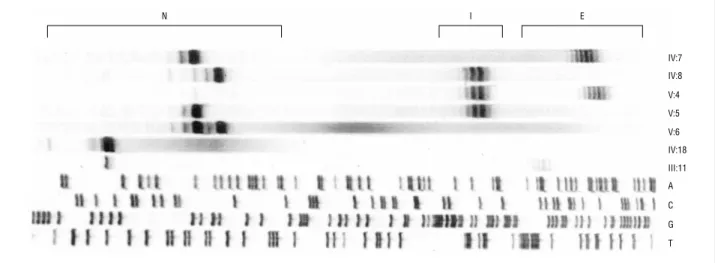
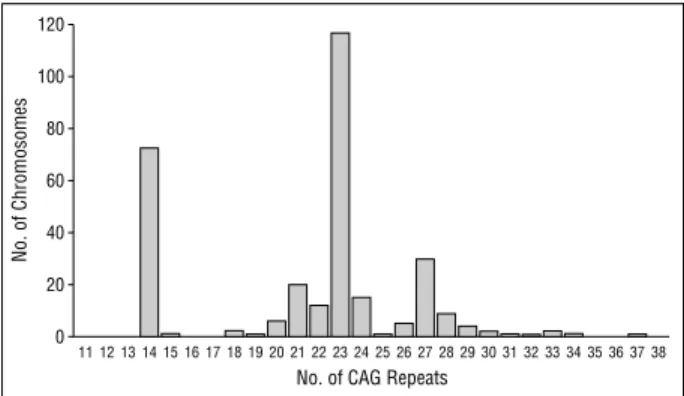
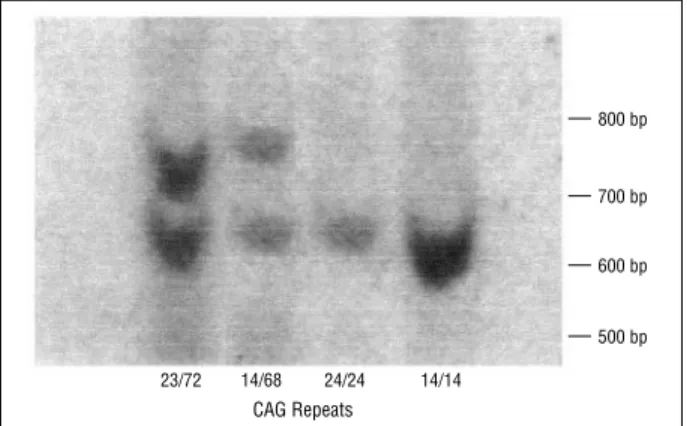
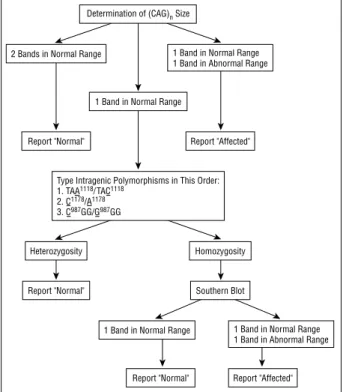
Improvement in the molecular diagnosis of Machado-Joseph disease
Texto
Imagem
Documentos relacionados
[r]
Considerando a formação dos clusters após a criação do IQM é possível perceber que, para os municípios do estado de São Paulo, não é possível afirmar que cidades
NT - Nº PA´s com Novas Técnicas – Para esta variável foi atribuído um peso de 5%, uma vez que se considera uma variável importante, mas na globalidade da avaliação
This work aims to demonstrate the processing and interpretation of multibeam bathymetric data, supported by geological samples collected in the area of Deception Island, South
Nessa condição, já não per- guntaríamos mais “o que é” a música ou a filosofia, mas reconheceríamos que há, no lugar da essência, apenas processos modalizados que,
With regard to the procedures performed, it was noted that the most prevalent one was that related to endodontic emergency, corresponding to 462 cases, with diagnosis of
Dessa forma, observa-se que “muitos dos valores da hospitalidade medieval ajustam-se aos dias de hoje, tais como o serviço amigável, a atmosfera amena e a abundância de
Dedicamos agora um capítulo autónomo ao direito de retenção, devido a uma inovação criada e importância dada pelo Decreto-Lei de 1980, em matéria de
