Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Patterns of T cell co-stimulation and co-inhibition in
tumor microenvironment conditions, in vitro
Dissertação de Mestrado orientada por:
Prof. Dra. Maria Margarida Telhada
Priv.Doz. Dr. med. habil. Frank Momburg
Dissertação
Luana Frias Guerra
Mestrado em Biologia Molecular e Genética
2015
Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Patterns of T cell co-stimulation and co-inhibition in
tumor microenvironment conditions, in vitro
Dissertação de Mestrado orientada por:
Prof. Dra. Maria Margarida Telhada
Priv.Doz. Dr. med. habil. Frank Momburg
Dissertação
Luana Frias Guerra
Mestrado em Biologia Molecular e Genética
2015
I
Acknowledgements
Aos meus pais, um obrigada imenso por me darem as ferramentas sem o livro de instruções ou o livro de instruções sem as ferramentas, mas nunca os dois em simultâneo.
À minha irmã, por ser parte de mim.
À família de cada dia, aos amigos de sempre e aos que se foram juntando a esse sempre, ao longo do caminho, os que me inspiram e guiam. Não seria metade sem vocês.
To my new friends, the ones that I was lucky to meet and share each moment of my new life, the reason why these months were pure bliss.
To Frank, who gave me the opportunity to work on what I am passionate about, for his guidance and insight.
To Alexander and Mostafa, who were utterly patient and without whom I would have not been able to complete this work.
À professora Margarida Telhada, por ter aceitado ser minha orientadora e pela sua disponibilidade e apoio.
E, finalmente, mas como se viesse em primeiro, à professora Margarida Gama Carvalho, por me ter iniciado nesta caminhada e por continuar presente e a acreditar.
II
Abstract
T cells (helper and cytotoxic) are present within the tumor microenvironment and constitute viable targets for immunotherapeutic approaches against cancer, either by adoptive T cell transfer or by the use of monoclonal antibodies. The activation, differentiation, function and survival of those T cells comprehends a complex process and it is regulated by co-stimulatory and co-inhibitory receptors which modulate the initial T cell receptor (TCR) signal, increasing or hindering their anti-tumor function, respectively. Furthermore, various factors secreted by tumor cells or non-lymphoid stromal cells are believed to mostly downmodulate T cell responses.
In this work the patterns of expression of co-signaling molecules on T cells were assessed using peripheral blood mononuclear cells (PBMCs) from healthy donors cultured with transforming growth factor (TGF-β), in order to mimic one of the major immunosuppressive stimuli of the tumor microenvironment. Noteworthy, all herein reported effects of TGF-β were entirely dependent on the subsequent T cell activation by an antibody against the CD3ε chain that is part of the TCR complex. While co-inhibitory receptors such as CTLA4 and PD1 were upregulated, PD-L1, TIGIT and TIM3 (also co-inhibitory) had their expression decreased. The transcription factor FOXP3, associated with the regulatory T cell phenotype, was upregulated by TGF-β. For co-activating receptors, the same condition upregulated 4-1BB, CD30 and CD28 expression, whereas it decreased OX40, ICOS and DNAM1 and did not it interfere with CD27 or LIGHT expression. Moreover, alongside TGF-β and anti-CD3, recombinant proteins, such as 4-1BB or OX40 ligand, were added, in order to simulate potential therapeutic strategies using agonistic 4-1BB or OX40 antibodies. It was shown that even though they could induce expression of co-stimulatory receptors such as OX40, CD27 and CD28, there was also CTLA4, PD-1 and TIGIT upregulation.
It seems crucial to unravel the details of the dynamics of the expression of co-signaling molecules on T cells to improve existent immunotherapies and design new approaches able to circumvent the immunosuppressive environment, created not only by tumor or stromal cells, but also by immune cells themselves.
Keywords: cancer, TGF-β, T cells, co-stimulatory receptors, co-inhibitory receptors,
III
Resumo
No microambiente tumoral, estão presentes linfócitos T (auxiliares ou citotóxicos) e estes constituem alvos viáveis em imunoterapias para o cancro, quer por transferência adotiva de linfócitos T, quer pela utilização de anticorpos monoclonais. A ativação, diferenciação, função e sobrevivência destas células T compreende um processo complexo e é regulada por recetores coestimuladores e coinibidores, os quais modelam o sinal inicial do recetor de células T (TCR), amplificando ou diminuindo a sua atividade antitumoral, respetivamente. Adicionalmente, fatores secretados por células tumorais e do estroma podem contribuir para a supressão das respostas das células T.
Assim, o padrão de expressão de moléculas cossinalizadoras dos linfócitos T foi estudado usando células mononucleares do sangue periférico (PBMC) de dadores saudáveis, colocadas em cultura com TGF-β para mimetizar os estímulos supressivos do microambiente tumoral. Todos os efeitos do TGF-β aqui reportados foram dependentes da subsequente ativação das células T por um anticorpo contra a cadeia ε de CD3, que faz parte do complexo TCR. Enquanto recetores coinibidores como CTLA4 e PD-1 tinham a sua expressão aumentada, PD-L1, TIM3 e TIGIT (também coinibidores) encontravam-se subexpressos. O fator de transcrição FOXP3 (associado ao fenótipo de células T reguladoras) estava aumentado na presença de TGF-β. Já no caso dos recetores coestimuladores avaliados, a expressão de 4-1BB, CD30 e CD28 aumentou nas células T, mas OX40, ICOS e DNAM1 encontravam-se subexpressos. Já CD27 e LIGHT não foram afetados pela adição de TGF-β. Adicionalmente, proteínas recombinantes, ligandos de 4-1BB e OX40, foram adicionadas, na presença de TGF-β e anti-CD3, para a simulação de potenciais estratégias imunoterapêuticas. Concluiu-se que, a diferentes níveis, apesar de se ter conseguido induzir a expressão OX40, CD27 e CD28 nas células T, a expressão de CTLA4, PD-1 e TIGIT estava também aumentada.
Revelar os detalhes da dinâmica da expressão de moléculas cossinalizadoras em linfócitos T é crucial para aperfeiçoar imunoterapias existentes e criar novas abordagens capazes de minimizar os efeitos dos estímulos imunossupressivos do ambiente tumoral, criado não apenas pelas células cancerígenas e do estroma, mas também pelas próprias células do sistema imunitário.
Palavras-chave: cancro, TGF-β, linfócitos T, recetores coestimuladores, recetores
IV
Resumo
O cancro é uma doença caracterizada por um crescimento anormal e não controlado de células que, ao sofrerem mutações no seu DNA, adquirem características que lhes conferem uma vantagem adaptativa de sobrevivência.
A função do sistema imunitário é contribuir para a homeostasia dos tecidos e, assim, os linfócitos T exercem um papel crucial no cancro ao reconhecerem e eliminarem células transformadas.
A ativação, diferenciação, função e sobrevivência das células T é um processo descrito por um modelo de dois sinais. Isto é, há uma ativação primária através da ligação do recetor de células T (TCR) ao complexo MHC (major histocompatibility complex) e este sinal é complementado por recetores coestimuladores e coinibidores. São estas moléculas que, colocalizadas com o complexo TCR-MHC na sinapse imunológica, ditam o destino das células T ao modular o sinal do TCR – amplificando o sinal de ativação (estimuladores) ou diminuindo-o (inibidores). O repertório destas moléculas cossinalizadoras é altamente versátil, temporal e espacialmente, e responde a mudanças no ambiente dos diferentes tecidos.
Os recetores coestimuladores e coinibidores pertencem, na sua maioria, a duas superfamílias: IgSF (immunoglobulin superfamily) e TNFRSF (tumor necrosis factor receptor superfamily). À IgSF pertencem correcetores estimuladores como CD28 e outros coinibidores, como TIGIT (T cell immunoreceptor with immunoglobulin and ITIM domains), para mencionar alguns exemplos. Já a superfamília TNFRSF, compreende membros como 4-1BB ou OX40, ambos coestimuladores.
Dada a capacidade destas moléculas cossinalizadoras modularem a ativação dos linfócitos T no microambiente tumoral, estes constituem já alvos clínicos de imunoterapias contra o cancro, como é o caso dos anticorpos monoclonais anti-CTLA4 (cytotoxic
T-lymphocyte-associated antigen) e anti-PD1 (programmed death protein 1), recentemente aprovados para
o tratamento de melanoma metastático. Ambos são recetores coinibidores e os anticorpos antagonistas utilizados diminuem o fenótipo imunossupressor, nomeadamente ao impedirem a ativação de células T reguladoras (Treg). No entanto, as possibilidades terapêuticas não se esgotam aqui. Com a variedade de recetores descritos, há a hipótese de criar novos tratamentos, recorrendo a combinações entre diferentes alvos, ou apenas melhorar a eficácia
V e o perfil de toxicidade das terapias existentes. Para isso, é crucial conhecer detalhadamente o fenótipo de ativação e inibição dos linfócitos T.
Assim, o objetivo deste trabalho é estudar os padrões de expressão de recetores coestimuladores e coinibidores presentes em células T, inseridas num microambiente tumoral,
in vitro.
As células tumorais encontram-se, normalmente, num ambiente imunossupressor. Produzem citoquinas, como TGF-β (transforming growth factor β), IL-10 ou prostaglandinas, que recrutam células imunossupressoras - Tregs ou MDSC (myeloid derived suppressor cells) - as quais impedem a função citotóxica das células T.
A estratégia experimental consistiu na utilização de células mononucleares do sangue periférico (PMBC) isoladas de sangue de dadores saudáveis. Estas células foram pré-cultivadas com TGF-β recombinante humano (rh), de modo a mimetizar o microambiente tumoral. Posteriormente, adicionou-se um anticorpo anti-CD3ε, a cadeia ε do complexo TCR, para que se desse a ativação policlonal das células T. A expressão de recetores (coestimuladores e coinibidores) na superfície dos linfócitos T foi posteriormente avaliada por citometria de fluxo. É de referir que todos os efeitos do TGF-β aqui descritos foram dependentes da adição da ativação dos linfócitos T pela adição de anti-CD3.
O ambiente supressivo foi criado in vitro: a presença de TGF-β induziu a expressão do fator de transcrição FOXP3 nas células T CD4+, o que está associado ao fenótipo de células T reguladoras.
Observou-se que a expressão dos recetores coinibidores PD1 ou CTLA4 se encontrava aumentada na presença de TGF-β, mediante a estimulação do TCR pela adição de anti-CD3, o que se coaduna com o perfil de exaustão dos linfócitos T no ambiente pro-inflamatório criado. Por outro lado, os correcetoresTIM3 (T cell immunoglobulin and mucin protein 3), TIGIT (T cell immunoreceptor with Ig and ITIM domains), também coinibidores, encontravam-se subregulados, enquanto a expressão de PD-L1 (programmed death-ligand 1) não foi modificada pela presença de TGF-β, apesar de TIM3, TIGIT e PD-L1 estarem descritos como sobrexpressos em linfócitos T infiltrados em tumores (TILs).
Já no que diz respeito a recetores coestimuladores, observou-se que membros da superfamília de TNFR, 4-1BB, CD30, LIGHT OX40 e CD27, têm uma sensibilidade distinta ao ambiente supressor criado in vitro. A expressão de 4-1BB e CD30 aumentou na presença de TGF-β e
VI anti-CD3, apesar de OX40 ter a sua expressão diminuída e LIGHT e CD27 não terem respondido à presença de TGF-β.
CD28, um dos principais recetores coestimuladores de células T, promove a ativação e sobrevivência destas células através do aumento da produção de IL-2. Com a pré-cultura dos PBMCs com TGF-β e adição do anticorpo anti-CD3, CD28 aumentou a sua expressão na superfície dos linfócitos T. Já ICOS (inducible T cell costimulator), também um recetor coestimulador, estruturalmente relacionado com CD28, diminuiu ligeiramente a sua expressão nas mesmas condições. A expressão de ICOS, ao contrário de CD28, não induz a superprodução de IL-2, mas sim de IL-10, descrita como pro-tumoral, podendo amplificar o ambiente inflamatório criado pelo TGF-β.
DNAM1 (ou CD226), recetor coestimulador, na presença de TGF-β e anti-CD3, também diminuiu a sua expressão.
Adicionalmente, para avaliar o efeito de potenciais imunoterapias que tenham como alvos moléculas cossinalizadoras das células T, foram adicionadas, na presença de anti-CD3 e após uma pré-cultura com TGF-β, proteínas recombinantes humanas, ligandos de recetores coestimuladores da superfamília de TNFR – rh4-1BBL e rhOX40L, os quais promovem a sobrevivência das células T. Sumariamente, observou-se que estas proteínas recombinantes induziram a expressão do recetor coestimuladores OX40, CD27 e CD28, apesar de TIGIT, CTLA4 e PD-1 e (coinibidores) terem também a sua expressão aumentada.
Evidentemente, o próximo passo deste trabalho seria realizar um estudo funcional. Avaliar-se-iam as citoquinas produzidas pelos linfócitos T e a sua relação com os padrões de expressão dos correcetores. Adicionalmente, poder-se-iam fazer, após a avaliação da expressão dos correcetores, ensaios de citotoxicidade, com diferentes tipos de células tumorais como alvo e, assim, relacionar a expressão destes com a eficácia das células T para cada tipo de tumor. Seria, também, de grande importância estudar a expressão dos mesmos recetores coestimuladores e coinibidores ex vivo, em TILs de doentes com cancro.
Concluindo, a soma destes resultados evidencia a importância de se conhecer o perfil de ativação/inibição das células T. Conhecer o padrão de expressão das moléculas cossinalizadoras – e poder compará-lo com o outcome funcional, anti- ou pro-tumoral, destes linfócitos – é uma ferramenta importante, não só no que toca a combinações terapêuticas, mas também, no que diz respeito a terapias específicas dirigidas a cada tipo de tumor, ou mesmo na medicina personalizado.
Index Acknowledgements ... I Abstract ... II Resumo ... III Resumo ... IV Introduction ... 2 Cancer - Hallmarks... 2
Cancer and the Immune System – a Paradox ... 3
T lymphocytes - co-stimulation and co-inhibition ... 5
Cancer Immunotherapy - Targeting T cell co-signalling ... 6
Aims ... 9
Materials and Methods ... 10
Isolation of Peripheral Blood Mononuclear Cells ... 10
PBMCs Culture ... 10
Flow Cytometry... 10
Results ... 12
A. Effect of TGF-β on the phenotype of CD4+ T cells from healthy donors’ PBMCs ... 12
B. Effect of TGF-β on co-stimulatory receptors expression on T cells from healthy donors’ PBMCs ... 13
B.1. Effect of rhOX40L and rh4-1BBL on the expression of co-stimulatory receptors on activated T cells cultured with TGF-β ... 16
C. Effect of TGF-β on co-inhibitory receptors expression on T cells from healthy donors’ PBMCs ... 20
C.1. Effect of rhOX40L and rh4-1BBL on the expression of co-inhibitory receptors on activated T cells cultured with TGF-β ... 22
Discussion and Conclusion ... 24
References ... 30 Annex ... A1 Index of Figures Figure 1 ... 13 Figure 2. ... 16 Figure 3. ... 19 Figure 4. ... 21 Figure 5. ... 23 Supplemental Figure 1. ... A2 Supplemental Figure 2 ... A3
2
Introduction
Cancer - Hallmarks
According to the World Health Organization, cancer is a leading cause of morbidity and mortality worldwide. In 2012, 14 million new cases were diagnosed and this number is expected to increase by 70% over the next two decades.3
Cancer is a disease characterized by an uncontrolled growth of transformed cells. Normal cells have a synchronized cell cycle which guarantees the homeostasis of cell number and, therefore, ensures the normal architecture and function of the tissue. On the contrary, due to a deregulation of the production and release of growth-promoting signals, cancer cells have the ability to sustain chronic proliferation. 4
The signals which orchestrate the entry into and the progression through the cell cycle are mainly growth factors that bind to receptors on the cell surface. Thus, this sustained proliferative signal might arise in different ways.4,5, For instance, transformed cells are able
to produce growth factor ligands that bind to cognate receptors on their own surface, which creates an autocrine loop.6 Moreover, cancer cells may stimulate normal stromal cells in the tumor microenvironment and those respond by providing additional growth factors.7 The rising in the number of receptor proteins expressed on the cancer cell surface or alteration of their structure (enabling ligand-independent signaling) constitute further ways of transformed cells to reach the threshold of proliferative signals with otherwise-limiting amounts of growth-factor ligand. A constitutive activation of components downstream of these receptors on the signaling pathways avoids the need of ligand-mediated receptor stimulation, which makes cancer cells independent of the stimuli of the surroundings.8
In addition to being able to induce and sustain proliferation, cancer cells are also programmed to evade growth suppressor mechanisms, many of them dependent on the action of tumor suppressor genes, such as retinoblastoma-associated (RB) and TP53 proteins.9,10 It is important to mention that the growth inhibition signal provided by cell-to-cell contact is abrogated throughout the tumorigenic process, which culminates in the loss of tissue integrity and contributes to the epithelial-to-mesenchymal transition (EMT).11-13
The activation of the EMT program by transformed cells is one of the key events in cancer progression, regulating invasion and metastasis.14 Epithelial cells, by activating a number of
3 of traits which allows them to invade local tissues and enter the blood or lymphatic vessels, to colonize distant tissues. Those cells loose adherens junctions and, as a consequence, their morphology is reshaped, they express matrix-degrading enzymes, increase their motility and become more resistant to apoptosis.16 However, the EMT program is not merely regulated in a cell-autonomous way17: crosstalk between cancer cells and cells from the stroma (such as tumor associated macrophages18, mesenchymal stem cells19 and fibroblasts20) are also implicated in the development of invasive characteristics.
It is clear that plasticity is one of the most important traits of high-malignancy tumors: cancer cells ought to reverse the mesenchymal traits so that they can form new tumor colonies for metastatic dissemination.21
Complementary to the capacity of maintaining a proliferative signaling, cancer cells are also resistant to cell death. These cells can circumvent apoptosis in several ways: loss of tumor suppressor genes, e.g. TP53 22, downregulation of proaptotic factors, e.g. Bax, or increased expression of antiapoptotic proteins, e.g. Bcl-2 23, and survival signals, e.g. Igf 1/224, and by hindering the ligand-induced death pathway, e.g. Fas/FasL25.
Moreover, when necrotic cell death occurs, it is a pro-inflammatory process, which might contribute to tumorigenesis as inflammatory cells (and the necrotic cells themselves) are able to induce proliferation, invasiveness and angiogenesis.26
It is worth mentioning that the outgrowth of tumors is facilitated by the fact that cancer cells have unlimited replicative potential due to the overexpression of telomerase, which enables them to resist senescence and apoptosis.27
This outgrowth is sustained by the angiogenic switch which occurs during tumor development.28 There is an increase of proangiogenic stimuli. Namely, vascular endothelial growth factor (VEGF) expression is upregulated, inducing neovascularization.29 The switch can occur via oncogenes that upregulate angiogenic factors28 or there might be an induction through immune inflammatory cells30. The formation of new vessels allows the cancer cells to have increased availability of nutrients essential for their growth.
Cancer and the Immune System – a Paradox
The mammalian immune system has evolved to maintain homeostasis of the tissues: to ensure protection against foreign pathogens while remaining tolerant to self-antigens.
4 Innate immune cells (dendritic cells (DCs), natural killer (NK) cells, macrophages, neutrophils, basophils, eosinophils and mast cells) constitute the first line of defense. When tissue homeostasis is disturbed, DCs, macrophages and mast cells (which are distributed through the different tissues) rapidly release soluble inflammatory mediators (cytokines, chemokines, proteases, reactive oxygen species (ROS) and histamine) to recruit more leukocytes to the damaged tissue. DCs take up foreign antigens and migrate to lymph nodes in order to present the antigens to adaptive immune cells.
As T and B cells are antigen-specific, the recognition of the cognate antigen presented by DCs and other professional antigen-presenting cells (APCs) in the lymph nodes results in clonal expansion of the lymphocytes specific to fight the threat. A subset of these lymphocytes differentiate to have a long-lasting memory phenotype, which guarantees a faster and more efficient response upon subsequent exposure to the same antigen.31
Although the immune system should eliminate cancer cells, it may play a counterintuitive role in tumor progression, as mentioned before. By promoting an inflammatory state within the tumor microenvironment, immune cells may supply the cancer cells with growth factors that maintain proliferation, survival factors that hamper cell death and proangiogenic factors and extracellular matrix-degrading enzymes that enable invasion and metastasis. 26, 32-35
Furthermore, cancer cells create their own way of evading immune destruction. They are able to secrete tumor growth factor β (TGF-β), IL-10 and prostaglandins36 and recruit
inflammatory cells that have an immunosuppressive phenotype, such as regulatory T cells (Tregs)37 and myeloid derived suppressor cells (MDSCs)38, hindering the actions of cytotoxic lymphocytes.
Additionally, there is a phenomenon named “immunoediting”, where highly immunogenic cells are constantly being eliminated by a competent immune system, leaving mainly the weakly immunogenic cells which have the ability to escape “immune surveillance” (recognition and destruction of transformed cells before they grow into tumors).39, 40
The expression of immune mediators and modulators, the activation state of the different immune cells within the tumor and the communication between the different cells that compose the tumor microenvironment are the features that dictate whether there is tumor promoting inflammation or if anti-tumor immunity will triumph.41
5 The most frequent immune cells invading the tumor microenvironment are tumor-associated macrophages (TAMs) and T cells.
TAMs are mainly associated with tumor growth, angiogenesis, invasion and metastasis18 and,
thus, high rate of TAMs infiltration is related with poor prognosis. 42
Mature T cells are divided into two major groups, based on the receptor (TCR) they express: γδ and αβ. αβ T cells have an additional classification according to their effector functions: CD8+cytotoxic T cells (CTLs) and CD4+ helper T cells (TH). The TH subsets include Th1,
Th2, Th9, Th17, Th22, natural killer T cells (NKT) and Treg cells.31 Dependent on their effector functions, T cells may have tumor-suppressive or tumor-promoting activity. On one hand, infiltration of the tumor by CD8+ T cells, Th1, Th2 and Th17 cells might be contributing to cancerogenesis.43-45 But, on the other hand, there are reports that increased CTL and Th1 cell numbers can also correlate with better survival in some cancers.46,47
T lymphocytes - co-stimulation and co-inhibition
CD8+ CTLs can exert their function in cancer by recognizing and killing potentially malignant cells that express tumor antigens (mutant cellular or oncogenic viral proteins) in association with class I MHC molecules. However, in most cases, specific responses of CTLs require cross-presentation of tumor antigens by professional APCs, such as dendritic cells, given the fact that co-stimulatory molecules necessary for T cell activation are normally expressed on APCs but not on cancer cells.48
T cell activation, subset differentiation, effector function and survival are orchestrated by the sum of two different signals –the “two-signal model” of T cell activation. The specific recognition through TCR of cognate antigenic peptides presented by MHC molecules on APCs is crucial. Nevertheless, co-signaling receptors (cell surface molecules) are required to transduce signals into T cells by positively (co-stimulatory) or negatively (co-inhibitory) modulating TCR signaling. These co-signaling receptors often co-localize with TCR molecules at the immunological synapse and co-stimulatory and co-inhibitory molecules are often expressed at the same time.49
The repertoire of co-signaling receptors expressed on T cells is extremely versatile and responsive to changes in the tissue environment. Co-signaling ligands and counter-receptors are most well characterized on APCs, as those are the primary drivers of T cell activation and differentiation in lymphoid organs.50,51
6 The B7-CD28 interaction is the paradigm of the two-signal model. CD28 is constitutively expressed on the cell surface of naïve CD4+ and CD8+ T cells and is an essential co-stimulatory molecule for T cell growth and survival upon ligation by B7-1 (CD80), B7-2 (CD86) and B7-H2 (expressed on activated APCs). On the other hand, there is a co-inhibitory molecule, cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), which is induced upon T cell activation that binds the same ligands as CD28. When CTLA4 expression is increased, CD28 is downregulated and hence CTLA4 (which has greater affinity for B7 ligands than CD28) interacts with the cognate counter-receptors and induces their trans-endocytosis, abrogating co-stimulatory signaling and T cell responses.52
T cell activation is a highly dynamic process which provides multiple levels of spatiotemporal regulation in order either to promote responses against non-self antigens or to limit aberrant and autoreactive T cell responses. This regulation can occur at many levels: modulation of cell surface expression, differential expression patterns of receptor-ligand pairs or distinct interaction through multiple interfaces (reflects binding competition).50, 53
Most co-signaling molecules belong to the immunoglobulin superfamily (IgSF) and tumor necrosis factor receptor superfamily (TNFRSF). 50
IgSF includes members from the families: CD28 and B754, type I transmembrane (or T cell)
immunoglobulin and mucin (TIM)55 as well as CD2/signaling lymphocytic activation
molecule (SLAM)56. Lymphocyte activation gene 3 protein (LAG3)57 and T cell
immunoreceptor with immunoglobulin and ITIM domains (TIGIT) are co-inhibitory molecules that belong to IgSF. On the other hand, DNAX accessory molecule 1 (DNAM1, CD226) and cytotoxic and regulatory T cell molecule (CRTAM) are examples of co-stimulatory members of the IgSF.58
TNFRSF comprises also a broad range of members: herpes virus entry mediator (HVEM), death receptor 3 (DR3), CD40, 4-1BB, OX40, glucocorticoid-induce TNFR-related protein (GITR) are examples of elements with a co-stimulatory function that synergize with TCR signaling to promote cell cycle progression, cytokine progression and T cell survival.59
Cancer Immunotherapy - Targeting T cell co-signaling
Mobilizing T cells for cancer therapies constitutes a particularly compelling method given the fact that T lymphocytes exert an antigen-specific function, are able to differentiate into a
7 memory phenotype and their response is adaptable and thus can accommodate tumor heterogeneity.60
T cell co-inhibitory molecules can also be named “immune checkpoints” and the expression of these immune checkpoints can be dysregulated by tumors as an important mechanism of immune evasion. The inhibitory ligands and receptors that regulate T cell effector functions (and not activation) are usually overexpressed on tumor-cells or on other cells in the tumor microenvironment. Membrane bound receptor-ligand co-signaling molecules are “druggable” by the use of agonistic antibodies to enhance co-stimulatory pathways and antagonistic antibodies to hinder inhibitory signals.61
CTLA4 was the first co-inhibitory receptor to be clinically targeted. It is exclusively expressed on T cells, thus the strategy had the potential to work in different tumors. It seems to play a major role not on activated CD8+ T cells but on the CD4+ subset – decreases helper T cell activity and enhances Treg immunosuppressive function (as CTLA4 is linked to the transcription factor FOXP3).62 Ipilimumab (anti-CTLA4 monoclonal antibody) was approved by the US Food and Drug Administration (FDA) for the treatment of advanced melanoma in 2011.63 Alongside with the mean survival benefit, blocking of CTLA4 has a critical effect on long-term survival, with recent reports stating a survival of ten years or more for a subset of patients.64 The finding of ongoing responses and survival after the completion of the treatment support the concept that immune-based therapies can re-educate the immune system and enhance immune surveillance.60
It is important to mention that, in contrast with conventional chemotherapeutic agents, response to immune checkpoint blockers is slower and delayed (up to 6 months after the beginning of the treatment).61,65
Another co-inhibitory molecule-blocking strategy was recently approved by the FDA. Pembrolizumab and nivolumab, antagonistic antibody against programmed death protein 1 (PD1), are being used in patients with metastatic melanoma and refractory or metastatic lung cancer, respectively.66,67
PD1 is expressed on activated T cells. Its major role is to limit the activity of T cells in peripheral tissues but only at the time of an inflammatory response. PD1 engagement with its ligands (PDL1 and PDL2, both members of the B7 family) inhibits the TCR signal, which can decrease the duration of T cell-APC or T cell-target cell contact, downmodulating T cell activity.68, 69 PD1 is highly expressed on the Treg subset and its blocking is crucial in tumors
8 vastly infiltrated with these type of cells.70 PD1 is more broadly expressed than CTLA4,
therefore its blocking might also have effects on NK and B cells activity, stimulating the production of antibodies specific for the tumor.71,72
PD1 and CTLA4 regulate different inhibitory pathways on T cells. In this way, a combinational therapy using antibodies targeting both molecules has already advanced for phase I clinical trials and the reports claim tumor regression in 50% of the melanoma patients treated. 73
The blockade of the CTLA4 and PD1 axis is claimed to be the tip of the iceberg in the realm
of potential targets that can enhance anti-tumor responses.60 Several other immunological pathways on T cells may be targeted (as monotherapy or in combination) for cancer treatment, not only the inhibitory but also co-stimulatory molecules.
For LAG-3 (a co-inhibitory molecule), there is a fusion protein and an antibody in clinical trials with some promising results.74 TIM-3 (co-expressed with PD1 on tumor-infiltrating lymphocytes) has been subject to preclinical studies, where it is shown that combinational therapies aiming these two pathways improves anti-tumor immunity.75 Other co-inhibitory targets are being evaluated, such as B7-H3 (already in phase I clinical trial)76, B7-H477 or V-domain Ig-suppressor of T cell activation (VISTA)78.
With regard to co-stimulatory molecules, efforts are being directed into agonistic antibodies targeting OX40 (already in phase I clinical trial, with evidence of anti-tumor responses), 41BB (with phase I/II studies in multiple cancers), both with an acceptable safety profile.79,80 Finally,
inducible co-stimulator ICOS, a member of the CD28/B7 family and whose expression is increased upon T cell activation, is also being investigated. It is expected that ICOS can not only serve as pharmacodynamics biomarker to indicate efficacy of anti-CTLA4 targeting (ICOS+ effector T cells are increased upon treatment with anti-CTLA4, but not ICOS+ Treg81) but also provide an important pathway to amplify T cell activation.82
However, as it happens with other cancer therapies, immune checkpoint therapies (the ones clinically tested until now) may have side effects and toxicities, mainly related to inflammatory conditions: dermatitis, colitis, hepatitis and pancreatitis, to name a few. This was expected since this type of therapy does not elicit only tumor-specific responses. So far, it has been manageable with corticosteroid therapy which does not seem to interfere with the clinical benefit of the immune checkpoint blockade.83
9
Aims
In order to increase the efficacy of such therapies, improving their toxicity profile and design new approaches, it is of great importance to understand the subtle mechanisms of T cell co-signaling. It is essential to keep in mind that manipulation of a co-signaling molecule has distinct effects in specific cell subsets.
Therefore, the aim of this work is to assess the pattern of expression of co-signaling molecules present on T cells in conditions of the tumor microenvironment.
Thus, peripheral blood mononuclear cells (PBMC) from healthy donors were used and the addition of recombinant human TGF-β (rhTGF-β) to the cell culture allowed the reproduction of the tumor immunosuppressive environment resembling the tumor milieu where the immune cells phenotype and function are shaped.
Additionally, ligands of the co-stimulatory receptors from the TNFR superfamily, 4-1BB and OX40, were also used to induce co-activation of T cells within the generated suppressive environment. After a pre-culture with TGF-β, 4-1BB and OX40 ligands were added alongside anti-CD3 in order to test if activation of T cells could be enhanced and if it gave rise to a phenotypic change in these lymphocytes.
Evaluating the expression of co-stimulatory T cell receptors such as 4-1BB, CD30, LIGHT, OX40, CD27, ICOS, CD28 and DNAM1 or co-inhibitory surface molecules like TIM3, TIGIT, CTLA4, PD1 and PD-L1 may help to decipher how the stimulation and inhibition of these lymphocytes are finely structured through the modulation of co-signaling receptors. It may clarify which ones are more sensitive to the inhibitory cytokines of the tumor microenvironment or those worth targeting for therapies.
10
Materials and Methods
Isolation of Peripheral Blood Mononuclear Cells
Peripheral Blood Mononuclear Cells (PBMCs) were isolated from 50-100 ml buffy coats collected from healthy donors following gradient centrifugation with Ficoll LymphoprepTM (Axis-Shield, PoC, AS, Oslo, Norway). Cells were washed three times in phosphate-buffered saline (PBS) (Sigma-Aldrich).
PBMCs Culture
Isolated PBMCs were cultured in suspension in serum free X-VIVO 20 medium (Lonza) at high density and recombinant human IL-2 (Proleukin, Novartis) was added at 100U/ml, with a cell density of 2x106 cells/ml.
When stated, PBMCs were pre-cultured with rhTGF-β1 (Miltenyi Biotec, 130-095-067) at 20 or 50 ng/ml for 24h. Under some conditions, it was followed by PBMCs stimulation with anti-CD3ε antibody OKT3 (kindly provided by Dr. Gerhard Moldenhauer, DKFZ)172 at 1 μg/ml
and/or by the co-stimulation with rh4-1BBL, rhOX40L (R&D Systems) at 1 μg/ml both, or by the anti-CD28 antibody 15E8 (kindly provided by Dr. Gerhard Moldenhauer, DKFZ)173 at 1 μg/ml.
Flow Cytometry
Evaluation of the co-signaling molecules expression patterns on T cells was performed in PBMCs samples stimulated under different conditions. Matched combinations of anti-human mouse monoclonal antibodies (mAbs) were used conjugated with Alexa Fluor 488 (AF488), AF647, allophycocyanin (APC), APC-Cy7, Brilliant Violet 500 (BV500), phycoerythrin (PE), PE-Cy7, Violet 450 (V450).
The combinations used were: anti-CD3 conjugated with APC/Cy7 (HIT3a)or AF488 (HIT3a ), CD4 with BV510 (OKT4 ), CD8 with V450 (RPA-T8), 4-1BB (4B4-1), anti-OX40 (Ber-ACT35), anti-CD27 (M-7271), anti-ICOS (C398.4A), anti-CD28 (CD28.2) with PE/Cy7, anti-CD30 (BY88 ), anti-LIGHT (115520*), anti-CTLA4 (L3D10), anti-PD1 (EH12.2H7), TIM3 (F38-2E2) with APC, and PD-L1 (29E.2A3), anti-DNAM1(11A8), anti-TIGIT (MBSA43**) and. To gate out B cells and monocytes together with PI-labelled dead cells, anti-CD19 (HIB19) and anti-CD14 (HCD14) antibodies
11 conjugated with PerCP/Cy5.5 were used. Antibodies were purchased from BioLegend, *Becton Dickinson (BD) or **eBiosciences.
For surface staining, PBMCs were incubated with the antibodies, at 1μg/2x105 cells, for 30
minutes, in the dark, at 4ºC, washed 2x with Fluorescent-Activated Cell Sorting (FACS Buffer). FACS buffer was prepared with fetal bovine serum (FBS) (Biochrom AG) and PBS (Sigma- Aldrich) at 1:50, respectively. Live/dead discrimination was done by using propidium iodide (PI) (Sigma-Aldrich) at 1,6 μg/ml in FACS Buffer.
For FOXP3 intracellular staining the eBioscience FOXP3/Transcription Factor Staining Buffer set (eBioscience) was used after the surface staining. PBMCs were first stained with Live/Dead® Fixable Green Dead Cell Stain Kit (Life Techonologies) at 1 μl/ml FACS Buffer, for 30 min on ice, for live/dead discrimination. Afterwards, PBMCs were fixed during 30 minutes at 4ºC with Fixation/Permeabilization Buffer (eBioscience) followed by Permeabilization Buffer (eBioscience) buffer for 20 min. Buffers were prepared according to manufacturer’s instructions (eBioscience). It was followed by intracellular staining with antibody anti-FOXP3-PE (236A/E7) (eBioscience).
For flow cytometry a BD FACScan II flow cytometer (BD Biosciences) and BD FACS DIVA software™ (BD Biosciences) was used. A total of 30000 cells/samples were acquired. Lymphocytes were gated followed by selection against cell doublets and dead cells. Then, CD3+ cells were gated followed by gating for CD4+ or CD8+ T cells.
12
Results
A. Effect of TGF-β on the phenotype of CD4+ T cells from healthy donors’ PBMCs
TGF-β is a pleiotropic cytokine secreted by immune cells and nonhematopoietic cells and it is responsible for maintaining immune homeostasis by acting as an immune suppressor: inducing differentiation of Treg cells, inhibiting proliferation, differentiation, activation and effector function of immune cells. However, depending on the context, TGF-β may act also as a pro-inflammatory cytokine, constituting a potent chemoattractant for neutrophils, driving Th17 and Th9 cells differentiation (which are pro-inflammatory cells) and it can also inhibit Th22 responses.
In cancer, in the beginning of the tumorigenic process, TGF-β inhibits the proliferation of transformed cells. Nevertheless, once those become resistant to this cytokine, it starts inducing angiogenesis and immune evasion, contributing to tumor growth and to the metastatic process.84 Sources of TGF-β in tumors are cancer cells themselves85,86, mesenchymal cells,
platelets, Treg cells, neutrophils, NK cells, monocytes, macrophages and DCs.87 High levels
of this cytokine are found in the plasma of cancer patients and it is normally associated with a poor prognosis.88 Thus, TGF-β is already being studied as a target for cancer treatment. 89
In human PBMCs, TGF-β induces FOXP3 expression and promotes differentiation of T conventional to regulatory T cells.90 In vivo, TGF-β also converts CD4+ CD25- T cells to induced FOXP3+ Tregs, which have a suppressive phenotype.91 Tregs are also able to secrete TGF- β, hence the TGF-β signaling pathway is one of the mechanism through which regulatory T cells mediate suppression of T effector activity.92, 93
In the tumor microenvironment, enzymes such as nitric oxide synthase or arginase contribute to this suppressive effect; hypoxic conditions promote Treg expansion, which in turn induces TGF-β expression and increases the immunoinhibitory milieu, creating a suppressive loop.94
Human Tregs are difficult to phenotype given their great diversity and the scarcity of identified markers specific for different Treg subsets. Nevertheless, CD25 and FOXP3 still remain the most frequently used phenotypic signature for human Tregs.95
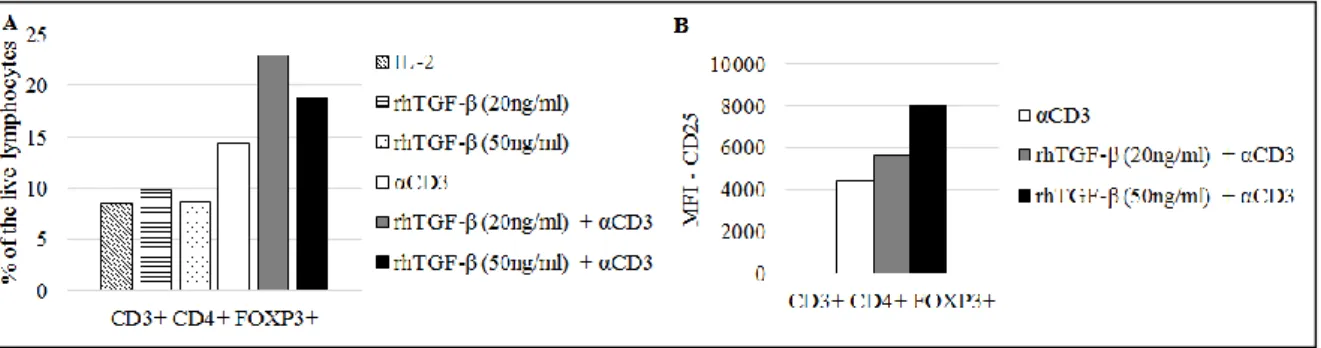
In the experiments reported in this work, in the presence of TGF-β and anti-CD3 antibody, CD4+ T cells upregulated the expression of the transcription factor FOXP3, as detected by intracellular stainings. The percentage of CD4+ FOXP3+ T cells cells increased in the culture (Fig. 1A). Moreover, it was observed that CD3+ CD4+ FOXP3+ cells had an augmented
13 expression of CD25 in the presence of TGF-β and anti-CD3 (Fig.1B). Thus, under the conditions used here, a suppressive environment was created, with induction of cells with a
bona fide Treg phenotype.
Interestingly, the augmenting effects exerted by TGF-β were completely dependent on T cell stimulation through CD3. Hereafter, it will be taken into account the conditions where rhTGF-β was added in conjunction with the anti-CD3 antibody.
Figure 1– A, CD4+ FOXP3+ T cells from healthy donors’ PBMCs cultured under different conditions B, Expression of CD25 on activated CD4+ T cells FOXP3+ from healthy donors PBMC cultured in the absence or presence of TGF-β
B. Effect of TGF-β on co-stimulatory receptors expression on T cells from healthy donors’ PBMCs
The co-stimulatory receptors evaluated are members of the TNFR superfamily (4-1BB, CD30, LIGHT, OX40 and CD27) or belong to the IgG superfamily (ICOS, CD28 and DNAM1). The expression of these co-receptors on T cells was firstly evaluated when PBMCs were cultured in the presence of TGF-β and anti-CD3.
As an important note, we have observed throughout that T cell preconditioning by TGF-β did not cause significant changes of co-receptors expression levels unless anti-CD3 was added. Thus, co-receptor expression levels in the presence of IL-2 and TGF-β (data not shown) resembled the expression levels reported for the condition “IL-2” in supplemental figures. 4-1BB is not detected on resting T cells, although its expression is induced upon T cell activation with a variety of agonists.96-98 Noteworthy, it was shown that 4-1BB can be induced in the absence of antigen-receptor signaling on memory but not naïve CD8+ T cells, through IL-2 and IL-15. This process is very important for CD8+ memory T cell survival after antigen clearance.99,100 However, 4-1BB might have a paradoxical role in the immune system. While there is evidence for its contribution on the induction of survival signaling in T cells, it was
14 shown that 4-1BB-deficient T cells are hyperproliferative.100-102 Moreover, 4-1BB can
mediate CD8+ T cell suppression and CD4+ CD25+ Tregs may express 4-1BB.103
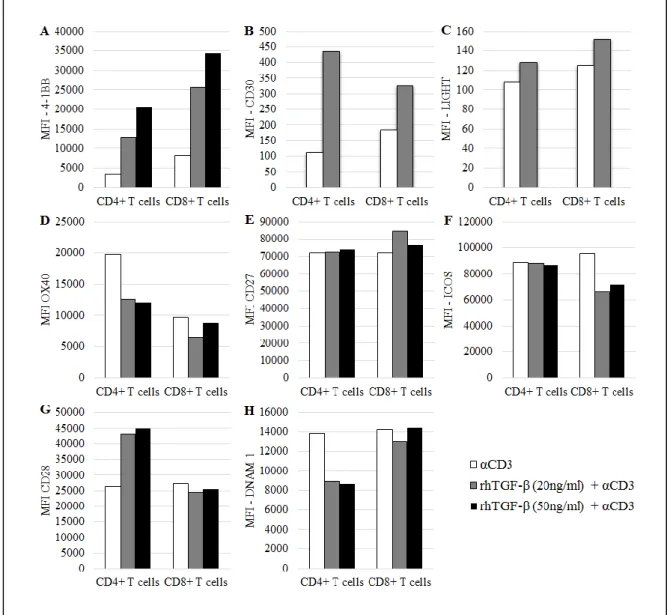
In our experiments, TGF-β-mediated induction of 4-1BB expression was dependent on the TGF-β availability on the medium, where the higher the concentration of TGF-β, the higher the expression of 4-1BB in both T cell subsets (Fig. 2A).
CD30 is not expressed on naïve T cells either – the peak of its expression happens after antigen encounter and the CD28:CD80/CD86 engagement is able to further induce the expression of this co-receptor.104 CD30 ligand is only expressed by professional APCs after activation.105 In vitro, crosslinking CD30 results in induction of cytokine production and proliferation of activated T cells. However, it was shown that signaling by CD30 is also able to inhibit Th1 driven responses.106
TGF-β pre-culture of PBMCs and TCR stimulation triggered an upregulation of CD30, particularly in CD4+ T cells (Fig. 2B).
With regard to LIGHT, besides being expressed on immature APCs and stimulating T cells through HVEM107, it works as a co-stimulatory receptor that is expressed on T cells upon activation.108 LIGHT was shown to play an important role on expansion of peripheral T cells by a T cell-T cell-dependent manner.109
LIGHT expression was not significantly changed in the presence of TGF-β and anti-CD3 (Fig. 2C).
OX40, like 4-1BB and CD30, is not detectable on resting naïve T cells, although it is induced on activated CD4+ and CD8+ T cells, as well as on Treg cells.110,111 TCR signals are sufficient to induce OX40 expression. However, it is the CD28/B7-1/2 interaction that sustains this expression and it can be modulated by T cell or APC-derived cytokines, such as IL-1, IL-2 and TNF.112 This sustained signal promotes antigen-specific T cell expansion and survival. Furthermore, it was shown that OX40 is able to antagonize FOXP3 induction in naïve CD4+ T cells, hindering the generation of inducible Treg cells.113,114 This concomitant effect of inducing effector activity and suppress immune suppression renders OX40 as a major player on tumor immune surveillance.115,116
In the presence of TGF-β and anti-CD3, OX40 surface expression was slightly downregulated, particularly in the CD4+ subset (Fig. 2D).
15 CD27 is expressed on naïve CD4+ and CD8+ T cells. But, unlike 4-1BB and OX40, its expression increases with activation but it is subsequently downregulated after several cell division cycles. This loss of CD27 is correlated with effector function of CD8+ T cells.117,118 CD27 is responsible for sustaining T cell survival, without influencing the cell division rate
in vitro.119,98
In the used conditions TGF-β had no additional effect on CD27 expression compared with the level reached when anti-CD3 was added alone (Fig. 2E).
Regarding the evaluated members, ICOS and CD28 are homologous co-receptors. While ICOS expression is restricted to activated T cells, CD28 is constitutively expressed in both naïve and activated T cells.120
In vitro, it was established that ICOS is important to sustain T cell proliferation and that it upregulates cell surface molecules and cytokine production. It also plays a role in T-cell dependent humoral immunity, given the fact that ICOS ligand is highly expressed on B cells. Upregulation of ICOS is strongly related with IL-10 production, thus, this receptor is also expressed on FOXP3+ Treg cells, as well as on effector-memory T cells, being able to control the pool size of both populations.121,122
In the used conditions, ICOS expression on CD4+ T cells was not affected with a TGF-β pre-culture, although it was slightly decreased in the CD8+ subset (Fig. 2F).
CD28, likewise ICOS, is implicated in T cell expansion, survival and differentiation and it is also necessary for proper IgG responses. The major difference between CD28 and ICOS is that the former upregulates not IL-10 but IL-2 production.120
CD28 expression, unlike ICOS, was more affected in the CD4+ subset, where it was increased in the presence of TGF-β and anti-CD3. On the contrary, CD28 expression on CD8+ T cells was not changed when PBMCs were pre-cultured with TGF-β (Fig. 2G).
DNAM1, also named CD226, is also a member of the IgG superfamily and it is expressed in both CD8+ and CD4+ T cells.123 The former constitutively express DNAM1, while the latter only express this co-stimulatory receptor upon activation.124
Under the conditions used, in the presence of TGF-β and anti-CD3, DNAM1 expression was diminished in the CD4+ subset, while TGF-β did not exert any effect on this co-stimulatory receptor expression on CD8+ T cells (Fig. 2H).
16 Figure 2 – Expression of co-stimulatory receptors on activated T cells from healthy donors’ PBMCs cultured in the absence or presence of TGF-β. MFI of A, 4-1BB+ T cells; B, CD30 + T cells; C, LIGHT+ T cells; D, OX40+ T cells; E, CD27+ T cells; F, ICOS+ T cells, G, CD28+ T cells, H, DNAM1+ T cells. Expression assessed after 7 days (4-1BB, OX40, CD27, ICOS, CD28 and DNAM1) or 13 days of culture (CD30 and LIGHT).
B.1. Effect of rhOX40L and rh4-1BBL on the expression of co-stimulatory receptors on activated T cells cultured with TGF-β
TNFR ligands (TNFRLs), such as OX40L and 4-1BBL were used as an approach to simulate potential cancer immunotherapies that use anti-OX40 and anti-4-1BB agonistic antibodies.
OX40 engagement by ligands expressed on dendritic cells increases proliferation, effector function and survival of T cells and preclinical studies showed that OX40 agonists are able to induce antitumor immunity and improve tumor-free survival. An OX40 monoclonal antibody has already been tested in a phase I clinical trial and it was proven to increase antitumor
17 reactivity on T and B cells in patients with late-stage melanoma, increased T and B cells responses to antigen immunization and drove a preferential upregulation of OX40 on FOXP3+ Treg cells, in TILs, with an acceptable toxicity profile.79
Regarding the importance of 1BB signaling pathway in cancer, it was already shown that 4-1BB knockout mice have a higher rate of mortality when treated with B16.F10 melanoma cells than 4-1BB wild-type (WT) mice. Furthermore, B16.F10-bearing 4-1BB WT mice treated with an agonistic 4-1BB monoclonal antibody had a prolonged survival, which was dependent on the effect of T cells and IFN-γ secretion. 4-1BB signaling, through agonistic monoclonal antibodies or soluble 4-1BBL, causes T cell expansion, cytokine induction, upregulation of anti-apoptotic genes and prevents activation-induced cell death. 96-98
The pattern of expression of T cell co-stimulatory receptors was assessed when PBMCs were cultured with TNFRLs in order to test if these co-stimulatory molecules were able to enhance T cell activation and if that was mirrored by phenotypic changes in these lymphocytes.
Firstly, the effect of recombinant proteins rhOX40L and rh4-1BBL was assessed in the absence of TGF-β in order to evaluate if these recombinant proteins were able to induce co-stimulation beyond the anti-CD3 antibody when the immunosuppressive stimuli were not present. Those two conditions were compared with the co-stimulatory effect achieved with the addition of anti-CD3 and anti-CD28.
Afterwards, rhOX40L and rh4-1BBL were added alongside anti-CD3 antibody after a pre-culture of the PBMCs with rhTGF-β and the expression of the same range of co-stimulatory receptors was evaluated.
The expression profile of the TNF members in response to the TNFR ligands (TNFRLs) was modified in a distinct pattern for each molecule.
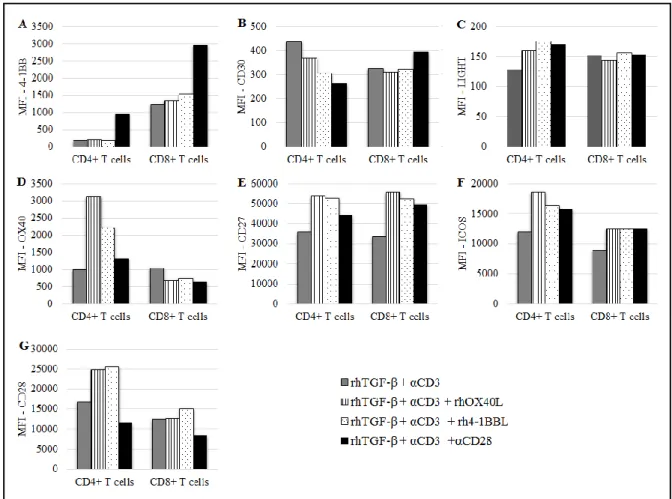
4-1BB expression on T cells was not changed when rhOX40L or rh4-1BBL were added alongside anti-CD3 but the use of anti-CD3 and anti-CD28 resulted in an increase of 4-1BB surface expression on CD4+ and CD8+ T cells (Sup. Fig. 1A, Annex).
In the presence of TGF-β (and anti-CD3), 4-1BB showed the same pattern of expression as in the absence of the suppressive cytokine: anti-CD28 was the only co-stimulatory molecule able to upregulate this marker, what was particularly clear in the CD4+ T cell subset (Fig. 3A).
In the absence of TGF-β, CD30 expression was increased on CD4+ T cells when the rhOX40L or rh4-1BBL were used (Sup. Fig. 1B, Annex). Interestingly, on the contrary, co-stimulation
18 with the anti-CD28 had no effect. In the CD8+ subset, the effects of the co-stimulation were minor.
In the presence of TGF-β, on CD4+ T cells there was a slight downregulation of CD30 when the CD28 antibody was added. On other hand, in the CD8+ subset, the addition of anti-CD28 resulted in a minor upregulation of CD30, while rhOX40L and rhOX40L had no effect on the expression of this marker (Fig. 3B).
In the absence of TGF-β, co-stimulatory molecules did to exert any significant effect on the expression of LIGHT on T cells (Sup. Fig. 1C, Annex).
Surprisingly, when the PBMCs were pre-cultured with rhTGF-β, the pattern of response of LIGHT to the TNFRLs and to anti-CD28 was changed in the CD4+ subset, where LIGHT expression was slightly upregulated (Fig. 3C). In the CD8+ T cells, the expression of this co-stimulatory receptor was not affected with the addition of co-co-stimulatory molecules.
OX40 expression was increased in the CD4+ subset upon co-stimulation with rhOX40L, rh4-1BBL, in the absence of TGF-β. In the CD8+ subset the co-stimulatory molecules seemed to exert no additional effect beyond anti-CD3 (Sup. Fig. 1D, Annex).
In the presence of TGF-β, TNFRLs were also able to upregulate OX40 expression in CD4+ T cells. Noteworthy, with the addition of the co-stimulatory molecules, expression of OX40 decreased in the CD8+ T cells (Fig. 3D).
Furthermore, in a conventional co-stimulatory setup without TGF-β, the co-stimulatory molecules used did not exert any significant effect on the expression of CD27 on T cells (Sup. Fig. 1E, Annex).
On the other hand, in the presence of TGF-β and anti-CD3, the addition of rhOX40L and rh4-1BBL induced CD27 expression on both T cell subsets, while the effect was anti-CD28 was less pronounced (Fig. 3E).
The same co-stimulatory molecules were tested for the IgG superfamily members CD28 and ICOS.
In the absence of TGF-β, rhOX40L and rh4-1BBL and anti-CD28 did not have a significant effect beyond CD3 stimulation on ICOS expression (Sup. Fig. 1F, Annex).
On the other hand, when PBMCs were pre-cultured with rhTGF-β, ICOS expression was only slightly upregulated in CD4+ T cells upon the addition of rhOX40L. In the CD8+ subset, the
19 expression of ICOS was modulated in the same way by the TNFRLs and the anti-CD28 but the effects were minor (Fig. 3F).
Figure 3 – Effect of rhOX40L, rh4-1BBL and anti-CD28 on the expression of co-stimulatory receptors on activated T cells from healthy donors’ PBMCs cultured in the presence of TGF-β. MFI of A, 4-1BB+ T cells; B, CD30+ T cells; C, LIGHT+ T cells; D, OX40+ T cells; E, CD27+ T, F, ICOS+ T cells, G, CD28+ T cells. Assessed after 13 days of culture.
CD28 expression is modulated in a distinct way by the different co-stimulatory stimuli used. In the absence of TGF-β, the TNFRLs did not exert any effect beyond anti-CD3 antibody on CD28 expression. Noteworthy, anti-CD28 antibody slightly downregulated CD28 expression in both T cell subsets (Sup. Fig. 1G, Annex).
On the contrary, in the presence of TGF-β, CD28 expression on CD4+ T cells is increased upon the addition of rhOX40L and rh4-1BBL. Once more, there was a reduction on CD28 expression on the surface of T cells, when anti-CD28 antibody was added (Fig. 3G). It happened in the absence and in the presence of TGF-β and seems to be mediated by epitope blockade or internalization of the CD28 molecule (Sup. Fig. 1G, Annex, and Fig. 3G).
20 DNAM1 expression was not evaluated in what regards the effects of the addition of co-stimulatory molecules, in the absence or presence of TGF-β.
C. Effect of TGF-β on co-inhibitory receptors expression on T cells from healthy donors’ PBMCs
Co-inhibitory receptors expressed on T cells were also evaluated within the suppressive environment created in vitro. TIM3, TIGIT, PD1, CTLA4 and PD-L1 were the co-inhibitory molecules whose expression was assessed when PBMCs were cultured in the presence of TGF-β and anti-CD3.
TIM3 is described as a negative regulator of IFN-γ-secreting CD4+ Th1 and CD8+ cytotoxic cells and it was shown to have an important role in CD8+ T cell exhaustion in cancer.148 Furthermore, it has a role in promoting MDSCs responses.125
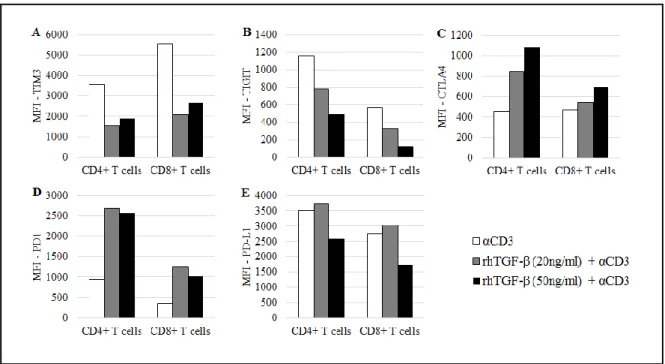
TIM3 was shown here to have its expression downregulated upon TCR stimulation and in the presence of TGF-β, in both T cell subsets (Fig. 4A).
TIGIT is expressed on activated T cells. Its suppressive function relies on its engagement with its ligand PVR (poliovirus receptor) on dendritic cells and subsequent induction of IL-10 production.126 TIGIT works as inhibitor of CD4+ T cell priming and CD8+ T cell anti-tumor effector function.127
Despite being a co-inhibitory receptor, TIGIT expression was downregulated upon TCR stimulation and in the presence of TGF-β. The effect seemed to be dependent on the TGF-β concentration: the higher it is, the less TIGIT is available on the T cell surface (Fig. 4B).
As mentioned earlier, the CTLA4:CD80/CD86 and the PD1:PD-L1 axis constitute the first approved targets of cancer immunotherapy. CTLA4 and PD1 have a distinct mechanism of action. While CTLA4 rises the activation threshold on T cells and attenuates proliferation of tumor-specific T lymphocytes, PD1 limits T cell effector function within the tumor microenvironment.128
CTLA-4 expression is minimal in resting T cells and, as it counteracts the CD28-mediated costimulatory signals, CTLA4 is enhanced through CD28/IL-2 co-stimulation after TCR engagement. Antigen-experienced CD4+ and CD8+ T cells, as well as CD4+ Tregs express CTLA4 constitutively.129
21 PBMCs pre-cultured with TGF-β and subject to TCR stimulation through anti-CD3 showed an increased surface expression of CTLA4 in T cells. This was particularly evident in the presence of higher concentrations of TGF-β in the CD4+ T cell subset (Fig. 4C).
PD1 is only expressed upon T cell activation on T cells and it is further enhanced when there is an inflammatory process ongoing.130
In the used conditions, PD1 expression was upregulated on T cells (CD4+ and CD8+) when PBMCs were pre-cultured with TGF-β and in the presence of anti-CD3 (Fig. 4D).
PD-L1 (B7-H1) is one of the PD1 ligands. PD-L1 is expressed constitutively on professional APCs, on B and T cells as well as on a wide range of nonhematopoietic cells, including tumor cells. PD-L1 expression on inflamed tissues promotes Treg differentiation.130 The PD1:PDL1 axis is discussed as a major immune evasion mechanism within the tumor microenvironment, where there is inhibition of T cell effector functions and enhancement of T cell suppression by Treg induction.128
However, in this experiment, in the presence of TGF-β and anti-CD3, the expression of PD-L1 was not significantly changed in neither of T cell subsets (Fig.4E).
Figure 4 - Expression of co-inhibitory receptors on activated T cells from healthy donors’ PBMCs cultured in the absence or presence of TGF-β. MFI of A, TIM3+ T cells, B, TIGIT+ T cells, C, CTLA4+ T cells; D, PD1 + T cells; E, PD-L1+ T cells. Assessed after 7 days of culture.
22
C.1. Effect of rhOX40L and rh4-1BBL on the expression of co-inhibitory receptors on activated T cells cultured with TGF-β
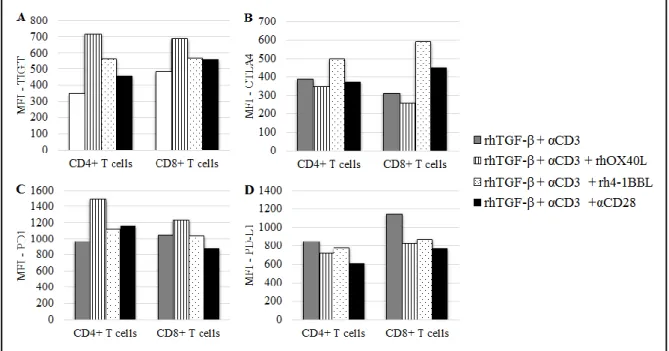
Similar to what was done for T cell co-stimulatory receptors, expression of the co-inhibitory receptors TIGIT, CTLA4, PD1 and PD-L1 was assessed when PBMCs were cultured in the presence of rhOX40L, rh4-1BBL or anti-CD28 antibody.
In the absence of TGF-β, TIGIT expression on CD4+ T cells was upregulated when anti-CD3 and rhOX40L or rh4-1BBL were added, although anti-CD28 had only a marginal effect. On the contrary, in the CD8+ subset the co-stimulatory effects of the TNFRLs were negligible and anti-CD28 addition resulted in upregulation of TIGIT on the surface of these cells (Sup. Fig. 2A, Annex).
Furthermore, when PBMCs were pre-cultured with rhTGF-β, TIGIT expression in CD4+ T cells was increased upon the addition of rhOX40L or rh4-1BBL, while anti-CD28 antibody did not further modulate TIGIT expression. In the CD8+ subset, the effects of the co-stimulatory molecules were minor (Fig. 5A).
In the absence of TGF-β, CTLA4 expression was upregulated upon the addition of rh4-1BBL, rhOX40L in both T cell subsets. This CTLA4 upregulation was particularly evident when anti-CD28 was added together with anti-CD3 (Sup. Fig. 2B, Annex).
However, after a pre-culture with TGF-β, co-stimulatory molecules did not seem to exert any additional effect on CTLA4 expression. There was only a slight upregulation of this co-inhibitory receptor on CD8+ T cells when rh-41BBL was added (Fig. 5B).
PD1 expression was upregulated on CD4+ T cells, when PBMCs were cultured without TGF-β and in the presence of the co-stimulatory molecules. On the other hand, in the CD8+ subset, rhOX40L and rh4-1BBL had only marginal effects and anti-CD28 antibody mediated PD1 downregulation (Sup. Fig. 2C, Annex).
In the presence of TGF-β, PD1 expression in only significantly affected in the CD4+ subset, where this co-inhibitory receptor is upregulated in the presence of rhOX40L (Fig. 5C)
Finally, PD-L1 expression on CD4+ T cells was not significantly affected by the addition of co-stimulatory molecules, neither in the absence of TGF-β (Sup. Fig. 2D, Annex) nor in the presence of this suppressive cytokine (Fig. 5D).
23 Figure 5 - Effect of rhOX40L, rh4-1BBL and anti-CD28 on the expression of the co-inhibitory receptors on activated T cells from healthy donors’ PBMCs cultured in the presence of TGF-β. MFI of A, TIGIT+ T cells, B, CTLA4+ T cells; C, PD1+ T cells; D, PD-L1+ T cells. Assessed after 13 days of culture.
24
Discussion and Conclusions
When PBMCs were pre-cultured with rhTGF-β for 24 hours and subsequently subjected to polyclonal activation by the addition of anti-CD3, the percentage of CD3+ CD4+ FOXP3+ cells increased, as well as the expression of CD25 on CD3+ CD4+ FOXP3+ cells. This result is consistent with induction of Treg cells, which are potentially able to inhibit anti-tumor immunity. Thus, a suppressive microenvironment was created in vitro.
The concomitant presence of TGF-β and anti-CD3 has distinct effects on co-stimulatory molecules, members of TNFRSF. While this condition upregulated 4-1BB and CD30 expression on T cells, LIGHT and CD27 did not significantly respond to the addition of TGF-β and OX40 was downregulated in the presence of this cytokine.
It was shown that important co-stimulatory molecules, such as ICOS and CD28, from the B7 family of the IgSF members, had also a different pattern of expression under the same conditions. In the presence of TGF-β and anti-CD3, CD28 expression was upregulated in CD4+ T cells, while ICOS showed the tendency to be downmodulated by TGF-β in CD8+ T cells. The CD28 signaling pathway induces IL-2 secretion, which might contribute to expand T cells with maintenance of their functional activity or it might contribute to Treg cells expansion, given the fact those need IL-2 to survive.95,131 On the other hand, ICOS expression on Treg cells is related with an increment in the production of the immunosuppressive cytokine IL-10.82,121 Thus, the expression levels of both co-receptors, CD28 and ICOS, might not be conclusive by T cell phenotyping only, given the highly dynamic and contradictory outcomes of cytokines whose secretion is induced by their signaling pathways. The inclusion of blocking antibodies for IL-2 and IL-10 needs to be explored in the presence and absence of TGF-β.
Moreover, in this suppressive environment, T cells upregulated exhaustion molecules, such as PD-1 and CTLA4, co-inhibitory receptors that were reported to be upregulated on TILs of cancer patients.132 However, TIM3 and TIGIT (also co-inhibitory) had their expression levels decreased and PD-L1 expression did not change in the presence of rhTGF-β, albeit these receptors are described as being expressed on T cells within the tumor microenvironment.75,127 Nevertheless, it remains to be explored how TGF-β suppresses TIGIT and TIM3 expression and does not interfere with PD-L1 expression on activated T cells.
25 These intriguing results might be explained by a different action of TGF-β, other than inducing Treg cells expansion, under the established conditions. It is described that TGF-β, besides promoting the expression of FOXP3 and inducing Treg cells expansion, is also able to drive Th17 cells differentiation.133,134 Additionally, it was reported that the capacity of TGF-β to abrogate FOXP3 expression is dependent on NF-κB activity. NF-κB is a transcription factor activated upon TCR/CD28 engagement and, at high doses of TCR stimulation, it can induce the production of IL-17 and inhibit FoxP3 expression, driving the differentiation of naïve T cells to Th17 cells, instead of induced Treg cells.135 Given the fact that we used high concentrations of TGF-β (20 ng/ml or 50 ng/ml) and 1µg/ml of anti-CD3, the possibility that a Th17 response was driven shall not be excluded.
Furthermore, Treg and Th17 subsets are plastic – there are reports in mice and in humans that identify Th17 cells that transit into induced Treg cells136 as well as Foxp3+ induced Tregs that have downmodulated RORγt and are able to transdifferentiate into Th17 cells, under pro-inflammatory conditions such as the presence of the cytokine IL-6.137
This Th17-driven response would explain the remarkable upregulation of CD30 under the presence of TGF-β (20ng/ml) and anti-CD3, given the fact that CD30:CD30L engagement was shown to play a critical role in Th17 differentiation.138 Furthermore, it would also be a reasonable explanation for a slight decrease on the percentage of FOXP3+ cells, when the concentration of TGF-β was increased (from 23% to about 19%).
Nevertheless, Th17 cells have also a role in cancer, although it might be ambiguous. There are reports on ovarian carcinomas-associated ascites that relate high Th17 cell density with better overall survival. On the contrary, in malignant tumors (such as hepatocellular carcinoma, colorectal cancer or pancreatic carcinoma), heavy infiltration of Th17 cells was correlated with a poor prognosis.139
This dual role of Th17 cells is explained by their contradictory effects on the tumor microenvironment. On one hand, Th17 responses are able to promote anti-tumor activity by downregulation of Tregs, promotion of MHC-I and II expression and induction of CTL activities. On the other hand, Th17 pro-tumor effects are likely underlined by the induction of pro-inflammatory cytokines, chemokines and matrix metalloproteinases – there is induction of MDSCs and inhibition of cytotoxic activity as well as promotion of angiogenesis.140 Thus, even if a Th17 response was driven, the suppressive microenvironment could have still been created in vitro.
26 However, this hypothesis shall be tested with the identification of a Th17 population under the established conditions and with the execution of a cytokine profile, by intracellular staining or ELISA. A titration of the TGF-β/anti-CD3 would allow to corroborate the results.
Concerning the approach to simulate immunotherapeutic approaches by the addition of costimulatory molecules such as rh4-1BBL, rhOX40L or anti-CD28 antibody, it is important to mention that it was not conclusive in the experiments shown here.
In summary, concerning the co-stimulation effect on co-stimulatory receptors, in the presence of TGF-β and anti-CD3, LIGHT, OX40 and CD28 expression were particularly upregulated on CD4+ T cells, while CD27 was increased in both subsets. On the other hand, 4-1BB, CD30 and ICOS expression had minor fluctuations with the addition of the TNFR ligands. Noteworthy, 4-1BB expression was clearly upregulated in CD4+ T cells in the presence of the anti-CD28 antibody, TGF-β and anti-CD3.
Regarding the co-inhibitory receptors tested, TIGIT was generally upregulated in the presence of co-stimulatory molecules, TGF-β and anti-CD3, and CTLA4 expression was increased on CD8+ T cells when rh4-1BBL was added. On the other hand, PD-1 and PD-L1 did not significantly respond to the presence of the co-stimulatory molecules.
This inability to restore a full-fledged activation phenotype with the addition of TNFRLs might rely on the late assessment of the expression of the co-receptors on T cells – most of the flow cytometry evaluations were done after 7 or 13 days of culture. This long incubation period with TGF-β/anti-CD3/rhOX40L/rh4-1BBL/anti-CD28 may result in T cell exhaustion induced by all the activation stimuli present in the culture medium. Thus, it would be important to study the kinetics of the expression of the co-signaling molecules and how they influence the outcome of the immunotherapies. Furthermore, assessing the same markers also with standard immunotherapies, monoclonal antibodies against CTLA4 and PD1, would allow: first, a comparison between therapies considered effective and potentially new ones, with regard to the expression of T cell co-receptors; second, a valid comparison between the experiments and the available literature and, finally, a phenotypical characterization of T cell responses under the established therapies.
It is worth mentioning that, although it has not been systematically studied, donor variability may be an important issue. PBMCs were collected from different donors and donor variability influences the pattern of expression of each molecule assigned.