur. J. Med. Chem. 69 (2013) 678. [33] A.A. Vieira, J.B. Azeredo, M. Godoi, C. Santi, E.N. da Silva Jr., A.L. Braga, J. Org.
Chem. 80 (2015) 2120.
[34] F.H. Allen, O. Kennard, D.G. Watson, L. Brammer, A.G. Orpen, R. Taylor, J. Chem. Soc. Perkin Trans. 2 (1987) S1.
[35] D. Cremer, J.A. Pople, J. Am. Chem. Soc. 97 (1975) 1354.
[36] E. Perez-Sacau, R.G. Díaz-Pe~nate, A. Estevez-Braun, A.G. Ravelo, J.M. Garcia-Castellano, L. Pardo, M. Campillo, J. Med. Chem. 50 (2007) 696.
[37] E.H.G. da Cruz, P.H.P.R. Carvalho, J.R. Corr^ea, D.A.C. Silva, E.B.T. Diogo, J.D. de Souza Filho, B.C. Cavalcanti, C. Pessoa, H.C.B. de Oliveira, B.C. Guido, D.A. da Silva Filho, B.A.D. Neto, E.N. da Silva Jr., New. J. Chem. 38 (2014) 2569. [38] (a) E.N. da Silva Jr., B.C. Cavalcanti, T.T. Guimar~aes, M.C.F.R. Pinto, I.O. Cabral,
C. Pessoa, L.V. Costa-Lotufo, M.O. de Moraes, C.K.Z. de Andrade, M.R. dos Santos, C.A. de Simone, M.O.F. Goulart, A.V. Pinto, Eur. J. Med. Chem. 46 (2011) 399;
(b) E.H.G. da Cruz, C.M.B. Hussene, G.G. Dias, E.B.T. Diogo, I.M.M. de Melo, B.L. Rodrigues, M.G. da Silva, W.O. Valença, C.A. Camara, R.N. de Oliveira, Y.G. de Paiva, M.O.F. Goulart, B.C. Cavalcanti, C. Pessoa, E.N. da Silva Jr., Bioorg. Med. Chem. 22 (2014) 1608;
(c) G.A.M. Jardim, T.T. Guimar~aes, M.C.F.R. Pinto, B.C. Cavalcanti, K.M. de Farias, C. Pessoa, C.C. Gatto, D.K. Nair, I.N.N. Namboothiri, E.N. da Silva Jr., Med. Chem. Commun. 6 (2015) 120.
[39] J.J. Pink, S.M. Planchon, C. Tagliarino, M.E. Varnes, D. Siegel, D.A. Boothman, J. Biol. Chem. 275 (2000) 5416.
[40] E.A. Bey, M.S. Bentle, K.E. Reinicke, Y. Dong, C.-R. Yang, L. Girard, J.D. Minna, W.G. Bornmann, J. Gao, D.A. Boothman, Natl. Acad. Sci. U S A 104 (2007) 11832.
[41] E.A. Bey, K.E. Reinicke, M.C. Srougi, M. Varnes, V.E. Anderson, J.J. Pink, L.S. Li, M. Patel, L. Cao, Z. Moore, A. Rommel, M. Boatman, C. Lewis, D.M. Euhus, W.G. Bornmann, D.J. Buchsbaum, D.R. Spitz, J. Gao, D.A. Boothman, Mol. Cancer Ther. 12 (2013) 2110.
[42] F.H. Fry, A.L. Holme, N.M. Giles, G.I. Giles, C. Collins, K. Holt, S. Pariagh, T. Gelbrich, M.B. Hursthouse, N.J. Gutowski, C. Jacob, Org. Biomol. Chem. 3 (2005) 2579.
[43] V. Nascimento, E.E. Alberto, D.W. Tondo, D. Dambrowski, M.R. Detty, F. Nome, A.L. Braga, J. Am. Chem. Soc. 134 (2012) 138.
[44] G.I. Giles, N.M. Giles, C.A. Collins, K. Holt, F.H. Fry, P.A.S. Lowden, N.J. Gutowski, C. Jacob, Chem. Commun. (2003) 2030.
[45] I. Dalle-Donne, G. Aldini, M. Carini, R. Colombo, R. Rossi, A. Milzani, J. Cell. Mol. Med. 10 (2006) 389.
[46] E. Cabiscol, J. Ros, Oxidative damage to proteins: structural modifications and consequences in cell function, in: I. Dalle-Donne, A. Scaloni, D.A. Butterfield (Eds.), Redox Proteomics: from Protein Modifications to Cellular Dysfunction and Disease, John Wiley&Sons, Inc, Hoboken, 2006, pp. 399e471. [47] V.F. Ferreira, Quim Nova na Esc. 4 (1996) 35.
[48] T. Mosmann, J. Immunol. Methods 65 (1983) 55.
[49] H. Buss, T.P. Chan, K.B. Sluis, N.M. Domigan, C.C. Winterbourn, Free Radic. Biol. Med. 23 (1997) 361.
[50] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, J. Biol. Chem. 193 (1951) 265.
[51] M.G. Salgo, W.A. Pryor, Arch. Biochem. Biophys. 333 (1996) 482. [52] Enraf-Nonius, Collect. B.V. Nonius, Delft, The Netherlands, 1997e2000.
[53] Z. Otwinowski, W. Minor, in: C.W. Carter, R.M. Sweet (Eds.), Methods in Enzymology, vol. 276, Academic Press, New York, 1997, pp. 307e326. [54] G.M. Sheldrick, SHELXS-97. Program for Crystal Structure Resolution,
Uni-versity of G€ottingen, G€ottingen, Germany, 1997.
[55] G.M. Sheldrick, SHELXL-97. Program for Crystal Structure Refinement, Uni-versity of G€ottingen, G€ottingen, Germany, 1997.
[56] L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565. [57] L.J. Farrugia, J. Appl. Crystallogr. 32 (1999) 837.