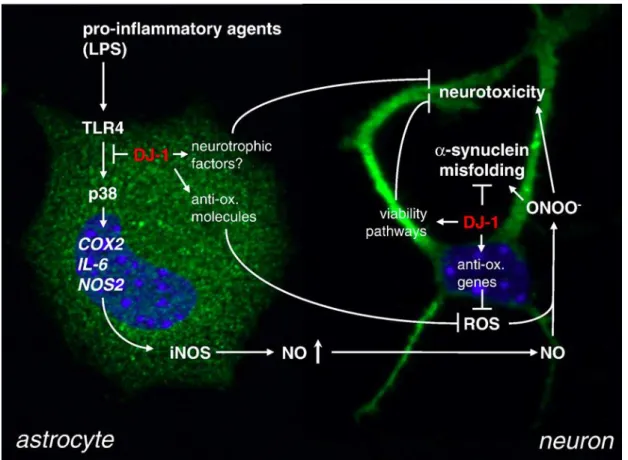
Regulation of DJ-1 Expression by Neuron-Astrocyte Interaction
68
0
0
Texto
Imagem

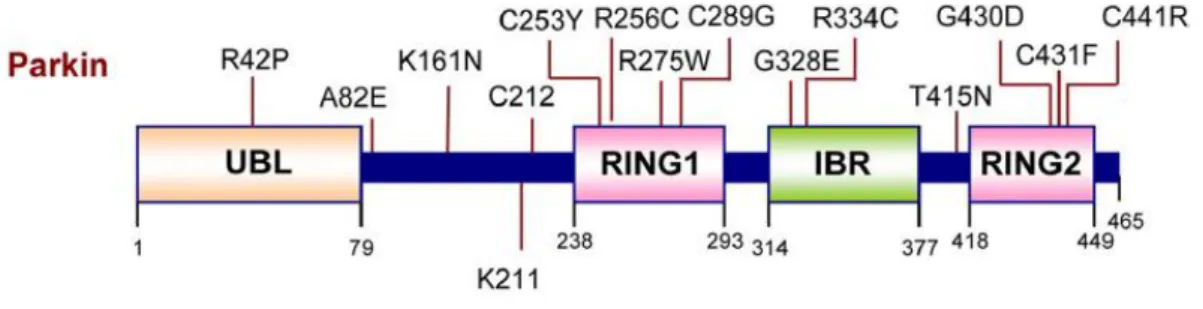
![Table 1.2 | Susceptibility Genes/Loci for PD [6].](https://thumb-eu.123doks.com/thumbv2/123dok_br/18915345.936761/23.892.160.774.163.396/table-susceptibility-genes-loci-pd.webp)

+7



Documentos relacionados