IDENTIFICAÇÃO DE ALVOS NA TERAPIA ANTI-CANCERÍGENA UTILIZANDO TÉCNICAS DE MODELAÇÃO MOLECULAR
Texto
Imagem
![Figura 2: Esquema representativo dos processos de proliferação celular e da apoptose. [8]](https://thumb-eu.123doks.com/thumbv2/123dok_br/15680079.625555/40.892.248.707.396.675/figura-esquema-representativo-dos-processos-proliferação-celular-apoptose.webp)
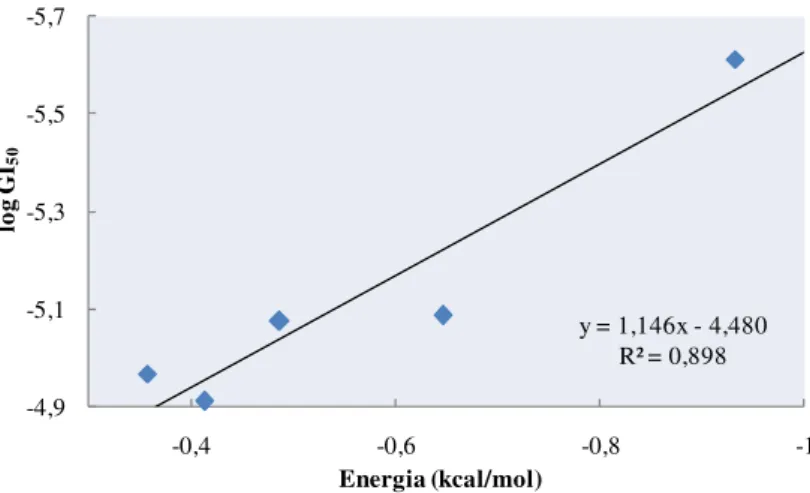
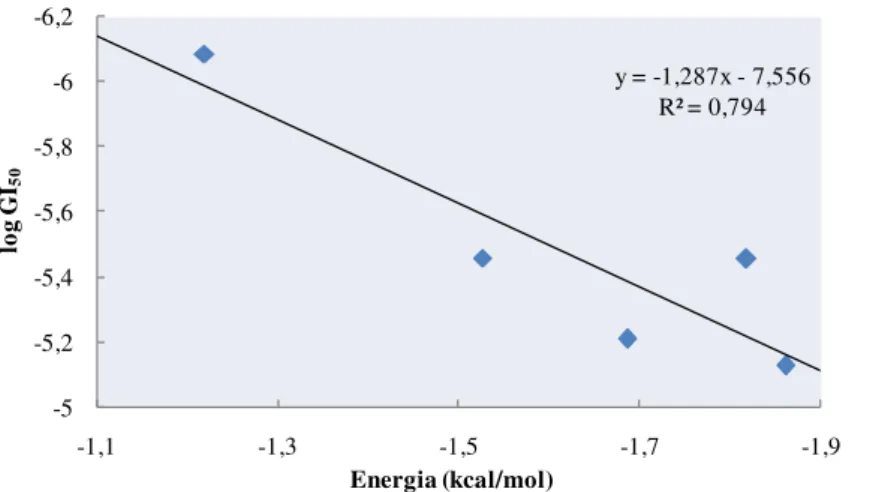
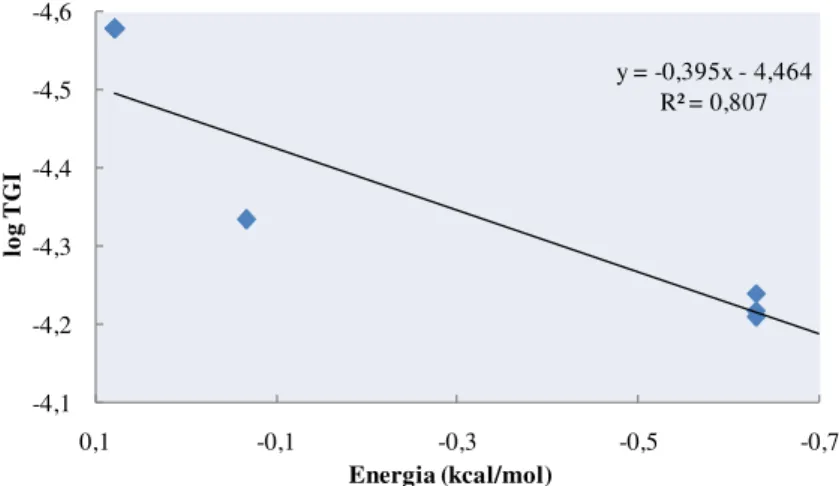
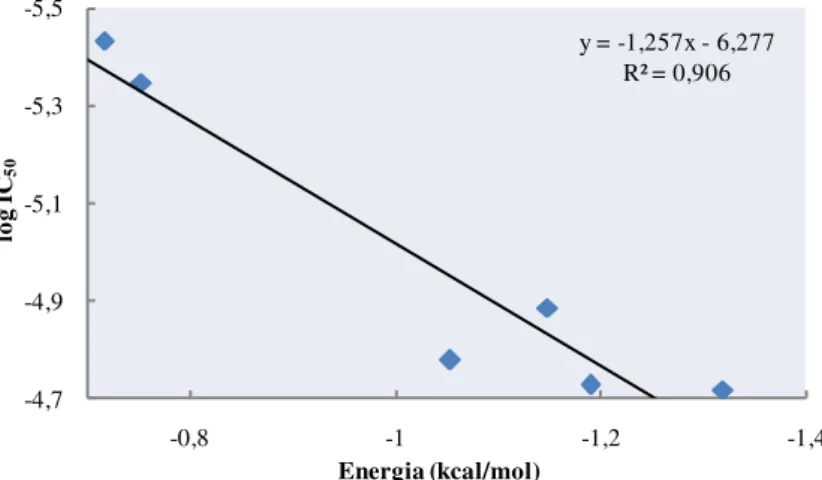
Outline
Documentos relacionados
hospitalizados, ou de lactantes que queiram solicitar tratamento especial deverão enviar a solicitação pelo Fale Conosco, no site da FACINE , até 72 horas antes da realização
Ficou com a impressão de estar na presença de um compositor ( Clique aqui para introduzir texto. ), de um guitarrista ( Clique aqui para introduzir texto. ), de um director
dois gestores, pelo fato deles serem os mais indicados para avaliarem administrativamente a articulação entre o ensino médio e a educação profissional, bem como a estruturação
O objetivo do curso foi oportunizar aos participantes, um contato direto com as plantas nativas do Cerrado para identificação de espécies com potencial
(2019) Pretendemos continuar a estudar esses dados com a coordenação de área de matemática da Secretaria Municipal de Educação e, estender a pesquisa aos estudantes do Ensino Médio
No código abaixo, foi atribuída a string “power” à variável do tipo string my_probe, que será usada como sonda para busca na string atribuída à variável my_string.. O
Use a auto hipnose para se libertar dos seus medos e fobias. A auto A auto sabotagem é sabotagem é uma constante uma constante em sua em sua vida? Sempre vida? Sempre que você
Quando Goffman (1985) fala em palco e cenário, atores e platéia, papéis e rotinas de representação, necessidade, habilidades e estratégias dramatúrgicas,
