UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIASDEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Blocking HIV at cell entry: innovative exploitation of the interaction
between membranes and next generation drugs - single domain
antibodies, peptides and photoactivatable oxidants
Tiago Nascimento Figueira
Dissertação
Mestrado em Bioquímica
(Bioquímica Médica)
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIASDEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Blocking HIV at cell entry: innovative exploitation of the interaction
between membranes and next generation drugs - single domain
antibodies, peptides and photoactivatable oxidants
Tiago Nascimento Figueira
Dissertação
Mestrado em Bioquímica
(Bioquímica Médica)
Dissertação orientada pelo Professor Doutor Miguel A. R. B. Castanho e
pela Professora Doutora Ana I. A. Coutinho
I
AGRADECIMENTOS
A realização deste trabalho contou com a participação e apoio de pessoas cujo contributo se tornou essencial tanto intelectual como animicamente.
Ao Professor Doutor Miguel Castanho deixo um especial agradecimento pela oportunidade de integrar o seu grupo de investigação, pela confiança na qualidade do meu trabalho e pela aposta no meu futuro académico e profissional. Agradeço igualmente à Doutora Ana Salomé Veiga pela dedicação ao projeto, pela presença e orientação durante todas as fases do meu estágio e pelo seu modelo de qualidade e integridade científica. À Professora Ana Coutinho não posso deixar de agradecer a preocupação no meu percurso e o seu permanente apoio.
Agradeço ainda ao Professor Doutor Nuno Santos e à sua unidade, em especial ao Doutor Axel Hollmann, pela disponibilização de instalações e equipamentos e pela introdução a técnicas laboratoriais.
Àqueles que hoje são meus colegas de laboratório agradeço a forma como me integraram e assistiram diariamente. Ao João Freire agradeço por ter partilhado a sua intuição e ideais científicos. À Doutora Diana Gaspar agradeço a paciência e os valiosos concelhos. À Antónia agradeço toda a simpatia e ajuda no laboratório.
Finalmente, um obrigado aos meus pais que me apoiaram sempre e incondicionalmente até este momento do meu percurso académico. À Alexandra Pousinha agradeço a sua dedicação e suporte aos meus esforços durante a realização deste trabalho.
II
RESUMO
Desde a sua descoberta, o Vírus da Imunodeficiência Humana (VIH) tornou-se uma das principais preocupações da comunidade médica e científica mundial. Mais de 34 milhões de pessoas encontram-se infectadas em todo o mundo, sob o risco de contrair o Síndrome da Imunodeficiência Adquirida (SIDA). Este estado imunocomprometido resulta de dois pontos críticos da patogenicidade lentiviral: a deplecção de linfócitos T CD4+ circulantes para níveis incapazes de combater infecções oportunistas e a persistência de reservatórios virais em macrófagos e células dendríticas..
O ciclo de vida do VIH revela alvos favoráveis no combate à progressão infecciosa. Ao entrar em contacto com as células do hospedeiro, o reconhecimento de receptores e co-receptores específicos desencadeia a fusão das membranas celular e viral com libertação do conteúdo para o citoplasma das células do hospedeiro. No interior da célula, o material genético é processado e integrado no genoma do indivíduo, a partir do qual poderão ser traduzidas novas proteínas virais. Da membrana do hospedeiro emergem novos viriões posteriormente activados por maturação proteolítica. Cada vírus apresenta um lipidoma sob a forma de bicamada lipídica que engloba e envolve o proteoma e genoma viral. Apesar de já se encontrarem disponíveis inibidores para as diversas fases do processo de infecção, as potencialidades terapeuticas destas drogas não permitem a eliminação total do vírus, mesmo sob a elevada carga farmacológica dos regimes de terapia antiretroviral (TARV).
Os inibidores de entrada do VIH são uma classe de antiretrovirais direccionados para a fase primária da infecção, que guia o reconhecimento, acoplamento e entrada do vírus. Uma vez que inibem o avanço do ciclo viral antes da invasão celular, estes inibidores possuem claras vantagens perante outros antivirais que actuam em estágios tardios. Por não ser necessária a sua presença ao nível do citoplasma, possuem ainda vantagens farmacológicas que favoreceram a aposta no seu desenvolvimento para aplicação terapeutica. Devido à natureza do processo de entrada, a gama de potenciais alvos permite uma variedade de mecanismos inibitórios e espécies moleculares, o que dinamiza as estratégias de combate ao vírus e atrasam o fenómeno de resistência. As proteínas do complexo do envelope (Env), gp41 e gp120, são os mais comuns alvos de inibição. Devido à sua variabilidade conformacional, intimamente relacionada com as
III
propriedades fusogénicas, estas proteínas tornam-se vulneráveis à acção de moléculas que estabilizem umaa estrutura tridimensional específica, competindo na dinâmica de infecção. Anticorpos monoclonais neutralizantes, fragmentos de anticorpo e pequenos péptidos foram já implicados neste tipo de mecanismo inibitório. O envelope viral é também um potencial alvo de inibidores de fusão. O processo de fusão membranar é sensível a variações nas propriedades físicas da bicamada lipídica como a fluidez e a curvatura, que podem ser modificadas pela acção de pequenas moléculas. Considerando que a membrana provém da célula do hospedeiro e não está sujeita a variabilidade genética, é um alvo conservado entre estirpes que não adquire resistência a fármacos.
Apesar do interesse demonstrado na última década, suportado por ensaios clínicos promissores, a comercialização e aplicação terapeutica de inibidores de fusão em TARV encontra-se ainda atrasada relativamente a outros inibidores. Apenas um inibidor de fusão, o péptido Enfuvirtide, se encontra actualmente disponível no mercado o que ilustra a necessidade de apostar no melhoramento das propriedades farmacologicas e citotoxicidade. A aplicação de conceitos modernos de biodistribuição, biodisponibilidade aliados a sistemas de libertação controlada recentemente descritos são uma via estratégica no melhoramento da farmacoterapeutica destes inibidores. A versatilidade e diversidade dos inibidores de fusão compatibilizamnos com novas modalidades de direccionamento do fármaco.
Independentemente do alvo molecular final, foi já sugerido que a interacção inespecífica com membranas lipídicas desempenha um papel activo no modo de acção de alguns inibidores de entrada Esta interacção gera um aumento de concentração favorável aos mecanismos inibitórios junto dos locais de acção, nomeadamente as membranas viral e da célula hospedeira. De igual forma, se esta interacção for explorada para o desenvolvimento de sistemas lipídicos de transportes de fármacos, as propriedades inibitórias bem como o próprio direccionamento dos inibidores de fusão para os locais de interesse serão consideravelmente aumentados. A possibilidade de promover direccionamento e transporte através do reconhecimento de ambientes membranares poderá ser uma resposta à baixa compatibilidade terapeutica dos inibidores de fusão.
Seguindo esta premissa, o principal objectivo deste trabalho foi compreender de que forma diferentes inibidores de fusão, de pesos moleculares e propriedades químicas distintas, poderiam explorar a interacção inespecífica com membranas lipídicas de modo a melhorar o seu direccionamento para os locais de acção e as suas propriedades
IV
farmacológicas no geral. Recorremos a três inibidores de fusão: um fragmento de anticorpo, F63; um péptido, sifuvirtide; e uma pequena molécula oxidante, LJ001. Tanto o F63 como o sifuvirtide actuam sobre a proteína gp41, enquanto que o LJ001 inibe a entrada do VIH através da rigidificação do envelope viral. A nossa abordagem consiste em duas frentes com o mesmo princípio base. Por um lado, pretendemos determinar de que forma o fragmento de anticorpo F63 interage com membranas lipídicas que mimetizam a membrana viral (rica em colesterol) e da célula hospedeira (maioritariamente fosfolipídica), tentando correlacionar as observações experimentais com o seu modo de acção. Estudámos também a interacção de duas versões modificadas do F63, F63-MPR e F63-CRAC, nas quais se adicionaram motivos de reconhecimento de colesterol, com os mesmos modelos membranares. Numa outra linha de trabalho, avaliámos a compatibilidade de dois inibidores de fusão, com modo de acção e propriedades moleculares distintas, para o transporte simultâneo na membrana de um sistema de biodistribuição lipossomal catiónico. Através de composições simples com carga e fluidez variável, procurámos identificar a formulação lipídica ideal para uma interacção compatível de ambas as moléculas com a mesma membrana, idealizando um veículo/vector sinérgico não só no modo de acção consertado em diferentes alvos mas também no processo de transporte.
Recorremos a técnicas de espectroscopia de fluorescências para analizar a interacção entre os inibidores e as membranas lipídicas. Seguimos a fluorescência intrínseca dos resíduos de triptofano, assim como a fluorescência intrínseca da molécula de LJ001, para traçar o perfil de partição dos três inibidores entre a fase aquosa e lipídica. Com o mesmo intuito, usou-se uma sonda potenciométrica sensível a alterações no potencial dipolar membranar para caracterizar a interacção com modelos membranares. As técnicas de anisotropia e a extinsão de emissão de fluorescência foram também utilizadas para, respectivamente, monitorizar a fluidez membranar e a acessibilidade do fluoróforo à molécula extintora aquando a adsorção/inserção do inibidor no ambiente membranar.
O fragmento de anticorpo F63 particiona para membranas lipídicas, independentemente do seu conteúdo em colesterol, o que sugere uma interacção não selectiva com a superfície celular e viral in vivo. As versões modificadas F63-MPR e F63-CRAC não apresentaram aumento de afinidade na presença de colesterol apesar dos domínios de reconhecimento membranar. Pelo contrário, a alteração do F63 parece ter prejudicado ligeiramente a dinâmica de interacção, possivelmente por destabilização
V
da sua estrutura ou pela introdução de um local adicional de ligação à membrana. As nossas observações sugerem que modificações conformacionais poderão explicar a estabilização na superfície membranar, suportadas pela flexibilidade estrutural deste tipo de domínios proteicos.
Tanto o sifuvirtide como o LJ001 interagem com lipossomas catiónicos, exibindo preferência para bicamadas em fase ordenada ou fase fluida, respectivamente. Um aumento da carga catiónica traduz-se num aumento da partição do sifuvirtide, que apresenta carga aniónica, mas não parece afectar a partição da molécula de LJ001. Os vectores lipossomais catiónicos de fase ordenada e fluida tiveram sucesso no carregamento das duas moléculas à superfície tanto individual como simultaneamente. Apesar da necessidade de optimização, os resultados são uma prova primária da possibilidade de desenvolver lipossomas como vectores de biodistribuição duais, com modo de acção sinérgico. No entanto, as técnicas de fluorescência foram insuficientes para monitorização eficiente de ambas as moléculas no mesmo plano espacial e temporal, devido à sobreposição das suas propriedades espectrais. Será necessária uma abordagem biofísica de maior complexidade, recorrendo a técnicas capazes de resolver os perfis de interacção individuais, para um mesmo veículo lipossomal.
Palavras-chave: VIH; inibidores de fusão; membranas lipídicas; interacção; biodistribuição
VI
ABSTRACT
Since its discovery, the Human Immunodeficiency Virus (HIV) has become one of the major concerns of the scientific and medical community. More than 34 million infected people worldwide bear the risk of contracting the Acquired Immunodeficiency Syndrome (AIDS), due to limited access to therapy. The available antiretroviral drugs struggle to effectively deplete viral loads and require administration in heavy therapeutic regimens.
Fusion inhibitors are a class of antivirals capable of blocking viral progression before cell invasion. Though the initial interest was reinforced by promising clinical results, their therapeutic relevance has fallen in recent years. Still, their versatile molecular properties enable the application of novel concepts of drug targeting and delivery. Following this premise, the aim of our study was to understand how HIV fusion inhibitors of different molecular weight could exploit lipid membrane interactions to enhance their antiviral potency and pharmacological properties. Through a biophysical approach, we analyzed the interaction of the inhibitory single-domain antibody (sdAb) F63 with host and viral model membranes, as well as two modified versions containing cholesterol-targeting domains. We used the same approach to characterize the loading profiles of two fusion inhibitors, sifuvirtide and LJ001, to cationic liposomal membranes, as a primary description of a combined delivery system.
F63 has high affinity to lipid model membranes, regardless of cholesterol content, suggesting a potential interaction with both host and viral membranes in vivo. The modified versions of this sdAb failed to enhance the interaction with cholesterol-rich membranes. Sifuvirtide and LJ001 were successfully loaded to cationic membranes, supporting the concept of dual liposomal delivery.
VII
SYMBOLS AND ABBREVIATIONS
<r> Steady-state anisotropy CRAC Cholesterol recognition amino acid consensus
ΔΨ Transmembrane potential CXCR4 C-X-C chemokine receptor type 4
γL Lipid molar volume DC Dendritic cell
Ψd Membrane dipole potential DHSM Dihydrosphingomyelin
ΨS Surface potential di-8-ANEPPS 4-(2-[6-(Dioctylamino)-2- naphthalenyl]ethenyl)-1-(3-sulfopropyl)pyridinium inner salt 1 O2 Singlet oxygen
6HB Six-helix bundle DMSO Dimethyl sulfoxide
Abs Absorbance DNA Deoxyribonucleic acid
Ac Acetyl group DOTAP N-[1-(2,3- Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate
AIDS Acquire Immunodeficiency Syndrome
ART Antiretroviral therapy DOTMA 1,2-di-O-octadecenyl-3-trimethylammonium propane CA Capsid protein DPH 1,6-diphenyl-1,3,5-hexatriene
CCR5 C-C chemokine receptor type 5 DPPC 1,2-dipalmitoyl-sn-glycero-3-phosphocholine
CD Cytoplasmic domain EDPPC 1,2-dipalmitoyl-sn-glycero-3-ethylphosphocholine
CD4 Cluster of differentiation 4 Env Envelope glycoprotein complex
CD8 Cluster of differentiation 8 EPOPC
1-palmitoyl-2-oleyl-sn-glycero-3-ethylphosphocholine cDNA Complementary DNA ER Endoplasmatic reticulum
CDR Complementary determining
region fb
Accessible fluorophore fraction
Chol Cholesterol FP Fusion peptide
VIII
gp41 Glycoprotein 41 Nef HIV-1 negative factor
gp120 Glycoprotein 120 NHR N-terminal heptad repeat
gp160 Glycoprotein 160 NiV Nipah virus
HAART Highly Active Antiretroviral
Therapy NK Natural killer cell
HEPES N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid PBMC Peripheral blood mononuclear cell
HIV Human Immunodeficiency
Virus PC Phosphatidylcholine
I Fluorescence intensity PE Phosphatidylethanolamine
I0
Fluorescence intensity in the
absence of quencher PIC Pre-integration complex
IL Fluorescence intensity at
saturating lipid concentrations POPC
1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine
IW
Fluorescence intensity in the
absence of lipid PR Protease IC50
Half maximal inhhibitory
concentration PS Phosphatidylserine IgG Immunoglobulin G RAFI Rigid amphipathic fusion
inhibitors
IN Integrase Rev Regulator of gene expression
KD Binding constant RNA Ribonucleic acid
Kp Partition constant ROS Reactive oxygen species
KSV Stern-Volmer constant RT Reverse Transcriptase
LUV Large Unilamelar Vesicle RTC Reverse Transcriptase complex
MHC Major Histocompatibility
Complex sdAb Single-domain antibody
MLV Multilamelar vesicles SIV Simian Immunodeficiency Virus
MPER Membrane proximal external
region Sft Sifuvirtide
MPR Membrane proximal region
derived consensus SM Sphingomyelin NC Nucleocapsid protein ssRNA Single stranded RNA
IX SU Surface protein
Tat Trancriptional activator
TAD Trimmer association domain
TD Transmembrane domain
TM Transmembrane protein
tRNA Transfer RNA
Trp Tryptophan
Vif Viral infectivity factor
Vpr Viral protein R
X
INDEX
1.INTRODUCTION ... 1
1.1.THE HUMAN IMMUNODEFICIENCY VIRUS –AN OVERVIEW ... 1
1.1.1 HIV Pathogenesis ... 1
1.1.2 Virion Structure ... 2
1.1.3 HIV Life Cycle ... 5
1.2.FUSION,ENTRY AND THE ENV COMPLEX ... 7
1.2.1 The Membrane Fusion Step ... 7
1.2.2. gp120 and gp41 ... 9
1.3.FUSION INHIBITION AND INHIBITOR MOLECULES ... 11
1.3.1. Mode of action: Targeting and Inhibition ... 12
1.3.2. F63 – a single domain antibody ... 13
1.3.3. Sifuvirtide – a peptide ... 14
1.3.4. LJ001 – a small photoactivatable oxidant ... 15
1.4.LIPID MEMBRANES:TARGETS AND CARRIERS ... 16
1.4.1. Targeting the membrane through cholesterol recognition ... 16
1.4.2. Liposomal delivery of two fusion inhibitors ... 17
2. THEORETICAL BACKGROUND ... 19
2.1. PHASE PARTITION ... 19
2.2. MEMBRANE ELECTROSTATIC POTENTIALS ... 21
3. MATERIALS AND METHODS ... 24
3.1. REAGENTS AND CHEMICALS ... 24
3.2. INSTRUMENTATION ... 24
3.3. SAMPLE PREPARATION ... 25
3.4. PARTITION TO MEMBRANES ... 26
3.5. MEMBRANE DIPOLE POTENTIAL SENSING ... 26
3.6. ACRYLAMIDE FLUORESCENCE QUENCHING ... 27
3.7. MEMBRANE FLUIDITY ASSAY ... 28
4. RESULTS AND DISCUSSION ... 29
4.1.SINGLE-DOMAIN ANTIBODIES F63,F63-MPR AND F63-CRAC INTERACT WITH HOST AND VIRAL MODEL MEMBRANES ... 29
4.1.1 Interaction of F63, F63-MPR and F63-CRAC with model membranes ... 29
4.1.2 sdAb induced membrane dipole potential perturbation ... 31
4.1.3 Fluorescence quenching of F63, F63-MPR and F63-CRAC in the presence of model membranes ... 32
4.2.SIFUVIRTIDE AND LJ001 SHARE COMPATIBLE LOADING PROPERTIES TO CATIONIC LIPID MEMBRANES ... 35
4.2.1 Sifuvirtide and LJ001 membrane interaction ... 37
4.2.2 LJ001 membrane ordering effect ... 39
4.2.3 Combined partition to liposomes ... 40
5. CONCLUSIONS AND PERSPECTIVES ... 45
1
INTRODUCTION
1.1. The Human Immunodeficiency Virus – An Overview
Thirty years ago, the isolation of a human T lymphotropic virus1, later named Human Immunodeficiency Virus (HIV), marked a crucial discovery in the race to identify the Acquired Immunodeficiency Syndrome (AIDS)-responsible pathogen. The finding stood on the shoulders of an extensive effort from the scientific community2, setting the pace for the following years of research. From the first reported cases of AIDS3,4 we have walked a long path towards the understanding and efficient treatment of this unsettling disease affecting more than 34 million people in all continents5. Still, a cure remains utopian due to the characteristics of HIV infection, absence of highly conserved targets and viral adaptation to available treatment strategies such as Highly Active Antiretroviral Therapy (HAART), which will be further discussed in this introduction.
1.1.1 HIV Pathogenesis
HIV is the result of an evolutionary adaptation of the Simian Immunodeficiency Virus (SIV)6,7, a primate-targeting lentivirus, but unlike its simian directed species it is able to infect cells from the human immune system, more specifically CD4 expressing T cells, macrophages and dendritic cells (DC)8. It does so by recognition of specific receptors and co-receptors on the surface of these cells, which enables viral content entry to the cytosol. The interplay between CD49,10 and chemokine receptors CCR511,12 and CXCR4 drives stable cell surface docking, fundamental for the viral entry in HIV’s viral life cycle (which we will further discuss in section 1.1.3). Co-receptor preference by different strains of HIV-1, the most pathogenic subtype, guides a differentiated cell subset invasion in the various phases of host infection13. While CCR5 binds the macrophage-tropic viral strains (R5), CXCR4 binds the T cell-tropic strain (X4). Consequently, R5 viruses mediate mucosal and intravenous transmission14 whereas X4 viruses are responsible for late stages of infection and viral set-point15.
Independently of the cell type, one of the major problems associated with HIV pathogenesis is the establishment of latent reservoirs upon infection, even in G0 resting
1
2
cells16. These pools of non-replicative HIV present a barrier to viral eradication in patients under heavy HAART treatment17, capable of leading to undetectable blood viraemia. Mechanisms for generation of HIV latency comprise transcriptional interference, epigenetic silencing, inefficient elongation of HIV transcripts, unproductive splicing and host innate antiviral defenses18. Although cells are incapable of expressing stable viral particles in these specific conditions, the reservoirs can be elicited through host immune cell activation, external factors and pharmacological approaches19. These drug based approaches appear as a possible strategy to increase antiretroviral efficiency of existing therapies20. Even if hard to achieve, purging latent reservoirs21,22 combined with an aggressive antiretroviral treatment (ART) should increase total viral eradication through reduction of the overall latent content.
Latency may be responsible for the maintenance of host infection levels but how does HIV achieve the advanced state of human impaired immunity known as AIDS? AIDS is characterized by a progressive loss of immune competence, evidenced by the rapid turnover of CD4+ T cells, which ultimately leads to opportunistic infections. Indeed the low CD4 expressing T cell count has been regarded as the hallmark of HIV infection leading to AIDS23. These cytopathic effects on specific cells work in combination with immune system dysfunction by aberrant activation and depletion of CD8+ T cells, B cells, natural killer (NK) cells and other nonlymphoid cells24. Soluble factors like pro-inflammatory cytokines (TNF-α, IL-1, IL-12, IL-6) and chemokines (CXCL10) are also present following HIV host invasion, as one of the first lines of innate immunity. HIV infection (and consequently AIDS) proceeds as the interplay between abnormal immune system activation, rapid T cell turnover with an acquired long term vulnerability to opportunistic pathogens, maintaining latent viral reservoirs.
1.1.2 Virion Structure
As a member of the lentivirus genus, HIV shares some common characteristics with other retroviruses such as SIV, visna virus and Jembrana disease virus. It possesses a complex genome, exhibits a cone shaped capsid and is enveloped in a host derived lipid bilayer25. Therefore, virion structure can be separated into its genetic, protein and lipid components. Due to its relative significance for the present thesis, HIV RNA will not be discussed in comparable detail (see references 26 and 27 for comprehensive reviews on the subject).
3
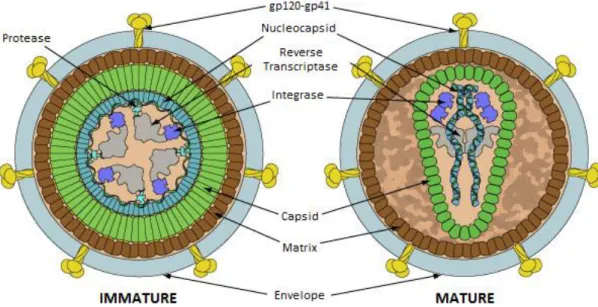
Structural, accessory and envelope proteins are translated within the host cell environment and, for the most part, incorporate the budding virion. The 55 kDa Gag polyprotein is cleaved in the maturation process (Figure 1, left to right) to the matrix, capsid and nucleocapsid proteins, responsible for most of HIV’s morphology. Its membrane interaction properties are capable of inducing viral particle formation independently of other viral protein components29. Matrix protein p17 (MA) assembles into trimmers that interact directly with acidic lipid membranes30 through myristoylated amino acid residues31. This property is critical for efficient Gag targeting to the plasma membrane rather than organelle membranes32. After maturation (Figure 1, right), capsid proteins p24 (CA)33 assemble the multimeric cone-shaped capsid34 encasing ssRNA, nucleocapsid, accessory proteins and remaining enzymes. As expected, CA protein rearrangement can only be observed after cleavage of the MA-CA bond in Gag35. The last of the structural proteins, nucleocapsid p7 (NC) acts both in recognition and packaging of the viral genome36, which includes tRNALys. A zinc finger-like structure is
responsible for genome stabilization37 and further upon maturation/infection the NC interaction primes reverse transcription of ssRNA. Thus, structural proteins are remarkably vital for virion infectivity maintaining a supporting functional activity.
Figure 1 – HIV Viral Particle Structure. Each virion is a structured assembly of ssRNA and viral proteins enveloped by a lipid bilayer. The matrix, capsid and nucleocapsid are structural proteins present in both mature and immature virions. Reverse transcriptase, integrase and protease (driving force of maturation) compose the viral enzymes. Accessory proteins (not represented) can be found within viral particles (Vif, Vpu and Vpr) or inside infected cells (Rev, Nef, Tat). The envelope contains high cholesterol content and displays the Env glycoprotein complex, responsible for viral fusion. Adapted from reference 28.
4
Each individual HIV virus requires protease, reverse transcriptase and integrase activity to be fully infectious. This is achieved through viral genome encoded enzymes. Protease (PR) is a part of the Gag-Pol complex, cleaved by an autocatalytic mechanism in immature viral particles (Figure 1, left). It is functionally active as a homodimer38 responsible for the cleavage of viral precursor proteins into 9 different catalytic products, required for maturation32. Reverse transcriptase (RT) is active towards ssRNA in the cytosol of infected cells. Cleavage of Gag-Pol by PR ensues two p66 molecules containing both a polymerase and RNase H domains. In full matured viral particles, it is present as a p66-p55 RT heterodimer39 (p55 results from proteolysis of a RNase H domain from one of the p66 molecules). Finally, HIV integrase (IN) is the fundamental player in HIV genome incorporation to target cell chromosomes. This enzyme comprises three structural/functional domains: a zinc binding domain, implicated in oligomerization, the central catalytic core, coordinating two Mg2+ ions, and a C-terminal DNA binding domain40. The assembled complex exerts both a 3’ processing and DNA strand transfer. These enzymes have highly optimized catalytic activities and retain their functional properties inside each virus.
Accessory proteins Nef, Rev, Tat, Vif, Vpu and Vpr are key regulatory factors during the HIV infectious cycle. The first three are expressed shortly after infection and are not recruited during assembly. Nef p24 stimulates CD4 and MHC I downregulation and enhances virion infectivity41. Rev p19 inhibits splicing events and enables nuclear transport of incompletely spliced and unspliced transcripts to the cytoplasm42. Tat p14 enhances viral transcription that terminates prematurely due to abortive elongation43. Vif p23 is an antagonist of human APOBEC3G, a human restriction factor that inhibits RT activityand Vpu ensures gp120 export to the plasma membrane44. Vpr p15 induces G2 cell cycle arrest and leads the PIC to the nucleus45. Although more small proteins are
coded in the viral genome, related functions are not completely understood and their overall importance for infection can be overlooked for the range of this introduction.
Finally, the envelope holds the Env complex, a protein of extreme relevance for the present work. Section 1.2 will cover the subject in more detail, describing the related glycoproteins and their function in the viral entry.
All of the discussed viral content evades the host cell through co-localized membrane budding and particle release. The resulting lipid envelope (100-140 nm) is, much like the viral structural proteins, a functional as well as morphological component of the HIV virion. Although derived from the host membrane, it presents a rather
5
particular and characteristic composition. Independent studies more than a decade apart achieved similar results46,47, enlightening the major differences between host and HIV membranes. From an average 296 000 lipid molecules/virion, more than 40% is cholesterol (Chol)47. This is approximately two fold higher than what is observed in host cell membranes. Concerning the phospholipid composition, the virion membrane presents a decrease in PC and PE, and an increase in SM (includingDHSM) and PS, when compared to host cell membranes. From what is known of lipid rafts, both in model48,49 and in biological membranes50, envelope content points towards the existence of these domains in both host and viral membranes. Despite the controversy surrounding the subject51, HIV Chol domains have been observed and identified as crucial for infectivity and assembly52. Especially in the latter, if Env complex proteins do not co-localize with required soluble factors and enzymes at raft domains, nascent viral particles become non-pathogenic. Other required parameters such as envelope fluidity and charge are also fine-tuned by the lipid composition. Fluidity is a natural enhancer of fusion at biological temperatures and deeply hinders viral competence if negatively modulated. In section 1.3.1 we will evoke these concepts supporting the mode of action of some HIV inhibitor molecules.
Every component in HIV’s structure has keenly evolved to ensure optimized infectivity and consequent host pathogenesis. Having a collective understanding of viral components and structure has and will further increase the available therapeutic options as well as their effectiveness.
1.1.3 HIV Life Cycle
Lentiviral life cycles, such as HIV’s, are cell specific multistep pathways composed of vital checkpoints between invasion and evasion of newly assembled virions. We will mainly focus on the cellular stages of the HIV infectious cycle, not neglecting that invasion and transmission are also critical points for viral dissemination (see references 53, 54 and 55 for comprehensive reviews on these subjects).
The life cycle of any virus must begin with viral entry. Even though HIV can be endocytosed56, actual invasion only occurs after viral and host cell membrane fusion followed by viral content release to the cytoplasm. This is possible through recognition of CD49,10 receptors on the cell membrane surface and stable docking to one of two co-receptors, CCR5 and CXCR457, as already stated in section 1.1.1 (Figure 2, step 1).
6
Entry is driven by a concerted conformational change in both gp120 and gp41 viral membrane proteins that compose the Env complex (Figure 2, step 2). A more detailed description of the entry process is found in section 1.2.
Viral CA uncoating occurs simultaneously with reverse transcription of RNA into cDNA (Figure 2, steps 3 and 4). Seemingly, viral capsid disassembly accompanies the reverse transciptase complex (RTC) conversion into the pre-integration complex (PIC) in the cytosol, where actin filaments drive complex motility58. The PIC, containing only cDNA, is then directed to the nuclear envelope through these cytoskeletal structures, followed by nuclear import (Figure 2, step 5). Entering the nucleus is a unique characteristic of lentiviral infection, compared to other retrovirus, as it enables integration and replication in metabolically active non-dividing cells, independently of mitosis59. From matrix proteins60 to viral tRNA61, the list of implicated molecules in the nuclear transport process is vast.
Integration is the last step in the early phase of HIV cell infection (Figure 2, step 6). After entering the nucleus through PIC, integrase leads proviral insertion on distinct chromosomal locations, influenced by epigenetic factors such as chromatin accessibility Figure 2 – The HIV replication cycle. HIV recognition through CD4 binding and docking to co-receptors (step 1) induces the viral and host membrane fusion step (step 2). After uncoating (step 3), reverse transcription of viral RNA (step 4) precedes PIC nuclear import (step 5). Integration (step 6) is the final stage of the early phase. Late phase cell infection begins with viral mRNA transcription (step 7), nuclear export (step 8) and translation of viral proteins (step 9). Assembly, budding, release and maturation (steps 10, 11, 12, 13) of infectious viral particles close the cycle. Each one of these steps can be countered by specific antiviral molecules, normally combined in ART. Adapted from reference 32.
7
and nucleosome remodeling13. Dependent on these integration sites, gene expression may lead to higher or lower viral transcription rates (Figure 2, step 7), also regulated by stochastic fluctuations63. These transcripts can range from highly spliced (Tat, Rev and Nef) to mildly spliced/unspliced (Gag, Gag-Pol and Env), dependent on the relative availability of said proteins13,25. Respectively, transcripts can be either easily exported or led by regulatory viral protein Rev25 (Figure 2, step 8). Translation (Figure 2, step 9) yields all the necessary viral proteins, namely the cleavable Gag-Pol (11 proteins) and Env (2 proteins) complexes.
In the final steps of the late phase, expressed proteins assemble near the membrane. Env protein complex co-localizes in raft domains63 while the Gag subunits (to a total of 1200-2000 copies) begin to assemble a multimer based budding structure64 that encapsulates two copies of viral unspliced RNA65 (Figure 2, steps 10 and 11). Finally, PR cleaves the poliproteins to individual enzymes in immature virions. Their internal rearrangement through the maturation process forms infectious HIV viral particles (Figure 1 and Figure 2, step 13). These are fully infectious and capable of inducing new viral cycles.
1.2. Viral Entry and the Env Complex
Enveloped viruses (flavivirus, herpesvirus, hepatitis D, among others) find in their membrane bilayers the key for infectivity. Frequently, virion endocytosis precedes a pH dependent fusion step that occurs upon endosome acidification. For HIV, external pH is not a limiting factor enabling target cell infection at the plasma membrane level66. Surface fusion glycoproteins (classes I-III), although structurally distinct amongst these viruses, share the common purpose of bringing the two membranes closer together, terminally leading to hemi and full fusion67. After breaking the positive to negative curvature barrier, the envelope becomes part of the plasma membrane, releasing its content. In this section we will focus on the HIV entry, describing the structural and functional properties of the Env complex.
1.2.1 The HIV Entry Process
From all the stages in HIV’s infectious cycle, only two occur outside the host cellular environment, maturation and entry. Entry is a mechanically based event
8
dependent on host recognition, docking and membrane fusion. The lead role is played by the Env complex, a glycan shielded assembly of gp120 (SU) and gp41 (TM) homotrimers68. Both exhibit a high structural flexibility required for the trademark conformational triggers of HIV internalization. Additionally, envelope fluidity and membrane raft domains also modulate the successful progression of viral entry.
As already described in sections 1.1.1 and 1.1.3, the first events are dependent both on specific and unspecific interactions between the viral particle and the target cell. After the initial stochastic approach, followed by receptor-independent interactions (Figure 3, A), gp120 binds the CD4 transmembrane receptor (Figure 3, B). The induced conformational changes expose the co-receptor binding site towards CCR5 and CXCR4. HIV does not require further intracellular signaling pathways for successful invasion to occur69. The concerted double receptor interaction leads to gp41 fusion peptide insertion into the host membrane (Figure 3, C). This pre-fusogenic conformation prompts an interhelical rearrangement of the N and C-heptad repeats (NHR and CHR), turning gp41 trimers into a fusogenic six-helix bundle (6HB) (Figure 3, D)70,71,72. The entropically unfavorable membrane curvature shift meets its energy requirements through gp41 structural reorientation.
At physiological conditions, entry kinetics show an initial lag phase of 10-15 minutes attributed to CD4 binding. The stochastic diffusion of viral particles may be in the basis of the observed rate limiting reaction. Subsequently a relatively quick rise in fusion saturates after 50-100 minutes. This sigmoidal behavior is explained by the fusion cascade after co-receptor binding72. CD4 and Env membrane densities correlate with fusion saturation levels while co-receptor availability enhances the slope of the rapid phase73.
Figure 3 – Env mediated viral entry. (A) HIV and host lipid membranes initiate unspecific interactions. (B) gp120 binds CD4 receptors in immune cells leading to consequent conformational changes in the complex. (C) Co-receptor docking allows gp41 insertion in the plasma membrane. (D) The three gp41proteins form a six-helix bundle through NHR and CHR interaction, bringing both membranes close enough for lipid mixing and fusion to occur. Adapted from reference 70.
9
The required number of Env proteins to induce one single fusion event is unlikely condensed to a relative small number, like depicted in Figure 3. The correlating numbers of receptors/receptors for pore formation depend on a surface co-localization that could not be met by simple two-dimensional diffusion. Lipid clusters may be able to explain how receptors co-localize in the host cell74. Chol and SM raft domains have been showed to enable pathogen invasion in mammalian cells, and have also been implicated in HIV assembly and budding, as described in section 1.1.2. Microdomains that assemble for signal transduction pathways are enriched in CD475 but to a less extent in the respective co-receptors76. Thus, these CD4 containing rafts are fit for fusion priming (lag phase) through Env recognition, prior to docking and 6HB formation (rapid phase). Along with the high mutational variability of HIV glycoproteins, the possibility of targeting the host and viral lipid environment in ART is an attractive possibility. Section 1.3 will cover some principles of fusion inhibition and inhibitor molecules targeting both proteins and membranes.
1.2.2. gp120 and gp41
In the previous section we discussed the entry mechanism of the HIV virus but gave only minor detail on the viral glycoproteins. Both gp120 and gp41 are coded as part of the precursor gp160, further cleaved to the respective subunits in the trans-Golgi network. At the same time, more than 25 putative N-glycosylation sites, accounting for 47% of the total molecular weight, suffer post-translational modifications leading to a mannose-rich glycan complex. From there (Golgi), the non-covalent heterodimer of trimmers formed by the outer gp120 and the occluded gp41 is directed towards the membrane. Fusogenic properties of the meta-stable Env complex are dependent on the presence of both glycoproteins.
The structure of Env is debated even today77. Due to heavy glycosylation, unusual meta-stability and conformational plasticity, it has been extremely difficult to obtain a high resolution structural map. X-Ray crystallography can only be applied to modified or bound variants78 and other methods have proven unfit or unreliable for structural determination. Still, studies have clearly reached a considerable level of detail, more than enough to draw conclusions on the arrangement of the subunits and domains.
10
Each monomer of gp120 exhibits 5 variable segments (V1-V5) that interlude constant portions of the protein sequence (Figure 4). The three-dimensional display of V3, V4 and V5 loops cover the exposed outer domain of gp120 whereas the contiguous V1/V2 loop emanates from the inner domain to form the trimmer association domain (TAD)78,79. The TAD domain has yet to be resolved. Sequential binding to CD4 and CCR5/CXCR4 is a structural as well as functional event. Co-receptor sites are only available after primary receptor interaction with the inner-outer domain interface, a site bare of carbohydrates and matching in electrostatics80. Consequently, the inner domain redirects the V3 loop towards the host cell surface, revealing the secondary binding site. Further changes in gp120 release the meta-stable gp41 trimer from the Env core, oriented towards the cell membrane.
gp41 is the most conserved subunit of Env. It displays a cytoplasmatic (intra-viral) C-terminal domain (CD), a hydrophobic transmembrane portion (TD) adjacent to a membrane proximal external region (MPER) that targets Chol, two helical heptad repeats (NHR and CHR, closer to the N or C-terminus, respectively) connected by an unstructured loop, and finally an N-terminal fusion peptide (FP)81,82 (Figure 4). Like gp120, it associates in a trimeric fashion, anchoring the complex to the viral membrane, and also exhibits fusion related conformations83. The pre-fusogenic conformation occurs immediately after co-receptor docking. The increasing exposure of the hydrophobic fusion peptide to the aqueous environment works as the driving force for its insertion in the host lipid membrane (Figure 3, C)84,85. At this point, gp41 is a linear protein connecting the virion and the target cell. As the fusion peptide is stabilized, the Figure 4 – gp160 polypeptide chain and secondary structure. The glycoprotein 160 is translated as a single chain later cleaved to gp120 and gp41. The heavy glycosylated gp120 contains five variable regions (V1-V5) that exhibit little to no structure and confer three-dimensional flexibility to Env. gp41 anchors the complex to the viral membrane through TD and displays two helical regions (NHR and CHR) capable of interacting with each other for 6HB formation. The presented secondary structure graphical scheme and glycosylation sites are based on the UniProt entry P04578.
11
ectodomain reorients by antiparallel interaction of the two heptad α-helixes in each monomer. The resulting 6HB, coupled with cell FP harpooning, enables pore formation85,86. At the end of the fusion step, both the FP and the TD are localized within the same membrane and gp41 exhibits a rod-like shape87.
The fact that gp41 is the most conserved of the glycoproteins, exhibits a conformational dependent mechanism and is an extracellular protein close to the membrane surface makes it a very attractive target for viral fusion inhibition88. The relevance of this protein will be evoked as a main target of fusion inhibitor molecules, such as the ones described in the next section.
1.3. Fusion Inhibition and Inhibitor Molecules
Throughout this introduction we have hinted towards the potential of both viral proteins and envelope as targets for ART. In Figure 2, each step of the HIV life cycle shows the respective available inhibitors. It is not surprising that HAART is based on a cocktail of these molecules, combining the synergistic effects of infection blockade at different stages. But only through the understanding and optimization of each step’s inhibitory potential can we create bottlenecks of viral strain and reduce the drug load bared by HIV infected patients.
Our work is focused on the specific subgroup of antiviral fusion inhibitors89. These can range from antibodies, peptides and small molecules, all targeting one of two basic elements, the surface glycoproteins or the lipid envelope itself. Modes of action may vary but the outcome is identical, the virus is unable to form a fusion pore. By acting on an extracellular step of the cycle, HIV never actually infects cells and the likelihood of increasing latent reservoirs is reduced90. At the same time, because the virus is not destroyed in the process, it does not present a cure.
For the sake of simplification and relevance, gp120-CD4 attachment inhibitors and chemokine antagonists will not be addressed in detail in this introduction (see Reference 91 for an extremely detailed review on these molecules). We shall focus on fusion inhibitors that target the late phase of phase entry, ie viral fusion (Figure 3, D), describing their relative modes of action and preferred targets. A description of new approaches and strategies will contextualize the rational of present thesis.
12
1.3.1. Mode of action: Targeting and Inhibition
The targeting properties, size and overall design modulate how fusion inhibitors block virus-cell pore formation. While the most common targets are mainly gp41 and the viral envelope, the underlying antiretroviral mechanisms are diverse.
Most gp41-targeted inhibitors will act by blocking the fusogenic conformational changes that lead to 6HB formation. If the trimmer does not reorient, the viral and host cell membranes will remain apart, preventing lipid mixing, hemifusion and fusion. This effect can be reached through different types of interactions dependent on the molecular weight of the inhibitor. Let’s consider the case of the 2F5/4E10 broad neutralizing antibodies (Figure 5, A). They are human immunoglobulins (IgG) (aprox. 150 kDa) that target a concealed epitope in gp41’s MPER92, exposed in a fusion intermediate state93, inhibiting the progression of fusion through binding. The size of these molecules alone causes an extreme spatial restriction for any conformational change to happen. On a relatively lower molecular weight (<10 kDa), inhibitor peptides were some of the first gp41 targeting molecules94. Their elegant design mimics the natural interaction of the heptad repeats during the 6HB formation95. A peptide derived from the CHR coil, capable of interacting with the complementary NHR, blocks the conformational shift by competitive binding to the heptad motif (Figure 5, A), and vice-versa. Enfuvirtide96 and sifuvirtide97, respectively first and second generation CHR based inhibitor peptides, are prime examples of the potentials of the peptide mode of action.
Figure 5 – Glycoprotein and membrane-targeted fusion inhibition. gp41 targeting inhibitors (A) block conformational changes leading to 6HB formation. While 2F5/4E10 mAbs target the MPER, peptides like sifuvirtide competitively bind to the NHR domain. Membrane curvature (B) as well as fluidity (C) can both inhibit membrane at certain thresholds. Curvature is increased through insertion of RAFI molecules in the lipid environment while fluidity can be influenced by oxidants such as LJ001.
13
Unlike the HIV glycoproteins, the viral envelope does not suffer from the same mutational variability and is broadly conserved across different viral strains. This has led to an increasing interest in developing membrane targeted, lipophilic molecules capable of virion inactivation. There are supposedly two ways of inhibiting fusion through the membrane: by increasing the envelope curvature to a point where not even the gp41 fusogenic core can break the required energy barrier of fusion98 and by reducing the membrane lipid fluidity, “stiffening” the virus99. Rigid amphipathic fusion inhibitors (RAFI) act by inserting within the lipid environment and imposing a positive curvature increase100 (Figure 5, B). This is possible because of the unbalanced size ratio of their head and tail moieties. A larger polar head will induce lipid reorganization to occupy the free space, inducing the inhibitory effect. LJ001, on the other hand, is capable of modifying lipid tails in viral lipids changing their phase transition properties at physiological temperature101 (Figure 5, C). When fluidity is reduced to a certain threshold, the virus is inactivated.
Although these molecules seem quite capable of reducing the infectivity of HIV to extremely low levels, only one fusion inhibitor, Enfuvirtide, is commercially available. Most of this is due to the toxic effects of some of these inhibitors aided by the slow progression of clinical trials. In the meantime, it is up to the scientific community to devise novel strategies as arguments for pharmaceutical investment. Using three distinct fusion inhibitors, our study tried to revamp the available approaches using both a targeting and a combined delivery approach. A relatively brief description of each inhibitor is present in the next sections.
1.3.2. F63 – a single domain antibody
A novel group of single domain antibody (sdAb) fusion inhibitors, sharing the mode of action of peptides such as Enfuvirtide, have been recently studied against HIV isolates, with promising results (Ana C. C. Santos, unpublished results). sdAb consist in immunoglobulin variable (heavy or light chain) domains (aprox. 10 kDa), stable as an independent protein, that retain the binding specificity of IgG through the complementary determining region (CDR) loops102. Applying genetic engineering and phage display techniques, synthetic sdAb can be targeted towards a desired molecule or motif with therapeutic relevance103. Gp41 targeting implies the same principles of
14
peptide based inhibition but may bring the advantages of much higher affinity (low KD)
and increased spatial restrain.
F63 is one of these novel sdAbs (Figure 6). It is derived from the variable light chain of a camelid IgG, genetically modified to target the NHR coil of gp41. The structural flexibility of the variable domain allows an increased complementation to the target coil in a pre-fusogenic conformation. Unpublished results show a low half maximal inhibitory concentration (IC50), comparable to that of Enfuvirtide, making it an
interesting alternative to the current therapeutics.
1.3.3. Sifuvirtide – a peptide
This 36 amino acid residues anionic peptide (Figure 7) was designed as a solution to the drug resistance induced by the first generation peptide, Enfuvirtide104. Similarly to its predecessor, sifuvirtide is based on the HIV gp41 CHR region, complementary to the NHR region in pre-fusion conformation, where it exerts its inhibitory potential. However, it differs in a total of 22 amino acid residues. These modifications achieved increased stability, pharmacokinetics and antiviral potency105. Indeed the IC50 was reduced up to 20-fold in certain HIV isolates and maintained sub-
sdAb F63 E L V L T Q T P S S V P A A V G G T V T I N C S G G G S D S T D Y C K G Y A N Y S G G G S W Y Q Q K P G Q R P K L L I Y G A S D L A S G V S S R F K G S G S G T Q F T L T I S G V Q C A D A A T Y Y C S G G G S S T Y A Y A Y S S G A Y S G G G S F A F G G G T E L E I L S S G G G T S G Q A G Q H H H H H H G A Y P Y D V P D Y A S sdAb F63-MPR E L V L T Q T P S S V P A A V G G T V T I N C S G G G S D S T D Y C K G Y A N Y S G G G S W Y Q Q K P G Q R P K L L I Y G A S D L A S G V S S R F K G S G S G T Q F T L T I S G V Q C A D A A T Y Y C S G G G S S T Y A Y A Y S S G A Y S G G G S F A F G G G T E L E I L S G G L W Y I K G G G Q A G Q H H H H H H G A Y P Y D V P D Y A S sdAb F63-CRAC E L V L T Q T P S S V P A A V G G T V T I N C S G G G S D S T D Y C K G Y A N Y S G G G S W Y Q Q K P G Q R P K L L I Y G A S D L A S G V S S R F K G S G S G T Q F T L T I S G V Q C A D A A T Y Y C S G G G S S T Y A Y A Y S S G A Y S G G G S F A F G G G T E L E I L S G G V L N Y Y V W R G G G Q A G Q H H H H H H G A Y P Y D V P D Y A S
Figure 6 – Amino acid sequence of the F63, F63-MPR and F63-CRAC sdAb. The sdAb sequence (grey fill) contains three CDR loops: CDR1 (red), CDR2 (yellow) and CDR3 (green). The C-terminal region, added through the expression plasmid, contains an histidine tail (blue fill) and an antibody-binding region (yellow fill) for western-blot staining. F63-MPR and F63-CRAC have an additional cholesterol recognition consensus (blue) before the histidine tail.
15
micro molar values in Enfuvirtide resistant strains. Combination of Enfuvirtide and sifuvirtide resulted in a synergistic therapeutic efficacy106.
The membrane interacting properties have shown to play an important role in sifuvirtide inhibitory potential. Unlike Enfuvirtide, it does not exhibit the promiscuous interaction with different types of lipids107, screening gel phase and liquid ordered domains108. As already stated, the viral envelope contains a great amount of Chol and so do the plasma membrane microdomains implicated in receptor clustering. By partitioning to these lipid domains109, the concentration near target sites should increase the potential of inhibitory interactions, in what is described as a “membrane catalysis” model110. These interactions have also been described in peripheral blood mononuclear cell (PBMC) lipids which may work as scaffold for interaction with circulating HIV111. We wish to take advantage of these lipid interactions to improve the biodistribution and targeting of this peptide towards viral particles.
1.3.4. LJ001 – a small photoactivatable oxidant
LJ001 comes from a line of new broad spectrum antiviral against enveloped virus. It consists in a 302 Da aryl methyldiene rhodanine derivative (Figure 8), found during a high throughput screening of Nipah virus (NiV) entry inhibitors112. At first glance, the inhibitor activity shows a membrane dependent mechanism which correlates with its activity towards a large list of enveloped virus (HIV, West Nile Virus, Hepatitis C virus, among others) but not towards non enveloped ones. Viral infection is blocked before entry in an irreversible manner and at the same time cell toxicity is low despite the observed interaction with the molecule112. The last two observations hinted towards
Figure 7 – Structure and amino acid sequence of Sifuvirtide. Side (A) and top (B) views of the peptide sifuvirtide in its inhibitory conformation. In both the structure (A and B) and the sequence (C) the hydrophobic amino acid residues are colored in blue, the non charged polar are colored in green and the charged polar are colored in red. The PDB entry for sifuvirtide is 3VIE. Adapted from reference 109.
16
Figure 8 – Chemical structure of LJ001. The sulfinyl group (S=C) seems to play a critical role in lipid damaging. Taken from reference 112, supplemental information.
the inhibitory mechanism of LJ001 based on irreversible lipid damaging. This would explain why cells were not affected, seeing that lipids could be replaced in the plasma membrane but not in viruses.
In fact, it was later proven that LJ001 is capable of generating reactive oxygen species (ROS) implicated in lipid peroxidation, by photoactivation of the compound101. Singlet oxygen (1O2) targets unsaturated carbon bonds in the fatty acid chains,
stabilizing them and reducing the amplitude of movement. As lipid carbon chains become less flexible, membrane packing increases and fluidity decreases (Figure 5, C). This represents a completely new antiviral mechanism that not only affects enveloped viruses as a hole but does so in an extremely potent and irreversible fashion.
The effects of LJ001 on the lipid membrane synergize with sifuvirtide’s rigid domain selectivity. By combining the two inhibitors, coupled interaction and dual targeting would increase their global antiviral activity.
1.4. Lipid Membranes: Targets and Carriers
Membranes have an undisputed role in the biology and biophysics of HIV. Enveloped viruses depend on it to scaffold the fusion proteins and to contain the infectious genome/proteome. At the same time, it is the main barrier of host cells towards the highly adapted mechanisms of viral entry. Inevitably, the knowledge on its physical and chemical properties makes way for new strategies taking advantage of lipid structures, natural or synthetic, to improve viral targeting and delivery of inhibitor molecules. To conclude this introduction we will describe the two approaches used to exploit the potential of membranes and membrane interactions for HIV fusion inhibitor therapeutics.
1.4.1. Targeting the membrane through cholesterol recognition
Any free diffusing molecule in circulation is bound to interact with supramolecular membrane structures. These interactions can range from simple Brownian collisions to adsorption or insertion in the lipid environment. In some
17
particular cases, molecules benefit from the partition between the two phases, moreover if they interact with membrane bound proteins such as receptors or transporters113. The concept of membrane-assisted diffusion as a means to facilitate receptor-ligand interactions has been discussed for the past century. In short, any molecule will have more probability of encountering its respective membrane target if it first interacts with the lipid surface and then diffuses in a two-dimensional plane114.
HIV fusion inhibitors that require interaction with the membrane bound gp41 in its pre-fusogenic conformation have a short time window to effectively bind to its target. It would be extremely difficult to achieve the observed low inhibitory concentrations if these molecules had to diffuse in the blood stream to reach the intended binding site. Unsurprisingly, many gp41 fusion inhibitors107,108 show interaction with lipid membranes which further supports the “membrane catalysis” scenario.
Our hypothesis considers that by introducing a Chol recognition amino acid consensus (CRAC) in the structure of a novel fusion inhibitor sdAb, F63, we would increase its interaction with HIV particles that have high Chol content, thus increasing its antiviral potency. We adopted a biophysical fluorescence spectroscopy approach to test the interaction of F63 and two derivatives, F63-MPR and F63-CRAC, with host (POPC) and viral (POPC:Chol 2:1) model membranes. MPR (LWYIK) and CRAC (VLNYYVWR) are two cholesterol recognition sequences, respectively from the gp41 MPER115 and the mouse peripheral-type benzodiazepine receptor116.
1.4.2. Liposomal delivery of two fusion inhibitors
Since the discovery of liposomes117, the therapeutic interest in lipid nanostructures opened the field of drug delivery to novel biomaterials increasingly adapted for improved biodistribution, targeting and availability of specific drugs118. These adaptations are partly due to the versatility of synthetic lipids, modified to the researcher’s desire. Charge modification, pegylation and other molecular attachments are some of the possibilities.
Their broad applicability turns liposomes into potential carriers for antiretroviral drugs against HIV. Up to sixteen different liposomal based delivery concepts have been designed as vaccines, gels, and delivery vectors119. Yet, very few directly target the HIV virion. In 2011, cationic liposomes were mentioned as potential carriers for adsorbed
18
sifuvirtide120. The charged vesicles would directly target the viral particle, leading the inhibitor to the envelope surface, near its active site.
We wish to improve this conceptual design by combining two HIV fusion inhibitors, sifuvirtide and LJ001, that display a compatible mode of action and synergistic loading properties. By reducing the fluidity of the membrane, LJ001 should increase the interaction of sifuvirtide towards the liposomal vector. Using a fluorescence spectroscopy approach we monitored the loading of both molecules to the membrane of different fluid and gel phase cationic liposomes.
19
Figure 9 – Partition of a molecule (M) between an aqueous and a lipid phase. The simplified scheme illustrates the partition equilibrium of a molecule moving from an aqueous (MW) to a lipid (ML) environment. The subset ML can be
composed of molecules adsorbed, inserted and even directly bound to the lipid molecules. The partition constant (Kp) describes the extent of partition through the ratio of the two concentrations.
THEORETICAL BACKGROUND
Throughout the following sections we will resort to biophysical concepts to describe the interaction between molecules and lipid membrane structures. As most experimental approaches will rely on such concepts to link data and conclusions, they will be defined here as the theoretical background for the remaining work.
2.1. Phase Partition
Partition can be defined as the concentration distribution of a single molecular solute between two immiscible phases, at equilibrium. The two considered phases are normally water and a hydrophobic medium (e.g. octanol) that remain separated and retain molecules differently based on their chemical properties. It can be a measure of relative solubility as well as a measure of more complex interactions if the phases are as complex as biological membranes.
In a lipid membrane, partition is the result of electrostatic accumulation/adsorption or effective binding/insertion of the considered molecules. Both events may guide a concentration shift towards the lipid environment, ie an increase of the mole-fraction ratio of lipid-interacting to water-diffusing molecules (Figure 9). This ratio is the partition constant, given by121,122:
p,x ,L L ,L , , (1)
2
20
where nW and nL are the moles of water and lipid, respectively, and nS,i are the moles of
solute in each phase (i = W, aqueous phase; i = L, lipid phase). The ratio can be simplified considering the moles of water and lipid are significantly higher than the relative amount of solute in both phases:
p,x ,L
L
, (2)
Instead of using the quantities of water and lipid, the partition constant is normally presented as a function of the phase volumes, Vi:
p ,L VL , V (3)
that can be converted to the mole-fraction partition constant (Kp,x) by the lipid and water
molar volumes, respectively γL and γw:
p p,x
γ
γL (4)
From equation 2 it is possible to relate the fraction of membrane bound solute molecules (XL) to Kp,x:
L p,x
L
p,x L (5)
where [L] and [W] is the concentration of lipid and water molecules, respectively. Equation 4 allows further simplification to equation 6:
L p
γL L
1 pγL L (6)
At this point, equation 6 can already be used to determine experimental Kp
values in centrifugation and chromatography techniques, among others. This is because the value of XL can be directly obtained from the experiments123. However, if the
21
experimental parameter p varies upon membrane interaction as a linear combination of the molecules in the water (pW) and lipid (pL) phases, the resulting equation is as
follows:
pγL L L
1 pγL L (7)
Our fluorescence spectroscopy approach takes advantage of the variations in the fluorescence emission intensity of membrane interacting molecules to determine their respective Kp values. The membrane hydrophobic environment favors a decrease in the
non-radiative decay rates of most fluorophores, which consequently leads to an increase in the emission quantum yield124. As we will see in the course of this work, there are specific interactions that may lead to negative variations in the fluorescence intensity. Nonetheless, as long as there is a variation of a fluorescence property, equation 7 can be modified to:
pγL L L
1 pγL L (8)
where IW and IL are the fluorescence intensities in the aqueous and lipid phases,
respectively. This equation can also be used with absorption spectrophotometry, fluorescence lifetime and, to some extent, fluorescence anisotropy data.
Measuring partition gives a bulk quantitative description of the molecular separation, preferential interaction and chemical compatibility used to predict the behavior of drugs in a biological scenario. We have used it to determine how fusion inhibitors interact with lipid model membranes of different compositions.
2.2. Membrane Electrostatic Potentials
The electrostatic properties of biological lipid membranes can be described as the contribution of three membrane-born potentials: the transmembrane potential (ΔΨ), the surface potential (ΨS) and the membrane dipole potential (Ψd)125 (Figure 10).
Transmembrane potentials are generated by selective ion movement across the membrane which leads to imbalanced charge densities in the two sides. Surface potentials, on the other hand, are the product of charge interaction between the
22
phospholipid head groups and the adsorbed ions. Finally, the membrane dipole potential originates from the alignment of the dipolar residues of lipid and water molecules in the internal carbon chain environment, between the bulk water phases.
Both the surface and dipole potentials are very sensitive to molecular interactions122. Adsorption and insertion lead to displacement and/or reorientation of charges and dipoles, disturbing the electrostatic equilibrium of the membrane. Therefore, effectively measuring these variations can be used to probe interactions with lipid bilayers. Because of the associated experimental artifacts of direct measurements, many indirect approaches have been considered when studying these electrophysical properties, depending on the model used126,127. Due to its actual relevance for our experimental design, emphasis will be given to the membrane dipole potential.
Voltage-sensitive lipophilic probes are a common solution to indirectly measure the variations in the membrane dipole potential. Their fluorescence properties are responsive to spatial and temporal variations in the internal electrical field of the membrane. On the basis of this potentiometric behaviour, the transition from ground to excited state induces an electronic redistribution within the chromophore donor-acceptor couple. As their position is normally on opposite sides of the rod-shaped probe, the local dipolar potential affects the energetic transitions as follows126:
(9)
where h is Plank’s constant, Δυ is the spectral shift, E is the electric field, |E| is the magnitude of the electric field, and Δμ and Δα are the variation in the dipole moment Figure 10 – The Membrane Electrostatic Potentials. Biological lipid membranes exhibit three electrostatic potentials: the transmembrane potential (ΔΨ), the surface potential (ΨS)
23
and polarizability upon excitation, respectively. To maximize the observed energy shift, it is very important that the probe inserts perpendicularly to the membrane and that the electronic charge density suffers considerable redistribution (substancial Δμ). Although both the emission and excitation spectral shift can be used, emission shifts are distorted by solvent relaxation effects, making them less reliable128. Blue and red shifts may occur, respectively from an increase or decrease in the membrane dipole potential.
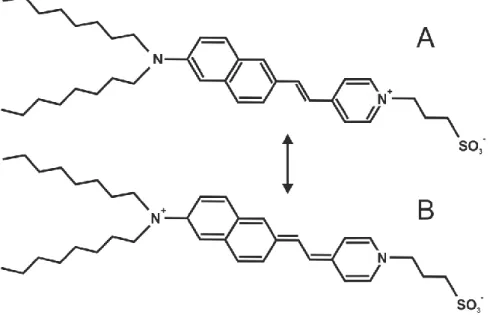
Potentiometric membrane reporters are convenient alternatives to monitor lipid-drug interactions when other biophysical approaches based on intrinsic properties are not applicable. We have used the styryl dye di-8-ANEPPS129 (Figure 11) to probe the interaction of the fusion inhibitor sdAb with lipid model membranes, complementing the results from intrinsic fluorescence.
Figure 11 – Chemical structure of the ground state and excited di-8-ANEPPS probe. The relative population of ground state (A) and excited (B) forms of the probe is highly dependent on the relative Ψd within the lipid membrane, where it inserts. Upon Ψd
perturbation, this equilibrium is displaced leading to variations in the observed fluorescence properties.