Universidade de Aveiro 2011
Departamento de Química
Ana Rita Dias Araújo
Antifungal drug resistance driven by mistranslation
in yeast
Resistência a antifúngicos gerada por erros na
tradução em levedura
Universidade de Aveiro 2011
Departamento de Química
Ana Rita Dias Araújo
Antifungal drug resistance driven by mistranslation
in yeast
Resistência a antifúngicos gerada por erros na
tradução em levedura
Dissertação apresentada à Universidade de Aveiro para cumprimento dos requisitos necessários à obtenção do grau de Mestre em biotecnologia molecular, realizada sob a orientação científica do Doutor Manuel António da Silva Santos, Professor associado do Departamento de Biologia da Universidade de Aveiro e co-orientação do Dr. Tobias Franz Anton Ludwig Weil, investigador pós-doutorado do Departamento de Biologia da Universidade de Aveiro.
Dedico o final desta etapa ao meu pai, que vai finalmente poder ir de férias sem ter de contar os “trocos”.
o júri
presidente Doutora Etelvina Maria de Almeida Paula Figueira professora auxiliar, Universidade de Aveiro
Doutor Vítor Manuel Vieira da Costa professor associado, ICBAS, Universidade do Porto
Doutor Manuel António da Silva Santos
professor associado com agregação, Universidade de Aveiro
Doutor Tobias Franz Anton Ludwig Weil investigador pós-doutorado, Universidade de Aveiro
agradecimentos Gostaria de agradecer, primeiramente, ao professor Manuel Santos por me ter dado a oportunidade de trabalhar no seu laboratório. Foi uma experiência completamente nova para mim. Aprendi muito.
Agradeço imenso ao Dr. Tobias Weil por ter supervisionado o meu trabalho ao longo do ano.
Estou extremamente agradecida a todos os membros do laboratório de biologia RNA, quer os actualmente presentes, quer os que já seguiram com as suas vidas. O bom humor que existe todos os dias no laboratório é graças a eles. Em especial, agradeço à Rita Bezerra e ao João Simões, que tinham sempre um tempinho no meio das suas experiências. Aos meus colegas de mestrado estou grata pelo companheirismo.
Agradeço aos meus amigos, que ouviram demasiadas vezes a palavra tese nos nossos últimos encontros.
Tenho de agradecer ao meu pai, que quando me via em baixo ou com vontade de barafustar, tentava sempre dar-me alento à sua maneira.
E finalmente agradeço ao Telmo, que esteve sempre comigo, sempre me apoiou, ouviu e, claro, aturou. Infelizmente, o tempo passa demasiado depressa e nem sempre tive tempo para aqueles de quem gosto.
palavras-chave erros de tradução, resistência a antifúngicos, RNA de transferência.
resumo A resistência a antifúngicos é, hoje em dia, um problema sério a nível clínico, pelo que há necessidade de descobrir novos alvos que possibilitem o desenvolvimento de novos antifúngicos. Investigação a decorrer no nosso laboratório indica que a ambiguidade no reconhecimento de codões em
Candida albicans, um patogénio humano, acelera a resistência a antifúngicos. O presente trabalho teve como objectivo elucidar se a ambiguidade em leveduras não patogénicas aumenta a resistência a antifúngicos. Para tal foi induzida artificialmente ambiguidade no reconhecimento de diferentes codões em Saccharomyces cerevisiae. As estirpes resultantes possuem um plasmídeo de replicação reduzida contendo um tRNAUGA
Ser
de C. albicans sujeito a mutagénese dirigida, de modo a mutar o anticodão. Os novos anticodões reconhecem codões de diferentes aminoácidos mas o tRNA mantém os elementos de reconhecimento, sendo acilado com serina. Os tRNAs mutantes vão competir com os nativos, formando-se um proteoma estatístico. Para verificar se estas estirpes apresentam um fenótipo mais vantajoso em resposta a variados antifúngicos, foram expostas a diferentes classes dos mesmos. Adicionalmente, foram analisados microarrays de estirpes não expostas a qualquer stress adicional, de modo a perceber se as mesmas apresentam já tendência para responderem de modo diferente perante os diferentes antifúngicos.
keywords mistranslation, antifungal drug resistance, transfer RNA
abstract Antifungal drug resistance has become a severe clinical problem and new targets for the development of new antifungal drugs need to be discovered. Ongoing work in our laboratory indicates that codon mistranslation due to codon ambiguity accelerates antifungal drug resistance in the human pathogen
Candida albicans. The present work aimed to elucidate if non pathogenic yeasts behave similarly. Therefore, mistranslation was artificially induced in
Saccharomyces cerevisiae strains by the expression of chimeric tRNAs. Each of the constructed strains carried a low-copy number plasmid, containing a C. albicans tRNAUGA
Ser
gene, whose anticodon was changed by site-directed mutagenesis, in order to replace it by several other anticodons. As the identity elements of the tRNA remained unchanged it was still acylated with serine. These mutant tRNAs are expected to compete with the native ones and have an impact on the proteome. To verify if mistranslation leads to an advantageous phenotype regarding antifungal drug resistance, cells were exposed to different antifungals. Additionally, microarray analyses were performed on non-exposed mutant strains in order to detect a possible pre-disposition to resist antifungal exposure.
i
List of contents
List of Abbreviations ... iii
List of figures ... iv
Thesis outline ... v
Introduction ... 1
1. The genetic code ... 1
1.1 Transfer RNA ... 2
1.2 Aminoacyl-tRNA synthetases ... 4
2. Protein translation in eukaryotes ... 5
2.1 Initiation ... 5
2.2 Elongation ... 6
2.3 Termination ... 8
2.4 Recycling and re-initiation ... 8
3. Mistranslation ... 10
3.1 tRNA aminoacylation errors ... 11
3.2 Decoding errors ... 13
4. Translation stress-induced mutagenesis ... 15
5. Antifungal drug resistance in fungi ... 16
5.1 Resistance mechanisms ... 18
5.2 ABC and MFS transporters ... 19
Materials and Methods ... 22
1. Strains and Growth conditions ... 22
2. Growth curves ... 23
3. Antifungal susceptibility testing ... 23
4. Evolution of antifungal drug resistance – forced evolution ... 24
5. Development of translational-induced stress along with generation increment ... 24
6. Phenomics study ... 24
7. Microarray analysis ... 25
Results ... 26
ii
2. Evolution of antifungal drug resistance ... 26
2.1 Antifungal susceptibility – Broth dilution test ... 26
2.2 Forced evolution ... 28
3. Antifungal susceptibility – Etest assay ... 29
4. Development of translation-caused stress along with generation increment .. 31
5. Phenomics of mistranslating yeasts ... 32
6. Microarrays ... 36
Discussion ... 39
1. Antifungal susceptibility – Etest assay ... 39
2. Evolution of antifungal drug resistance ... 41
3. Development of translation-caused stress along with generation increment .. 42
4. Phenomics study and microarray analysis ... 43
Conclusion and Outlook ... 47
Bibliography ... 49
iii
List of Abbreviations
aaRS – tRNA-aminoacyl synthetase aa-tRNA – aminoacyl-tRNA
AMP – adenosine monophosphate AP – amphotericin B
ATPase – adenosine triphosphatase CTZ – clotrimazole
CS – caspofungin
DHA – drug H+ antiporter
DMSO – dimethyl sulfoxide DNA – deoxyribonucleic acid eEF – eukaryotic elongation factor eIF – eukaryotic initiation factor eRF – eukaryotic release factor FDR – false discovery rate FLZ – fluconazole
GTP – guanosine 5‟-triphosphate IRES – Internal ribosome-entry site MCZ – miconazole
MDR – multidrug resistance mRNA – messenger RNA
NBD – nucleotide binding domain PDR – pleiotropic drug resistance PPi – pyrophosphate
ROS – reactive oxygen species RNA – ribonucleic acid
rRNA – ribosomal RNA
TMD – transmembrane domain tRNA – transfer RNA
TSM – translation stress-induced mutagenesis
iv
List of Figures
Figure 1 – The standard genetic code ... 2
Figure 2 – Structure and domains of yeast tRNA ... 3
Figure 3 – Classes and subclasses of aminoacyl tRNA synthetases ... 4
Figure 4 – Translation initiation in eukaryotes ... 6
Figure 5 – Translation elongation in eukaryotes ... 7
Figure 6 – Translation termination and recycling ... 8
Figure 7 – “Closed-loop” mRNA model ... 9
Figure 8 – Sources of errors in eukaryotic protein synthesis ... 10
Figure 9 – Aminoacylation – quality control steps ... 12
Figure 10 – The ergosterol pathway ... 17
Figure 11 – Azole resistance mechanisms ... 18
Figure 12 – ABC transporter ... 19
Figure 13 – MFS transporter ... 20
Figure 14 – Single copy plasmid based on pRS315 ... 22
Figure 15 – Growth curves and growth rates of mistranslating yeast ... 26
Figure 16 – Antifungal susceptibility – broth dilution test for fluconazole ... 27
Figure 17 – Antifungal susceptibility – broth dilution test for miconazole ... 27
Figure 18 – Forced evolution of S. cerevisiae under miconazole exposure ... 28
Figure 19 – S. cerevisiae cell morphology under miconazole exposure ... 28
Figure 20 – Antifungal susceptibility – Caspofungin Etest results ... 29
Figure 21 – Antifungal susceptibility – Amphotericin B Etests results ... 30
Figure 22 – Antifungal susceptibility – Itraconazole Etest results ... 30
Figure 23 – Antifungal susceptibility – Itraconazole Etest – examples ... 31
Figure 24 – Antifungal susceptibility – Flucytosine Etest – examples ... 31
Figure 25 – Antifungal susceptibility – Etest - comparison between generations ... 32
Figure 26 – S. cerevisiae cell morphology with generation increment ... 32
Figure 27 – Phenomics of mistranslating yeast strains ... 33
Figure 28 – Phenomics of mistranslating yeast strains – relative growth rates ... 35
Figure 29 – Microarray analysis – one class SAM – genes related to amphotericin B and caspofungin response ... 37 Figure 30 – Microarray analysis – one class SAM 2 – genes related to azoles response . 38
v
Thesis outline
This work was aimed to elucidate if mistranslation due to codon ambiguity accelerates antifungal drug resistance in Saccharomyces cerevisiae, as it was previously tested for Candida albicans (data not published).
S. cerevisiae is a good model organism to study the antifungal action upon
mistranslation. It is well characterized, its genome was fully sequenced and it is closely related to C. albicans, a major opportunistic pathogen.
Experiments were performed using mutant S. cerevisiae strains, in which artificial mistranslation was induced by the expression of a mutant Candida
albicans serine tRNA. This mutant tRNA was previously subjected to site-directed
mutagenesis in order to change its anticodon to several other anticodons. The anticodon of serine-tRNAs is not an identity element and therefore, aminoacylation is not compromised. These mutant tRNAs will compete with the native ones by recognizing different amino acid codons but inserting always serine into the nascent polypeptide. A fraction of mutant proteins will not fold or will be unstable. These unstable proteins are expected to either unfold, have impaired function or eventually gain new functions. As each of the different strains carries a unique amino acid substitution (e.g. replacement of threonine by serine), severe consequences are expected to happen especially when very different chemically and structurally amino acids are exchanged.
First, we accessed the growth rate of the mistranslating strains. Then, an evolution experiment was carried out in order to investigate if mistranslation accelerates antifungal drug resistance and if translational stress-induced mutagenesis (TSM) occurs also in eukaryotes. To check for differences in antifungal drug resistance susceptibility tests were performed using different classes of antifungals (e.g. azoles, echinocandins, pyrimidine analogs and polyenes). Additionally, a long term experiment was performed using these strains, aimed to answer the question whether mistranslation over time increases mutation rate on a genome wide view. Therefore, whole genome sequencing will be applied in order to check for single nucleotide polymorphisms, indels and chromosomal rearrangements, among others. Finally, microarray analyses of the mistranslating strains were used to support the data obtained in a phenomics study in which several antifungals and translation inhibitors were used.
1
Introduction
1. The genetic code
The genetic code represents a well thought but degenerate system to translate nucleic acid sequences into proteins. Initially, 20 amino acids were described (Crick, 1966b), but a few years later, selenocysteine and pyrrolysine were discovered (Bock et al., 1991; Hao et al., 2002).
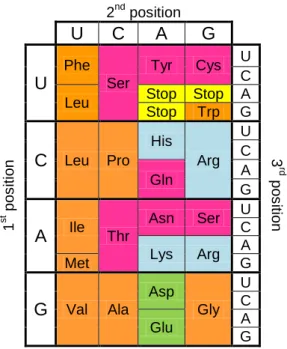
DNA sequences consist of four deoxyribonucleotides which can be purines - adenine (A) and guanosine (G) – or pyrimidines – cytosine (C) and thymine (T). In eukaryotes, these sequences are transcribed into non processed messenger RNAs (mRNA) by RNA polymerase II and after maturation are translated into proteins by ribosomes. mRNA sequences consist of four ribonucleotides - adenine (A), uridine (U), guanosine (G) and cytosine (C) - which are organized in triplets, enabling a set of 43 =64 possible codons. Those codons are either specifically assigned to amino acids or lead to translation termination (figure 1). Furthermore, the genetic code is considered to be degenerate, as with the exception of methionine and tryptophan, every amino acid can be decoded by two or more synonymous codons. Those codons similar to each other are assigned to chemically similar amino acids. This biased display of triplets is very important in order to reduce the incorporation of dissimilar amino acids into a protein. Hence a near cognate amino acid can be misincorporated but rarely will a non-cognate one. Due to their chemical differences the introduction of dissimilar amino acids could cause major consequences (e.g. improper protein folding). Another important feature is that most synonymous codons have the same ribonucleotide at the same position, usually the second one. Such feature also contributes to reduce the consequences of misincorporation as errors happen more easily by misreading of the third ribonucleotide position known as the wobble position.
Actually, the wobble hypothesis predicted that ribonucleotides could be displaced and form a non-Watson-Crick base pair (Crick, 1966a). Recent research has confirmed this statement, verifying that the wobble position often contains a modified base, which can have more than one correspondent ribonucleotide (Agris et al., 2007).
Another aspect to be considered is the correlation between sets of codons and the two groups of tRNA-aminoacyl synthetases. Group I synthetases deal with codons containing uridine in the second position (with the exception of the phenylalanine codon), while group II synthetases handle all codons containing cytosine in this position (Wetzel, 1995).
Thus, the organization of the genetic code contributes to the improvement of translation accuracy.
2
1.1 Transfer RNA
Besides the wobble hypothesis, Crick postulated his adaptor hypothesis, which states that adaptor molecules possibly link nucleotides to amino acids(Crick, 1958). These adaptor molecules, which recognize codons in the mRNA were discovered to be transfer RNAs (tRNA). Due to the wobble position mentioned above, tRNAs exist in isoacceptor families, and each family is recognized by a cognate aminoacyl-tRNA synthetase (aaRS).
tRNAs‟ secondary structure resembles a cloverleaf and their tertiary structure is L-shaped. The cloverleaf shape forms due to base pairing and consists of a stem with a loose 3‟-CCA end, called acceptor stem as it binds the corresponding amino acid, and three stem loops: the D loop, the anticodon loop and the T loop. Some tRNAs have an extra variable loop located between the anticodon and the T loops. Regarding the tertiary structure, the anticodon is located on the first major domain of the “L” shape, while on the second domain the correspondent amino acid is attached (figure 2).
In eukaryotes, post-transcriptional processing of tRNAs is required to allow fine tuning of structure and identity in order to obtain diverse tRNA types (Hopper et al., 2010). Part of this process is the customization of nucleotides in specific positions. Some modifications are quite complex, requiring more than one modifying enzyme. A well known example is the ubiquitous pseudouridine (ᴪ), the first discovered tRNA modification. So far, 81 modifications concerning tRNA are known and from those, 47 belong to the Eukarya domain (the RNA modification
2nd position U C A G 1 st po s iti on U Phe Ser Tyr Cys U 3 rd po s iti on C Leu Stop Stop A Stop Trp G C Leu Pro His Arg U C Gln A G A Ile Thr Asn Ser U C Lys Arg A Met G G Val Ala Asp Gly U C Glu A G
Figure 1 - Standard genetic code: Pink-polar uncharged; blue- positively charged;
3
database). These contribute to the stability and recognition of the carrying molecule, and tend to be conserved within phylogenetic domains. In particular, they contribute to tRNA folding, Mg2+ binding, intron removal, protein recognition, codon recognition and fidelity of the translational reading frame (Agris, 2008). Variation in modification level appears along with disease, under exposure to stresses such as starvation and drug exposure, and seem to be age and localization dependent (Dirheimer et al., 1995).
Besides such nucleoside modifications, transfer RNAs are subjected to further post transcriptional processing, such as splicing and trimming of 5‟- and 3‟-ends by specific RNAses (reviewed by Hopper et al., 2010; Nakanishi and Nureki, 2005). Unlike in many bacteria, eukaryote tRNA genes do not encode the 3‟-CCA tail, thus, it has to be added enzymatically. An important role of this loose end is to protect the tRNA from degradation due to its function as an anti determinant against the 3‟end ribonucleases (Phizicky and Hopper, 2010).
tRNA identification by aaRS owes less to modified nucleotides than to structural elements along the tRNA. However, if located at the wobble position, the modified residue can contribute both for precise codon-anticodon pairing and the recognition of the tRNA. One example is the hypermodified nucleoside mnm5s2U at the wobble position of tRNALys and tRNAGlu (Nakanishi and Nureki, 2005). tRNAs may interact with aaRSs at several regions (e.g. Lenhard et al., 1999; McClain et al., 1998; Sekine et al., 1996; Senger et al., 1995), but mainly with the discriminator base (position 73), the acceptor stem pairs 1-72, 2-71 and 3-70, and the anticodon (McClain, 1993). These identity elements serve not only specific recognition purposes but also can inhibit aminoacylation by non-cognate aaRSs (antideterminants or negative elements) (McClain, 1993).
Figure 2 - The structure and domains of tRNA. The cloverleaf secondary structure (A) is color coded to identify
the structural domains of the crystal structure: amino acid accepting stem, or aminoacyl-stem (AA) in red; dihydrouridine stem and loop domain (DSL) in black; anticodon stem and loop domain (ASL) in green; variable or extra loop (EL) in orange; and the ribothymidine, or TYC, stem and loop (TSL) in light blue. The positions of the (almost) invariant U33 and the amino acid accepting 3‟-terminus (C74, C75 and A76) are shown. The three-dimensional structure of tRNA is represented by the crystallographic structure of yeast tRNAPhe(B). Adapted from Agris (2004).
4
1.2 Aminoacyl tRNA synthetases
Aminoacyl tRNA synthetases catalyze the 3‟- esterification of tRNAs with their cognate amino acid. Esterification mostly happens in a direct way, according to the following reactions:
Amino acid + ATP + aaRS aaRS(amino acid)-AMP + PPi aaRS(amino acid)-AMP + tRNA aaRS + (amino acid)-tRNA + AMP Although similar in action aaRSs can be divided into two evolutionary distinct classes. Each class gathers those containing a certain structural motif in their active site. Even though not all synthetases have been extensively studied, these two motifs seem to result in two different ways of binding ATP and approaching the cognate tRNA: Class I binds ATP in an extended conformation and approaches the tRNA‟s acceptor stem from its minor groove side, and class II binds ATP in a bent conformation and approaches the tRNA‟s acceptor stem from the major groove side (Ibba and Soll, 2000). Only LysRS can be found in both classes, depending on the organism.
Although evolutionary distinct, it has been suggested by Rodin and Ohno (1995) that they could be complementary to each other, giving rise to the idea that in early genomes both strands of RNA were used for protein synthesis. Further, each class can be divided in three subclasses (a, b, and c) representing closer related enzymes (Schimmel, 2008).
Class I Class II a MetRS SerRS ValRS ThrRS LeuRS AlaRS IleRS GlyRS CysRS ProRS ArgRS HisRS b GluRS AspRS GlnRS AsnRS LysRS-1 LysRS-2 c TyrRS PheRS TrpRS
Figure 3 - Classes and subclasses of aminoacyl tRNA synthetases. Subclasses include enzymes that are most closely related to each other in their sequences. Significantly, the subclasses also group aminoacyl tRNA synthetases according to their amino acid chemical types. Adapted from Schimmel (2008).
5
2. Protein translation in Eukaryotes
Messenger RNA (mRNA) is subjected to maturation resulting in a ready-to-go template that can be frequently used by the translation machinery, as long as it remains stable and is needed. The translation machinery consists of cytoplasmic ribosomes, tRNAs and translational factors, and is assisted by aaRS. Eukaryotic ribosomes are large complexes of proteins and ribosomal RNA (rRNA). They can be divided into a large 60S and a small 40S subunit. These two subunits bind to each other during translation. The complete ribosome 80S permits the binding of three tRNAs, whose binding sites are located on the 40S subunit. These are called aminoacyl (A site), peptidyl (P site) and exit (E site) sites.
Protein translation can be divided in three steps: initiation, elongation and termination. These steps are followed by the recycling of the ribosome.
2. 1 Initiation
Initiation is assisted by the initiation factors, which interact with each other, resulting in a solid and stable initiation complex, able to bind strongly to the mRNA. Many of these factors are also macrocomplexes.
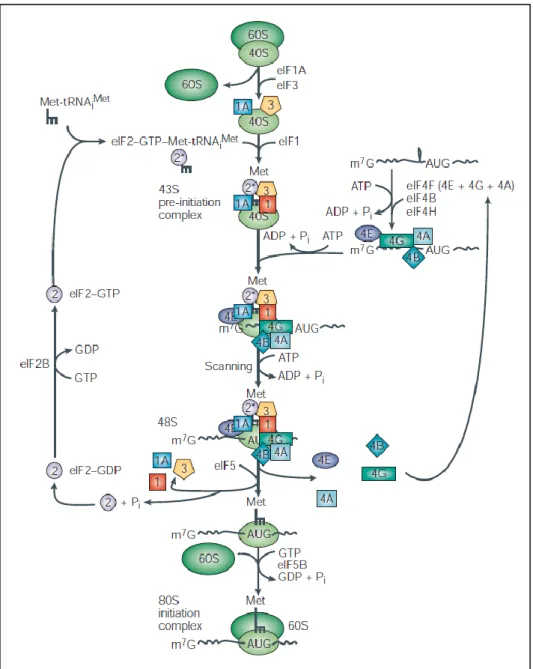
Initiation requires the formation of a ternary complex, which is formed by the initiator transfer RNA bound to methionine (Met-tRNAiMet), the eukaryotic initiation factor 2 (eIF2) and GTP. eIF2 is assisted by eIF2B which recycles GDP back to GTP. This complex binds to the small ribosomal subunit (40S) and to eIF1, 1A and 3, resulting in a larger complex, the 43S. These factors appear to help scanning the mRNA (eIF1 and 1A) and to avoid premature binding of the large ribosomal subunit (eIF3) (reviewed by Kapp and Lorsch, 2004).
Before the 43S complex attaches to the 5‟end of the mRNA, forming the 48S complex, unwinding of the mRNA‟s secondary and tertiary structure in 5‟-untranslated region (UTR) takes place and an anchor is assembled. The unwinding seems to be performed by the helicase eIF4A, stimulated by eIF4B (Grifo et al., 1982; Lawson et al., 1989; Ray et al., 1985; Rozen et al., 1990). The anchor assembly seems to be accomplished by the binding of eIF4E to the 5‟ 7-methylguanosine cap. The anchor eIF4E, together with eIF4A bind to eIF4G. These three factors together form the eIF4F complex. eIF4G functions like a hub and is thought to be very important in unusual cases of initiation, such as the absence of the 5‟ cap, the poly A tail, or both simultaneously, and in the presence of an internal ribosome-entry site (IRES) mediated translation which is cap-independent (Holcik and Sonenberg, 2005; Johannes et al., 1999). Finally, the 48S complex can start scanning the 5‟UTR for the initiation codon (AUG).
With the localization of the initiation codon, the 60S ribosomal subunit, binds to the small subunit, assisted by eIF5B. As ligation happens, eIF 1, 1A, 3 and 5 are released. Premature association of ribosomal subunits is thought to be blocked in part by the binding of eIF6 to the 60S.
6
2.2 Elongation
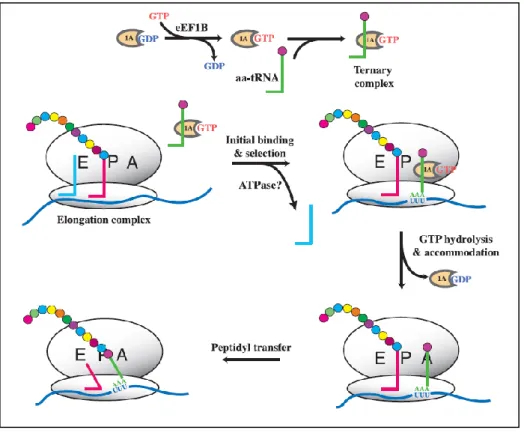
After the binding of the met-tRNAiMet anticodon to the initiation codon (AUG) elongation starts. Met-tRNAiMet binds at the P site while the following tRNAs bind to the A site of the ribosome. Elongation ternary complexes bind to the elongation factor eEF1A instead of eIF2. eEF1A triggers GTP hydrolysis, and eEF1AGDP releases the aminoacyl tRNA. Codon-anticodon base pairing and the GTP hydrolysis by eEF1A are part of the quality control steps (see below). The formation of a peptide bond between the incoming amino acid and the peptidyl aminoacyl-tRNA is carried out by the ribosomal peptidyl transferase center. Then, translocation takes place with the deacylated tRNA moving to the E site. During
7
translocation the tRNA passes through a hybrid state in which it is present in both sites. The same happens to the tRNA at the A site by first translocating its acceptor stem to the P site and only then the anticodon end. Translocation is possible due to GTP hydrolysis stimulated by the factor of the eEF2GDP complex. After translocation the A site is free for the next aa-tRNA to bind. However, eEF1AGDP has to be recycled and this is accomplished by the eEF1B complex, consisting of eEF1Bα and eEF1Bβ (Kapp and Lorsch, 2004). During translation the three ribosome sites will always be occupied. Once a deacylated tRNA is released from the E site, a new ternary complex binds to the A site. This process is repeated until a stop signal appears.
Particularly, in fungi a third factor, eEF3, exists which was shown to be essential in yeast survival (Qin et al., 1990). It interacts with eEF1A (Kovalchuke et al., 1998), and is required for every round of peptide bond formation. Two possible roles are described for eEF3: a) to aid in the release of the deacylated tRNA from the E site and b) to increase efficiency in the binding of eEF1AGTPaa-tRNA to the A site (Trianaalonso et al., 1995).
It was previously demonstrated that the rate-limiting step in elongation is tRNA selection and that different tRNAs exist in different concentrations leading to variation in the translation rate along the mRNA (Varenne et al., 1984). Thus, to maintain translation at a specific relevant rate (Reynolds et al., 2010) it is important that a set of correctly acylated tRNAs is available at any time. Otherwise the elongation complex will get stalled and/or a near-cognate tRNA, more
8
abundant at that moment, may eventually take the correct one‟s place, resulting in a mistranslated protein.
2.3 Termination
Once a stop signal enters the A site the release of the peptide is initiated. There are no cognate tRNAs to decode stop signals. Instead, a release factor with a similar 3-D structure binds to the ribosome. Only two release factors exist in eukaryotes. eRF1 decodes all three stop signals (UAA, UAG or UGA) while eRF3 is discussed to be a GTPase that stimulates the release of eRF1 from the ribosome, leading to consequent peptidyl tRNA hydrolysis. eRF3 is essential in eukaryotes and its interaction with eRF1 leads to an optimal termination efficiency in S. cerevisiae (Kapp and Lorsch, 2004).
2.4 Recycling and re-initiation
After termination the ribosomes can be used again for another translation round on the same mRNA (re-initiation) or reassembled on a new mRNA (recycling). Regarding recycling it is known that a post-termination complex (PoTC) remains, consisting at least of mRNA, tRNA and ribosome. Recently, eIF3, eIF1 and eIF1A have been suggested to cooperatively disassemble PoTCs into
9
free 60S subunits and mRNA and tRNA bound 40S subunits (Pisarev et al., 2007). In yeast eEF3 and ATP have been implicated in the disassembly of this complex, suggesting that they catalyze the simultaneous split of the ribosomal subunits and the detachment of mRNA and deacylated tRNA (Kurata et al., 2010). There is still much to unravel about recycling especially concerning which factors are involved and how.
The discovery of the interaction of poly-A binding protein (PABP) with eIF4G led to the assumption that mRNA circularizes and re-initiation of translation takes place. Hence, after termination, the 40S subunit might not be released, but instead transferred back to the 5‟ end via 3‟ and 5‟ translation factors (figure 7) (Tarun and Sachs, 1996).
Figure 7 - „Circular‟ or „closed-loop‟ mRNA model showing circularization mediated by poly(A)-binding protein (PABP) poly(A)-binding to both the poly(A) tail and eIF4G: (i) scanning of the 40S containing 43S initiation complex to the initiation codon, (ii) joining of the 60S ribosomal subunit to form a translation-competent 80S complex, (iii) protein elongation, (iv) termination of translation, and (v) re-initiation. Adapted from Mazumder et al. (2003).
10
3. Mistranslation
Translation is the most error-prone step of protein synthesis. The overall translational error rate is about 1 in 104 polymerized amino acids which is approximately the sum of transcription (∼10−4), aa-tRNA synthesis (∼10−4), and ribosomal decoding (∼10−4
) errors. Mainly for eukaryotes, the known mechanisms controlling protein translation are important to avoid wasting energy by producing unnecessary amounts of non-functional proteins. Actually, it has been estimated that translation can consume up to 50% of the cell‟s energy (Holcik and Sonenberg, 2005), and therefore, it is important for cells to have different possibilities to control translation (Gebauer and Hentze, 2004; Hinnebusch, 2005). In cases of stress global translation can be reduced, while selective translation can be activated in order to synthesize specific proteins that aid cell survival. Protein synthesis errors can arise from DNA mutations, during transcription and/or splicing, during the translation process or even during folding (figure 8). Only errors occurring during translation are considered in the following.
Mistranslation has a plethora of sources and contributes greatly to the production of non functional proteins. In general, mammalian cells have proven to be more sensitive to mistranslation than bacteria and yeast (Nangle et al., 2006).
Figure 8 - Sources of errors in eukaryotic protein synthesis. Adapted from Drummond and Wilke (2009)
11
3.1 tRNA aminoacylation errors
As mentioned, direct tRNA aminoacylation is a two-step process meticulously controlled by aaRS (e.g. “double-sieve” model proposed by Fersht (1977)), but still errors can occur at a significant error rate (Reynolds et al., 2010). The errors at this stage include:
1. Editing failures
2. tRNA misrecognition by aaRS 3. Amino acid misactivation
1) Editing is needed to amend amino acid misactivation. These types of failures are probably due to mutations. To date, the only known case which seriously affects mammals is caused by a heritable missense mutation in the editing domain of AlaRS in mice (Lee et al., 2006). This enzyme mischarges tRNAAla with serine, giving rise to a statistical protein population. As a result, the unfolded protein response (UPR) is triggered, leading to Purkinje cells (in cerebellum) degeneration and consequently, ataxia1. In vitro cultures of human cells, using an editing-defective ValRS have also shown increased degradation and apoptosis (Nangle et al., 2006). Regarding prokaryotic cells, heritable mutations in the editing site of an IleRS were verified to result in the induction of error-prone DNA polymerases (Bacher and Schimmel, 2007).
2) The structural diversity of different base combinations, and a complex network of sequence-specific aaRS-tRNA interactions, sometimes aided by binding enhancers, ensures the accurate selection of the cognate tRNA. Further, in vivo aaRSs compete for their cognate tRNAs. For these reasons, misrecognition of tRNAs seems to be rare compared to other errors and thus, there is no need for proofreading ability. In eukaryotes, despite some exceptions, tRNA transcription and maturation occur in the nucleus. This “boundary” guarantees to aaRSs that only functional tRNAs are exported to the cytoplasm (Ibba and Soll, 1999).
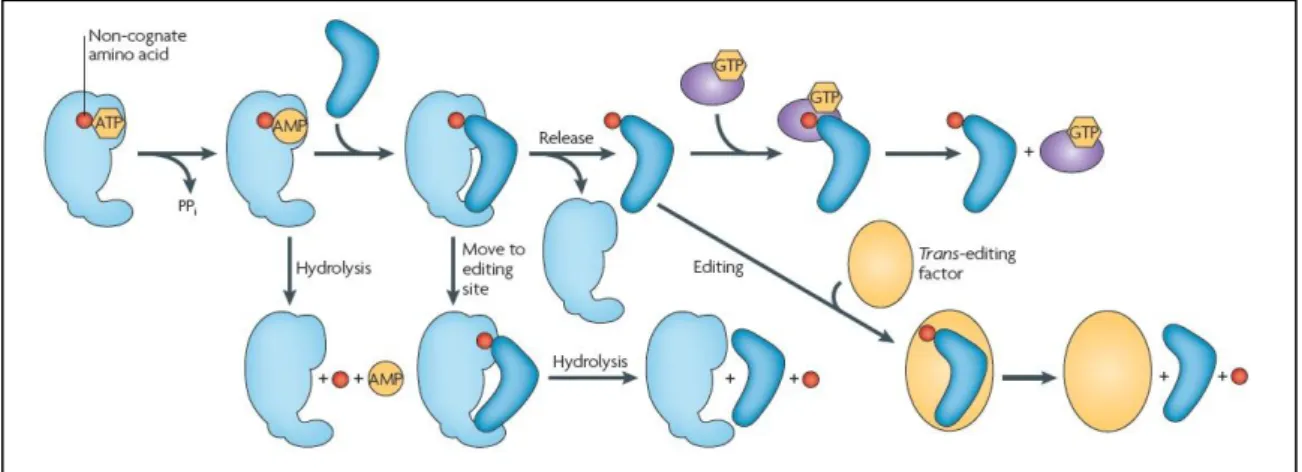
3) Amino acids are very small molecules and therefore pose a challenge to aaRSs. For instance, class I synthetases can deal with wrong attached amino acids in two ways (Cochella and Green, 2005; Reynolds et al., 2010):
Pre-transfer (before the linkage to the tRNA): First, the correct amino acid has to be selected by the aaRS. This is a grand challenge as some of them have similar chemical or physical properties and hence, must be distinguished mainly by their side chains. During selection amino acids with larger side chains are easily discarded but those with smaller ones can pass through the “sieve”. If the aaRS detects the erroneous selection, the misactivated amino acid is transferred to a second catalytic site, called editing-site (second sieve) in which, in contrast to
1
Ataxia is a non-specific clinical manifestation implying dysfunction of the parts of the nervous system that coordinate movement, such as the cerebellum.
12
the smaller misactivated amino acid, the cognate amino acid does not fit. There, the misactivated amino acid is hydrolyzed.
Post transfer (after linkage): If the aaRS does not detect the error, the amino acid will be linked to the tRNA, which still might be correctly selected due to its numerous identification elements. Misacylation can also be detected by the aaRS followed by the hydrolysis of the ester bound between the 3‟-CCA end of the tRNA and the amino acid in the editing site. If the misacylated tRNA is released it might be re-sampled by the aaRS or recognized by trans-acting factors (trans editing), such as D-aminoacyl-tRNA deacylases.
If all of these check points fail, there is still one last possibility of preventing the use of a misacylated tRNA during the formation of the ternary complex with GTP and eIF2 (or eEF1A). Both amino acid and tRNA contribute to the binding affinity to the ternary complex. If a non-cognate amino acid is attached to the tRNA, this binding affinity decreases immensely (up to 700-fold) (Asahara and Uhlenbeck, 2002).
Although not common for most organisms, the stereospecificity (D or L) of each amino acid can contribute to erroneous discrimination. In response to this problem one D-Tyr-tRNATyr deacylase has been found to act upon D-aminoacyl-tRNAs in bacteria, yeast and more recently human cells (Soutourina et al., 2000; Soutourina et al., 1999; Zheng et al., 2009).
Two commonly referred examples of misacylation are: 1) the selection of L-valine instead of L-isoleucine by E. coli IleRS, which differ only by one methyl group and 2) the selection of L-threonine by ValRS, which has a hydroxyl group instead of a methyl group like L-valine (Fersht, 1977; Fersht and Kaethner, 1976). So far, a second catalytic site, necessary for the double sieve model, has not been described for all aaRSs.
Also, regulated misacylation events are known, for instance, the transamidation pathway, which was described in bacteria and archaea. Here, GluRS is used instead of GlnRS to produce Gln-tRNAGln (Becker and Kern, 1998; Wilcox, 1969; Wilcox and Nirenber.M, 1968) and AspRS is used to produce Asn-tRNAAsn
Figure 9 - Quality control steps during the formation of a non-cognate aminoacyl-tRNA. Adapted from Reynolds et al. (2010)
13
(Curnow et al., 1996). Some organisms can only use this pathway (Curnow et al., 1997). Beside bacteria and archaea this pathway was also described for yeast mitochondria (Frechin et al., 2009) and other eukaryotes (Nabholz et al., 1997; Schon et al., 1988).
Other examples using different pathways are the production of cysteine which requires the O-phosphoseryl-tRNA synthase (SepRS) to acylate tRNACys and the formation of selenocysteine, which starts with the attachment of serine to tRNASec by a SerRS. It is worth noting that these examples, together with codon reassignments, show the evolutionary relevance of genetic code ambiguity (Moura et al., 2009).
3.2 Decoding errors
Decoding errors can happen mainly due to missense and nonsense errors and less frequently due to processivity errors. aa-tRNA selection has to be fast to achieve the optimum translation rate, leaving the translation machinery only a short time interval to explore the full discrimination potential between cognate and near cognate aa-tRNAs. Acceptance of near cognate tRNAs is avoided because of kinetic discrimination mechanisms. In a healthy organism the acceptance of non cognate tRNAs is rare. Besides codon organization the reciprocal relationship between E- and A-sites also contributes to tRNA discrimination. An occupied E site induces a low affinity A site and vice versa. So, the accommodation of the ternary complex will trigger the release of the tRNA in the E site (Nierhaus, 2006). The referred kinetic mechanisms are (Cochella and Green, 2005; Hopfield, 1974):
1. Kinetic proofreading: this strategy resembles the double-sieve editing mechanism but instead of two different selective steps, the same basic principle is repeated. The first selection step explores the ability of the ternary complex to hydrolyze GTP. This is an easy task for the cognate complex but problematic for near cognate complexes, as they will most likely dissociate. The second selection step scrutinizes binding differences between codon and anticodon, as the cognate tRNA will more easily accommodate. Non cognate aa-tRNAs are essentially excluded in the first step, while near cognate ones can pass this step with a frequency of approximately 1 in 30.
2. Induced fit: during initial selection, the rate of GTPase activation is significantly higher for the cognate aa-tRNA. During proofreading the accommodation rate is also higher. Summarizing, this strategy counts on the ability of cognate molecules to induce conformational changes on the enzyme and/or substrate, resulting in downstream effects on catalysis.
Missense substitutions are quite common errors, occurring approximately at a frequency of 4x10-4 per codon translated in prokaryotes (Parker, 1989). However, only 1 in 400 misincorporations completely inactivate the protein by affecting its folding, structure or function (Nierhaus, 2006). Such substitutions result from mischarging of tRNAs or codon-anticodon mismatches on the
14
ribosome. Their detection is difficult because the resulting protein has essentially the same size and composition as the native one. Also, missense substitutions, can lead to frameshifting, as described below.
Suppression of stop codons, known as translational readthrough, is another decoding error. The stop codon is misread either by a mutated tRNA or a near-cognate tRNA, which pairs with at least two bases of the codon. Here, the quality control mechanisms fail, and competition between the elongation and the termination machineries occurs. Nucleotide sequences surrounding stop codons can influence the readthrough efficiency. These context sequences can be quite complex (e.g. pseudoknots) (reviewed by Beier and Grimm, 2001). It is worth noting that premature stop codons lack this context sequences. In eukaryotes the nonsense-mediated mRNA decay mechanism can detect these premature codons and degrade the mRNA to avoid translation. Mutations like those in the yeast genes encoding eRF1 (SUP45) and eRF3 (SUP35), lead to suppression of the three stop codons (UAA, UAG, UGA) (Serio and Lindquist, 1999).
Processivity errors can be widely defined as those causing the release of the nascent polypeptide chain prior to its completion. The resulting truncated peptide can be toxic to the cell and will probably have an energetic cost, especially if highly expressed.
Frameshifts, which can be considered as processivity errors, are rare events (estimated error frequency of 3x10-5) that imply the alteration of the reading frame forward (+) or backwards (-) by one or more nucleotides. The most probable consequence is the production of truncated proteins due to early emergence of nonsense codons. For instance, tRNA slippage can happen if in the coding sequence the nucleotides before or after the target triplet are the same as the first and third of the latter, respectively. More rarely (10-15) the ribosome carrying a peptidyl-tRNA can slide along the sequence (Nierhaus, 2006). Further, the occupation of the E-site of the ribosome is also of great importance, because an empty one augments the probability of +1 frameshifts by 25% (Marquez et al., 2004). Although not common, there are specific cases where frameshifts can be programmed like in viral genes, and in a single case in E. coli (Craigen et al., 1985). A programmed frameshifting error has an efficiency of nearly 100%. Under these circumstances the mechanism controlling frameshifting seems to be switched off. In eukaryotes, programmed frameshifting has been observed as well (Manktelow et al., 2005; Weiss-Brummer et al., 1987; Wills et al., 2006).
Nonsense errors that arise during replication or transcription can originate frameshifts and ribosome drop-off during translation. Other types of premature termination, which are not due to the presence of stop codons are thought to happen in two different ways: 1) the growing peptide could be released from the ribosome, followed by hydrolysis of the bond attaching it to the peptidyl-tRNA, or 2) a release factor could misread a sense codon. It is hard to distinguish them experimentally and both are difficult to distinguish from frameshifts and ribosome pauses. In vivo, drop-off of peptidyl-tRNA from the ribosome seems to happen at frequencies between 3x10-3 and 10-4 per elongation event (Parker, 1989).
15
4. Translation Stress-induced Mutagenesis
The great amount of possible mistranslation errors arising in cells activates intrinsic mechanisms, as described above. However, to cope with different types of stress and/or accumulation of errors, cells may be forced to evolve faster, in order to survive. Usually, a certain accumulation level of aberrant proteins results in a decrease in fitness, culminating in cell death. However, below this limit, cells can present a statistical population of proteins, consisting of a minor part of mutant proteins together with a major part of wild-type proteins. The mutant proteome fraction is expected to include unfolded proteins but also unstable proteins which can misfold and form toxic aggregates. The subsequent overloading of protein control systems might result in the activation of stress responses and alteration of gene expression. Nonetheless, this minor proteome grants genetic and phenotypic diversity and if phenotypic advantage arises, fixation of mutation may happen (Moura et al., 2009).
If error-prone polymerases and defective DNA repair enzymes are produced they might originate hypermutagenic clones possibly exhibiting high adaptation potential (Moura et al., 2009). Recently, a hypermutagenesis phenotype associated with codon ambiguity was discovered in E. coli (Michaels et al., 1990) and named translation stress-induced mutagenesis (TSM). TSM has similarities with the SOS response (reviewed in Humayun, 1998), the best studied inducible mutagenic pathway in bacteria, as it also requires recABC and ruvAC (Ren et al., 2000). It is associated to two mutator loci named mutA and mutC which exhibit similar phenotypes. Compared to the wild-type strain they presented a transversion increase, namely the AT TA and GC TA changes, followed by the less frequent AT CG. These two mutators were found to encode glycine tRNAs, which mutations affected the anticodon (Slupska et al., 1996). The mutA allele consists of a tandem set of three identical tRNA genes (glyV), while mutC contains four copies of also identical tRNAs (glyW). Both alleles code tRNAs that misread the 5‟-GAU/C aspartic acid codon as glycine (5‟-GGU/C). In each case, only one copy is affected and so the existence of a statistical tRNA population, resulting in a low level of mistranslation was proposed. After comparison with
mutD mutations, it was suggested that the low mistranslation level might be due to
the action of an error-prone polymerase. The mutD/dnaQ gene mutations are implicated in the impairment of polymerase III (pol III) editing function, particularly the ε subunit, and implicate also the loss of an aspartic acid. DNA pol III was later established to be error-prone (Al Mamun et al., 2002; Dorazi, 2002). However, the exact changes of pol III are unknown.
A mutator phenotype was also observed when cells were exposed to streptomycin, a translation inhibitor (Ren et al., 1999). Later on, using this same stressor, it was proven that mistranslation in general could induce TSM (Balashov and Humayun, 2002), possibly through enhanced protein misfolding and turnover, and culminating in the expression of an error-prone DNA polymerase (Dorazi et
al., 2002).
Finally, there is evidence in the literature supporting the appearance of other mutator phenotypes due to translational stress (Connolly and Winkler, 1989; Connolly and Winkler, 1991).
16
5. Antifungal drug resistance in fungi
Microorganisms‟ resistance to drugs is a growing problem nowadays mainly as a consequence of treatment failures. In the present thesis we focused on antifungal drug resistance, in particular resistance to azoles, using S. cerevisiae as a model system. To date, most of the available information on this topic concerns the genus Candida and the antifungal fluconazole. Besides these two, other genera will only be mentioned briefly.
In the mid-80‟s a significant amount of reports on antifungal drug resistance emerged. These concerned mainly resistant clinical isolates of Candida albicans, that were attributed to prolonged treatment with miconazole and ketoconazole (Sanglard et al., 1998). Nowadays about 200 yeast species are associated with humans, which are either commensals or pathogens. Actually, fatal invasive fungal infections are mostly caused by the commensals Candida, Aspergillus and
Cryptococcus, which are opportunistic fungi. The costs associated with treatment
of fungal infections are considerably high, even for the most common infections such as candidiasis. Patients‟ survival (specially for immunocompromised patients) is often time dependent and usually a correct diagnosis needs more than the available time. Therefore, patients are not always treated with the right antifungal from the beginning, thus favoring resistance increment.
Currently, four antifungal classes are mainly used for treatment: echinocandins, fluorinated pyrimidine analogs, polyenes and azoles. Besides these, there are two other less used antifungal classes, the allylamines and the morpholines. The effectiveness of each antifungal depends on the fungus, the dose, the exposure time and the mechanism of action. Two types of drug-induced stresses, the short- and the long-term stress, can contribute to the survival of fungi.
Briefly, echinocandins are lipopeptide molecules that inhibit beta-glucan synthesis by impairing the enzyme complex beta-(1,3)-glucan synthase, located in the cell wall. The first echinocandin, anidulafungin, was discovered in 1974 and more than ten years later, caspofungin and its precursor micafungin were developed (Denning, 2003).
Fluorinated pyrimidine analogs such as 5-fluorocytosine, inhibit DNA and RNA synthesis by mimicking the structure of pyrimidines. This compound enters the cell via a permease and is converted into 5-fluorouracil by a cytosine deaminase. 5-fluoroacil can then be converted into 5-fluorouridylic acid, which is incorporated in RNA after phosphorylation or into 5-fluorodeoxyuridine monophosphate which interferes in DNA synthesis by inhibiting the enzyme thymidylate synthase (Ghannoum and Rice, 1999). It is used frequently in combination with polyenes.
Widely used, polyenes such as amphotericin B, bind to ergosterol, a component of the fungal plasma membrane and thereby cause the formation of membrane-spanning channels. These channels provoke leakage of ions with consequent loss of cell integrity. Resistance to polyenes seems to be related to low ergosterol content, probably due to defective ERG3 function like described for azoles (Cowen and Steinbach, 2008).
17
There are two strong reasons why azole drugs are the most commonly used antifungals nowadays: firstly because unlike other drugs such as amphotericin B (polyene), mammalian cells tolerate them quite well and secondly because azoles act against a wide spectrum of fungi. This class comprises two generations named imidazoles (first generation, e.g.: ketoconazole and miconazole) and triazoles (second generation, e.g.: fluconazole and itraconazole). The first generation comprises, among others, miconazole, used in the current study, and ketoconazole. The primary target of azoles, is the ergosterol biosynthesis, in particular the cytochrome P450 Erg11p or lanosterol-14α demethylase (CYP51) encoded by the Erg11 gene. Azoles mimic lanosterol and compete for enzyme binding. This step is rate-limiting in ergosterol biosynthesis (figure 10). Ergosterol is a key component of fungi plasma membranes, analogous to mammalian cholesterol.
Figure 10 - Part of the ergosterol pathway. Squares indicate azoles‟ target enzyme and the gene encoding it. Adapted from Bammert and Fostel (2000).
18
5.1 Resistance Mechanisms
Luckily, fungi do not generally spread their resistance determinants like bacteria, nor mate often. Also, resistant isolates in human patients have never been reported to pass to other hosts. Still, several resistance mechanisms are known and the combination of two or more can result in synergy, antagonism or addition (Anderson, 2005).
Membrane or osmotic stresses are short-term consequences of azole exposure, as cells are more and more depleted in their fundamental membrane molecule ergosterol. Briefly, such stresses can be fought by activation of conserved signaling pathways like the mitogen-activated protein kinase (MAPK) and the cyclic AMP-protein kinase A pathway (reviewed in Roman et al., 2007).
Further, the serine/threonine protein phosphatase calcineurin, has an opposite mechanism of action, and is known to play a role in membrane stress. Its inhibition in C. albicans turns azoles from fungistatic into fungicidal drugs. Acting in the same signaling pathway as calcineurin, heat shock protein 90 (Hsp90) was also shown to contribute to fluconazole resistance in S. cerevisiae and C. albicans (Cowen et al., 2006; Cowen and Lindquist, 2005). However, the precise mechanism behind azole tolerance remains to be discovered.
In most fungi, azoles cause growth arrest by reducing the ergosterol content of membranes and by accumulating toxic ergosterol precursors (e.g. 14α-methylergosta-8,24(28)-dien-3β,6α-diol). However, there are species for which some azoles are fungicidal (e.g. Arpergillus fumigatus) (Espinel-Ingroff, 2001; Manavathu et al., 1998; Manavathu et al., 2000). Resistance to azoles seems to be acquired due to multiple mechanisms. These are, among others, overexpression or point mutations (confirmed to reduce fluconazole binding) of
ERG11, efflux via ATP-binding cassette (ABC) and major facilitator superfamily
(MFS) transporters, tolerance to methylated sterols via mutation in ERG3, stress tolerance induction and aneuploidy (Cannon et al., 2009).
Figure 11 – Azole resistance: Three main forms of resistance to azoles can result from the upregulation of ABC and MFS transporters that remove the drug from the cell, through the mutation or overexpression of Erg11, which minimizes the impact of the drug on the target, or alterations in ergosterol biosynthesis, such as the loss-of-function mutation of Erg3, which blocks the accumulation of a toxic sterol intermediate that is produced when Erg11 is inhibited by azoles (Cowen, 2008).
19
A gain of fitness is often associated with the development of azole resistance and therefore resistant isolates may persist in the cell population after the cessation of azole therapy. Evolution of resistance strongly depends on the population size, as each propagule (any fungal structure capable of dissemination) represents an independent possibility of resistance acquisition (Anderson, 2005).
Yet to be precisely determined, certain levels of overexpression of drug efflux pumps, present in the plasma membrane, could reduce azoles to non toxic levels (Cannon et al., 2009). Lately, great interest has turned to these efflux pumps, especially in order to understand how these pumps bind and transport substrates, and to determine their “regular” substrates. Therefore, they will be described in more detail below.
5.2 ABC and MFS transporters
ABC proteins are primary transporters that require ATP hydrolysis and can be found in every organism‟s plasma membrane. Their basic structure consists of two nucleotide binding domains (NBD) and two transmembrane domains (TMD), which are arranged accordingly to the type of ABC protein. In S. cerevisiae ABC proteins can be divided in three subfamilies: pleiotropic drug resistance (PDR), multidrug resistance2 (MDR) and multidrug resistance-associated protein (MRP). The PDR subfamily is most frequently associated to antifungal resistance, with S.
cerevisiae‟s Pdr5p considered as an archetype.
As S. cerevisiae cells contain numerous ABC genes, a new system (Decottignies et al., 1998) was developed in a mutant species, which allowed background reduction. Using this system it was demonstrated that the genes
YOR1 and PDR5 share miconazole as common a substrate. A derived system
overexpressing CaCdr1p3 showed increased itraconazole resistance (1,000-fold) (Lamping et al., 2007).
2
The term Multidrug Resistance is also used as a broader concept, covering other subfamilies, such as PDR and MDRp.
3 CaCdr1p – “Ca” stands for Candida albicans, the “p” usually stands for protein.
20
Clinical isolates of C. albicans were found to have increased levels of CDR genes (Perea et al., 2001), known to confer resistance to multiple azoles. Disruption of CaCDR1 led to hyper susceptibility to azoles and deletion of both
CaCDR1 and CaCDR2 provoked even higher susceptibility (Sanglard et al., 1996;
Sanglard et al., 1997). CaCDR1 overexpression in S. cerevisiae was found to confer cross-resistance to different azoles, including fluconazole, itraconazole and ketoconazole (Sanglard et al., 1995). Regarding CDR proteins, there is evidence that CaCdr1p, 2p and 3p are involved in phospholipid transport, having some influence in membrane leaflets composition. Thus, it was suggested that overexpression can indirectly contribute to antifungal resistance (e.g. effects on membrane function or membrane protein activity). Further, C. glabrata azole resistance is also associated with PDR ABC pumps (CgCdr1p and CgCdr2p).
C. dubliniensis is another organism for which the genes CdCDR1 and CdMDR1 (MFS transporter) have been implicated in azole resistance. Contrary to C. albicans that preferentially uses CaCDR1 for drug efflux, CdMDR1 is directly
linked to fluconazole resistance. CdCDR1, is not as commonly used, although it has been found to efflux ketoconazole and itraconazole and its deletion showed increased susceptibility to both azoles (for more detailed information see Sullivan et al., 2004).
MFS pumps belong to protein superfamilies, that are widespread across phylogenetic domains. Their expression reveals a much weaker association with azole resistance than ABC transporters. They are secondary transporters, containing only TMDs that require a proton gradient and can be divided in two subfamilies regarding the number of transmembrane spans (TMS): DAH1 (12 TMS) and DHA2 (14 TMS).
The first MFS gene characterized was C. albicans’ CaMDR1. Later, the same gene was identified in fluconazole resistant mutants (Albertson et al., 1996) and clinical isolates of C. albicans (Perea et al., 2001; White, 1997). The same clinical isolates also showed increased levels of the ERG11 gene (White, 1997) as
Figure 13 - Arrangement of a MFS transporter in C. albicans ans A. fumigatus. (Cannon et al., 2009)
21
well as increased resistance to itraconazole, voriconazole, and posaconazole (Perea et al., 2001) Overexpression of CaMDR1 in S. cerevisiae conferred, I addition to fluconazole resistance, ressitance to ketoconazole (Lamping et al., 2007), while only very high levels of overexpression led to fluconazole resistance in clinical C. albicans isolates (Hiller et al., 2006). Additionally, CaMDR1 confers resistance to other drugs (e.g. cerulenin and brefeldin A).
Both types of efflux pumps are related to transcriptional factors that act as regulators. While exposed to antifungals, these can be subjected to gain-of-function (point) mutations. Elements acting upon the PDR ABC subfamily of S.
cerevisiae are among the best studied. Gain-of-function mutations have been
identified in the genes of two transcription factors, ScPdr1p and ScPdr3p (Carvajal et al., 1997; Nourani et al., 1997). ScPdr1p is required to induce compensatory upregulation of efflux pumps if individual efflux pump genes are deleted (e.g.
ScPDR5) (Kolaczkowska et al., 2008).
Also, aneuploidy might play a role in azole resistance. A duplicated arm of the chromosome 5 of C. albicans clinical isolates was discovered to contain duplicates of the transcription factor gene CaTAC1, which regulates CaCDR1 and
CaCDR2 (Selmecki et al., 2006). Transcriptional control mechanisms of the genus Candida, seem to be similar to those of S. cerevisiae. Finally, one other gene, CAP1, similar to S. cerevisiae‟s YAP1, was expressed in the latter and it was
shown that it could activate the transcription of FLR1, which has functional similarity to the C. albicans MDR1 gene (Alarco et al., 1997).
Solutions to efflux-mediated resistance are currently under investigation. Proposed solutions so far comprise 1) the use of antifungals that are not substrates of efflux pumps, 2) the development of systems that prevent efflux, 3) removal of the energy required by these pumps to function, and 4) attempt to shift the balance between antifungal uptake and efflux. Studies in S. cerevisiae have been carried out to identify pump inhibitors, as these possibly chemosensitize the cells to the antifungals (Cannon et al., 2009). Besides solving efflux-mediated resistance, other approaches under discussion, such as the development of stronger azole molecules and the discovery of new antifungal classes. Still, resistance will not disappear and more strains will evolve resistance more rapidly than expected. Research and drug use have to be directed in a way that channel resistance to a less harmful direction. The first attempt to channeling, currently in use, is combination therapy (two different antifungal drugs used simultaneously or in succession). Also, the development of new research tools (e.g. microarrays) can increase the knowledge available so far, providing more rapidly the information needed to find innovative solutions.
22
Materials and Methods
1. Strains and growth conditions
Mistranslating strains used in this work were constructed previously in our laboratory and are based on the S. cerevisiae BY4743 (MATa/αhis3Δ1/his3Δ1, leu2Δ0/leu2Δ0, LYS2/lys2Δ0, met15Δ0/MET15, ura3Δ0/ura3Δ0) strain. The control strain used has a single-copy plasmid, pRS315, while the mistranslating strains carry the same plasmid with a mutated tRNAUGASer inserted between the BamHI and SalI restriction sites. The tRNAUGASer anticodon was mutated by site-directed mutagenesis, resulting in several other anticodons (Table 1) (Mateus, 2011; Paulo, J. unpublished). Unlike the case of most aaRS, the SerRS recognition mechanism does not depend on the anticodons sequence, so acylation is not blocked.
Table 1 – List of plasmids carried by strains used in this work.
plasmid ID Description
pUA262 pRS315 with Ser-tRNACAGLeu
pUA263 pRS315 with Ser-tRNAGUATyr
pUA265 pRS315 with Ser-tRNACGUThr
pUA266 pRS315 with Ser-tRNACACVal
pUA268 pRS315 with Ser-tRNAUGCAla
pUA269 pRS315 with Ser-tRNAUCCGly
pUA809 pRS315 with Ser-tRNACAUMet
pUA810 pRS315 with Ser-tRNAAGGPro
Figure 14 - Single copy plasmid used for the expression of C. albicans tRNAUGA Ser
in S. cerevisiae BY4743. The UGA serine anticodon was mutated by site-directed mutagenesis to several other anticodons (see table 1)
23
All strains were grown at 30oC in minimal medium without leucine (MM-Leu) (0.67% yeast nitrogen base w/o amino acids, 0.2% amino acid drop out mix w/o leucine, 2% agar, 2% glucose (all from Formedium)).
Stressor agents were added to the medium when required. Miconazole, itraconazole, amphotericin and cycloheximide (all from Sigma) were dissolved in DMSO (Sigma). Geneticin (Formedium) and caspofungin (Merck) were dissolved in Milli-Q water.
2. Growth Curves
Cell cultures of mistranslating strains and the control were grown at 30oC until they reached a late stationary phase and re-inoculated into 10mL of selective media with a starting OD595 of 0.100. At various time points aliquots of the cultures were harvested and OD595 was measured. The growth rates of each mistranslating strain were calculated relative to the control by using exponential phase values. Growth rates were compared with a one-way ANOVA coupled with Dunnett‟s multiple comparison test, with a 95% CI relative to pRS315, using GraphPad Prism software (GraphPad, Inc.).
3. Antifungal susceptibility
In vitro susceptibility was determined according to the guidelines of the EUCAST4 method for the determination of broth dilution minimum inhibitory concentrations (MICs) of antifungal agents (Rodriguez-Tudela et al., 2008). Briefly, for the inoculum, strains were grown on MM plates. 3 to 5 colonies were resuspended in PBS 1x and diluted to an OD530 of 0.15. Sterile 96-well plates containing a two-fold dilution series with twice the final concentrations of miconazole or fluconazole were inoculated with 100μL of cell suspension (3-4replicas) or 100μL of PBS 1x (negative control). The final concentration of the antifungal ranged from 0.125 to 64mg/L. After 24h and 48h of incubation at 30ºC, the most suitable MIC values were determined (OD595).
For all other antifungal susceptibility tests, the Etest® assay for MIC determination was utilized according to manufacturer‟s instructions (Biomerieux). All strains were tested for amphotericin B, caspofungin, itraconazole and flucytosine (except for proline- and methionine-to-serine strains). Therefore cell suspensions were grown in liquid MM-Leu medium until exponential phase and diluted to an optical density of 0.05. 150μL of cell suspension was inoculated onto selective agar plates and spread evenly with glass beads. Plates were incubated at 30oC. MIC endpoints were determined after 24h, 48h and 72h of incubation. A minimum of three independent tests was performed.
4
24
4. Evolution of antifungal drug resistance
Cells grown until exponential phase were inoculated into 15mL Falcon tubes containing liquid selective media and a miconazole concentration of 0.5μg/mL. Once a cell suspension reached the exponential phase it was changed to two new Falcon tubes, one containing twice the last used concentration of miconazole and the other containing the last used concentration as a backup. The experiment was carried on until the cells stopped growing.
5. Development of translational-caused stress along with generation increment
Cells were inoculated into 100mL Erlenmeyer flasks containing 20mL of MM-Leu and ampicillin (100μg/mL, Sigma). As cultures reached the stationary phase they were changed to new media. MIC endpoints for the antifungals caspofungin and amphotericin were determined at the end of the experiment using the Etest® assay.
6. Phenomics of mistranslating strains
This experiment was based on the methods described by Homann (2009) and Kvitek (2008). BY4743 mistranslating strains were grown until exponential phase and 1x107 cells were collected and ressuspended in 1mL of PBS 1x. From this initial dilution, a series of 5 dilutions were made and transferred to 96-well plates. Six ten-fold serial dilutions were plated in MM-Leu agar plates supplemented with the following stressors:
stress compound concentration (μg/mL)
amphotericin B 0,5 caspofungin 0,05 itraconazole 0,25 miconazole 0,05 cycloheximide 0,06 geneticin 75
Cell suspensions were inoculated using a Sciclone liquid handling workstation and plates were incubated at 30oC. After 3 and 4 days photos were taken (Quantity One® software, Biorad) and areas of colonies were measured using Image J software (Abramoff, 2004). Due to the half time of antifungals, only data obtained at day 3 were considered for further analysis. Day 4 measurements were used in the case of the xenobiotic cycloheximide, as colonies sizes were too small to score after 3 days. The growth rates of each strain were calculated by dividing the average area of the triplicates of each strain exposed to the stressor by the average area of the triplicates of each strain grown in MM-Leu only. This ratio was calculated for all measurable dilutions. Statistics were made using GraphPad Prism software (GraphPad, Inc.).
25
7. Microarray analysis
Images of the microarray hybridizations were previously acquired using the Agilent G2565AA microarray scanner (Agilent). Fluorescence intensities were quantified with Quantarray v3.0 software (PerkinElmer). Using R2.2.1 limma software (Smyth and Speed, 2003) and BRB-ArrayTools v3.4.0 (developed by Dr. Richard Simon and BRB-ArrayTools Development Team), log2 intensity ratios were median normalized, to correct for differences in genomic labeling efficiency between samples. One-class (to compare each mistranslating strain with the control) and multiclass (for comparison between mistranslating strains) Significance Analysis for Microarrays (SAM), with a false discovery rate (FDR) <0.001, were performed using MultiExperiment Viewer (MeV) 4.7.4 software (Saeed et al., 2003). Individual hybridizations of two clones with two dye-swaps were used as the input data for each strain.