Joana Rita do Vale Pais Costa
Licenciada em Química Aplicada
Síntese de novas glico-macromoléculas
poliméricas
com potencial atividade
biológica
Dissertação para obtenção do Grau de Mestre em
Mestrado em Química Bioorgânica
Orientadora:
Maria Teresa Barros
Professora Associada Agregada
FCT-UNL
Júri:
Presidente: Prof. Doutora Paula Cristina de Sério Branco Arguente: Prof. Doutor Christopher David Maycock
Vogal: Prof. Doutora Maria Teresa Barros Silva
ii
Joana Rita do Vale Pais Costa
Licenciada em Química Aplicada
Síntese de novas glico-macromoléculas
poliméricas
com potencial atividade
biológica
Dissertação para obtenção do Grau de Mestre em
Mestrado em Química Bioorgânica
Orientadora:
Maria Teresa Barros
Professora Associada Agregada
FCT-UNL
Júri:
Presidente: Prof. Doutora Paula Cristina de Sério Branco Arguente: Prof. Doutor Christopher David Maycock
Vogal: Prof. Doutora Maria Teresa Barros Silva
iii
Direitos de cópia
Joana Rita do Vale Pais Costa
“A Faculdade de Ciências e Tecnologia e a Universidade Nova de Lisboa têm o direito, perpétuo
e sem limites geográficos, de arquivar e publicar esta dissertação através de exemplares impressos reproduzidos em papel ou de forma digital, ou por qualquer outro meio conhecido ou que venha a ser inventado, e de a divulgar através de repositórios científicos e de admitir a sua cópia e distribuição com objetivos educacionais ou de investigação, não comerciais, desde que
v
Agradecimentos
Esta página parece demasiado pequena para agradecer a todos os que me ajudaram durante esta dissertação, não é possível referir todo o apoio que recebi durante este desafio. A todos que estiveram presentes, e que de alguma forma contribuiram para que isto fosse possível: Obrigada!
Inicialmente quero agradecer à Prof.ª Maria Teresa Barros, por ter sido uma excelente orientadora. Não tenho palavras suficientes para agradecer todo o compromisso, compreensão e apoio que recebi. Foi sem dúvida alguma um prazer trabalhar com alguém tão conhecedor e tão disposta a transmitir esse conhecimento, sempre disponível a novas ideias e sugestões, tal como a esclarecer todas as dúvidas que pudessem existir.
Em seguida quero agradecer ao meu pai, Jorge Costa, e à minha mãe, Clara Pais, esta tese é para vocês, por serem os excelentes pais que são e por sempre me terem apoioado acreditando na minha pessoa. São a minha voz da razão e grande parte da minha motivação, sem vocês nada disto seria possível. Não podia deixar de agradecer ao meu querido irmão, Ricardo Costa, que sempre foi incansável e indispensável, com todo o apoio e carinho; à pequena, Carolina Costa, que sem sequer saber, foi quem mais me ajudou a desanuviar e abstrair quando os nervos ficavam mais à tona.
A todos os meus colegas do grupo de investigação, por me terem feito sentir bem vinda, por toda a compreensão e paciência, pela disponibilidade e apoio, por todas as sugestões, muito deste trabalho é deles também. Um enorme obrigada à Cláudia Raposo, por toda a ajuda, mesmo nas questões mais banais, pelo companheirismo, desabafos e por todas as palavras de consolo. Ao André Jorge, Sandro Belchior e Cristiano Conceição, pela atenção, apoio e todos os bons momentos, foram excelentes colegas de trabalho.
Aos meus amigos, Ricardo Silvestre, Diogo Morgado, João Ferreira, Ricardo Tavares, pelo ombro amigo sempre disponível quando precisei, apesar da distância, sempre estiveram disponíveis para me ajudar e levantar a auto-estima, acabando sempre comigo às gargalhadas.
vii
Resumo
Ao longo deste trabalho está descrita a estratégia usada na obtenção de macromoléculas com eventual atividade biológica, pelo que foi necessário usar unidades biodegradáveis e/ou biocompatíveis. Tendo por base este requisito, numa primeira abordagem, decidiu-se incorporar a unidade de glucose na macromolécula pretendida, por ser biocompatível, biodegradável e possuir elevada versatilidade e funcionalidade.
Outro objetivo consistiu no desenvolvimento de sondas químicas através da síntese de moléculas fluorescentes. Devido à presença de um marcador as moléculas podem ser facilmente seguidas ao longo do seu trajeto, quando usadas em testes ou terapias.
Os heterociclos contendo azoto são estruturas promissoras no desenvolvimento de novos fármacos. Assim, pretendeu-se construir macromoléculas contendo pirimidinas (nucleosídeos) e investiu-se em “click chemistry” usando a formação do anel triazole como ponte de ligação.
Por outro lado, a presença de unidades de cumarina (sendo estes fluoróforos), aumentará a intensidade da fluorescência, o que será uma grande ajuda na deteção das macromoléculas pretendidas.
Usando esta estratégia esperávamos obter novos bioconjugados que poderiam ser usados como sondas fluorescentes biodegradáveis. Por outro lado, incorporando uma unidade polimérica seria possível uma futura abordagem à obtenção de nanopartículas poliméricas, pelo que se optou por usar o PEG 2000, por ser biologicamente seguro e inerte, sendo frequentemente utilizado em aplicações farmacêuticas.
A timidina é um composto biocompatível e facilmente reconhecido pelo organismo devido à presença do nucleosídeo timina.
O sistema triazole tem sido admitido, através de vários estudos, que possui potencial interação com sistemas biológicos. Recentes estudos de relação estrutura-atividade mostram a importância de unidades triazole em compostos com atividade farmacológica.
ix
Abstract
This work describes the strategy used to obtain macromolecules with potential biological activity and in order to enhance their utility it was necessary to use biodegradable and/or biocompatible units. Based on this requirement, as a first approach, it was decided to incorporate a glucose moiety into the desired macromolecule, as it is biocompatible and biodegradable and has high versatility and functionality.
Another objective was the development of chemical probes by synthesizing fluorescent versions of these molecules. The presence of a label, permits them to be easily traced along their path when used in tests or therapies.
Nitrogen-containing heterocycles are promising structures in the development of novel drugs. Thus, it was intended to construct pyrimidine-containing (nucleoside) macromolecules and we invested in "click chemistry", forming triazol rings as binding bridges.
On the other hand, the presence of coumarin units (these being fluorophores) will increase the fluorescence intensity, which will be of great help in detecting the desired macromolecules.
Using this strategy we expected to obtain new bioconjugates that could be used as biodegradable fluorescent probes. On the other hand, incorporating a polymer unit would be a future possible approach for obtaining polymer nanoparticles, it was decided to use PEG 2000, because it is biologically safe and inert, and is often used in pharmaceutical applications.
Thymidine is a biocompatible compound that is easily recognized by the body due to the presence of the nucleoside thymine.
The triazol system has potential for interaction interaction with biological systems and recent studies of structure-activity relationships show the importance of triazol units in compounds with pharmacological activity.
xi
Índice de matérias
Resumo vii
Abstract ix
Índice de figuras xv
Índice de esquemas xvii
Índice de tabelas xix
Lista de abreviaturas xxi
Lista de compostos xxiii
Capítulo I – Introdução 1
I.1. Carboidratos 1
I.1.1. Glucose 3
I.2. Cumarinas 5
I.3. PEG 8
I.4. Timidina 9
I.5. Triazole 11
I.6. Métodos de análise 13
I.6.1. Espectroscopia Infravermelho 13
I.6.2. Espectrometria de Ressonância Magnética Nuclear 14
I.6.3. Polarimetria – Rotação Ótica Específica 14
Capítulo II – Parte Experimental 15
II.1 Preâmbulo 15
II.2 Reações de síntese de compostos 17
Síntese de 1,2,3,4,6-penta-O-acetil-D-glucopiranose 17
Síntese de 1-bromo-2,3,4,6-tetra-O-acetil-α-D-glucopiranose 17
Síntese de 1-O-(1-bromoetil)-2,3,4,6-tetra-O-acetil-β-D-glucopiranose 18
Síntese de 1-O-polietilenoglicol 2000-2,3,4,6-tetra-O-acetil-α-D-glucopiranose 18
Síntese de 1-azido-2,3,4,6-tetra-O-acetil-β-D-glucopiranose 19
Síntese de 1-O-metil-2,3,4,6-tetra-O-benzil-α-D-glucopiranose 19
Tentativa de síntese de 1-hidroxi-2,3,4,6-tetra-O-benzil-α-D-glucopiranose 20
Tentativa de síntese de 3-azido-7-hidroxicumarina 21
Síntese de (4,8-dimetil-7-O-propargil-3-cumarinil)-3-propanoato de etilo 21
Síntese de Ácido (4,8-dimetil-7-O-propargil-3-cumarinil)-3-propanóico 22
xii
Tentativa de síntese de Succinato de tosilPEG2000 23
Síntese de PropargilPEG2000 23
Síntese de 5’-O-tert-butildimetilsilil-timidina 24
Síntese de 3’-O-propargil-5’-O-tert-butildimetilsilil-timidina 25
Síntese de 3’-O-(pent-4-inoíl)-5’-O-tert-butildimetilsilil-timidina 25
Síntese de 3’-O-succinoíl-5’-O-tert-butildimetilsililtimidina 26
Síntese de 1-O-(succinoíl polietilenoglicol2000)-2,3,4,6-tetra-O-acetil-α-D-glucopiranose
27
Síntese de 5’’-O-tert-butildimetilsililtimidina-3’’-O-(1-O -[polietilenoglicol2000]-2,3,4,6-tetra-O-acetil-α-D-glucopiranosil) succinato 28
Tentativa de síntese de 3-[triazolil-4-(3’-O-propanoato-5’-O
-tert-butildimetilsilil-timidina)]7-hidroxicumarina 29
Síntese de 1-[triazolil-2’’-(ácido(4’,8’-dimetil-7’-O-etil-3’-cumarinil)-3’
-propanóico)]-2,3,4,6-treta-O-acetil-β-D-glucopiranose 29
Capítulo III – Discussão de resultados 31
III.1 – Esquemas gerais 31
III.2 – Funcionalização de PEG 34
Síntese de Tosilato de PEG 2000 (11) 34
Tentativa de síntese de Succinato de tosilPEG2000 (12) 36
Síntese de PropargilPEG2000 (13) 37
III.3 – Funcionalização de cumarinas 39
Tentativa de síntese de 3-azido-7-hidroxicumarina (8) 39
Síntese de Ácido (4,8-dimetil-7-O-propargil-2-cumarinil)-2-propanóico (10) 39
III.4 – Funcionalização da timidina 42
Síntese de 5’-O-tert-butildimetilsilil-timidina (14) 43
Síntese de 3’-O-propargil-5’-O-tert-butildimetilsilil-timidina (15) e 3’-O-(pent-4-inoíl)-5’-O
-tert-butildimetilsilil-timidina (16) 45
Síntese de 3’-O-succinoíl-5’-O-tert-butildimetilsilil-timidina (17) 48
III.5. Síntese de derivados da glucose 50
Síntese de 1,2,3,4,6-penta-O-acetil-D-glucopiranose (1) 50
Síntese de 1-bromo-2,3,4,6-tetra-O-acetil-α-D-glucopiranose (2) 51
Síntese de 1-O-(1-bromoetil)-2,3,4,6-tetra-O-acetil-β-D-glucopiranose (3) 52
Síntese de 1-O-polietilenoglicol 2000-2,3,4,6-tetra-O-acetil-α-D-glucopiranose (4) 54
Síntese de 1-O-(succinoíl polietilenoglicolil2000)-2,3,4,5-tetra-O-acetil-α-D-glucopiranose
(18) 56
Síntese de 5’’-O-tert-butildimetilsililtimidina-3’’-O-(1-O-[polietilenoglicol-2,3,4,6-tetra-O
xiii
Síntese de 1-deoxi-1-azido-2,3,4,6-tetra-O-acetil-β-D-glucopiranose (5) 60
Síntese de 1-[triazolil-2’’-(ácido(4’,8’-dimetil-7’-O-etil-3’-cumarinil)-3’
-propanóico)]-2,3,4,6-tetra-O-acetil-β-D-glucopiranose 62
Conclusões 65
xv
Índice de figuras
Figura 1.1
–
Representação esquemática do tipo de macromolécula pretendido.
1
Figura 1.2
–
Estrutura de vancomicina.
2
Figura 1.3
–
Estrutura de alguns exemplos de D-monossacáridos.
3
Figura 1.4
–
Representação da β
-D-Glucose.
3
Figura 1.5
–
Figura representativa do feijão tonka.
5
Figura 1.6
–
Estrutura base da cumarina.
6
Figura 1.7
–
Exemplos de cumarinas com atividade biológica.
6
Figura 1.8
–
Exemplos do efeito de substituintes na fluorescência.
7
Figura 1.9
–
Molécula de polietilenoglicol.
8
Figura 1.10
–
Composto PEG com diferentes massas.
8
Figura 1.11
–
Estrutura do AZT.
9
Figura 1.12
–
Estrutura da timidina.
10
Figura 1.13
–
Representação da molécula de DNA, em que se observa o emparelhamento entre
nucleosídeos.
10
Figura 1.14
–
Estrutura do 1,2,3-triazole e 1,2,4-triazole, respetivamente.
11
Figura 1.15
–
Exemplos de anéis de heterocíclicos presentes em fármacos.
11
Figura 3.1
–
Sobreposição do espectro do composto inicial PEG (espectro preto) com o
composto 11 (espectro vermelho).
36
Figura 3.2
–
Sobreposição do espectro do composto inicial PEG (espectro preto) com o
composto 13 (espectro vermelho).
38
Figura 3.3
–
Espectro RMN de protão do composto 10.
42
Figura 3.4
–
Molécula da timidina com os centros nucleofílicos assinalados.
42
Figura 3.5
–
Espectro RMN de protão da timidina protegida (14).
45
Figura 3.6
–
Espectro RMN de protão do composto 4, com expansão dos sinais do açúcar.
55
Figura 3.7
–
Espectro RMN de protão do açúcar protegido após azidação.
61
Figura 3.8
–
Espectro correspondente ao composto 21.
63
xvii
Índice de esquemas
Esquema 1.1 – Configurações possíveis da glucose, em condições de equilíbrio. 4
Esquema 1.2 –Representação do anómero α e β da glucose. 4
Esquema 1.3 – Efeito da presença de substituintes electroatractores no carbono anomérico. 5
Esquema 1.4 – Reação de acetilação da D-Glucose. 5
Esquema 1.5 – Rota biossintética da cumarina. 6
Esquema 1.6 – Possíveis métodos de síntese de cumarinas. 7
Esquema 1.7 –Processo químico de obtenção de uma poliamida (‘’nylon’’). 9
Esquema 1.8 – Comparação entre as reações de cicloadição azida-alcino de Huisgen e a
catalisada por cobre. 12
Esquema 1.9 – Mecanismo possível para o ciclo catalítico CuAAC. 13
Esquema 1.10 – Representação de uma reação CuAAC. Uma molécula funcionalizada com azida reage com uma molécula contendo um alcino terminal formando um conjugado via
triazole. 13
Esquema 3.1 – Esquema geral das reações de funcionalização da timidina. 31
Esquema 3.2 – Esquema geral das reações de funcionalização do PEG. 32
Esquema 3.3 – Reações de funcionalização da cumarina. 32
Esquema 3.4 – Esquema geral das reações de síntese de derivados da glucose. 33
Esquema 3.5 – Reação de proteção seletiva do PEG 2000 34
Esquema 3.6 – Mecanismo da reação de tosilação do grupo hidroxilo. 34
Esquema 3.7 – Reação de esterificação com anidrido succínico. 36
Esquema 3.8 – Reação de substituição com brometo de propargilo. 37
Esquema 3.9 – Mecanismo da reação de substituição. 37
Esquema 3.10 – Reações para a síntese de 3-azido-7-hidroxicumarina. 39
Esquema 3.11 – Reações de síntese para obtenção da cumarina 10. 39
Esquema 3.12 – Mecanismo da reação de substituição nucleofílica. 40
Esquema 3.13 – Reação de proteção da timidina com TBDMSCl. 43
Esquema 3.14 – Mecanismo proposto para a reação de inserção do grupo protetor (terc.butil dimetilsilil) na timidina. 43
Esquema 3.15 – Exemplo de reações de funcionalização da timidina. 45
Esquema 3.16 – Mecanismo da reação de substituição na posição 3’-O. 46
Esquema 3.17 – Mecanismo de esterificação utilizando o reagente de acoplamento DCC e o
catalisador DMAP. 46
xviii
Esquema 3.19 – Mecanismo de esterificação utilizando a base Et3N e o catalisador DMAP.
48
Esquema 3.20 – Reação de proteção dos grupos hidroxilo da glucopiranose com grupos
acetato. 50
Esquema 3.21 – Mecanismo possível para a reação com anidrido acético. 50
Esquema 3.22 – Reação de bromação da glucose protegida 1. 51
Esquema 3.23 – Mecanismo da reação de substituição do grupo acetilo por bromo. 52
Esquema 3.24 – Reação de substitição na posição anomérica. 52
Esquema 3.25 – Reação de substituição de bromo por PEG 2000. 54
Esquema 3.26 – Reação de esterificação com anidrido succínico 56
Esquema 3.27 – Mecanismo de esterificação utilizando a base Et3N e o catalisador DMAP.
56
Esquema 3.28 – Reação de esterificação 57
Esquema 3.29 – Mecanismo de esterificação utilizando o reagente de acoplamento DCC e o
catalisador DMAP. 58
Esquema 3.30 – Reação de azidação. 60
Esquema 3.31 – Mecanismo da reação de substituição. 60
xix
Índice de tabelas
Tabela 3.1 – Comparação entre os valores obtidos na análise do composto PEG e do seu
derivado tosilado, 11. 35
Tabela 3.2 – Comparação entre os sinais do composto inicial PEG e o produto obtido, 13. 37
Tabela 3.3 – Comparação entre os sinais obtidos no produto inicial e no intermediário. 40
Tabela 3.4 – Comparação entre os espectros de RMN do composto inicial (éster) e do produto obtido (ácido carboxílico). 41
Tabela 3.5 – Comparação entre os resultados obtidos e os descritos na literatura relativamente
ao composto. 44
Tabela 3.6 – Comparação entre os sinais obtidos nas duas reações de funcionalização da
timidina. 47
Tabela 3.7 – Comparação entre os sinais do derivado da timidina inicial, 14, e do composto
obtido. 49
Tabela 3.8 – Comparação entre os dados de RMN e IV obtidos da glucose e da glucose
per-acetilada. 51
Tabela 3.9 – Comparação entre os espectros dos compostos 1 e 3. 53
Tabela 3.10 – Comparação entre os dados espectrais dos compostos 1 e 4. 54
Tabela 3.11 – Comparação entre os espectros do composto 4 e 18. 56
Tabela 3.12 – Comparação entre os dados espectrais dos compostos 18 e 19. 58
Tabela 3.13 – Comparação entre os dados obtidos para os compostos 1 e 5. 60
xxi
Lista de abreviaturas
13C-RMN Ressonância magnética nuclear de carbono
1H-RMN Ressonância magnética nuclear de protão
ט Frequência de absorção (cm-1)
δ Desvio químico
Ac Acetilo
AcOEt Acetato de etilo
Bn Benzilo
c Concentração
c.c.f. Cromatografia em camada fina
CuAAC Copper-Catalyzed Azide-Alkyne Cycloaddition = Cicloadição azida-alcino
catalisada por cobre
d Dupleto
DCC N,N’-diciclohexilcarboxilato
DCM Diclorometano
dd Dupleto de dupletos
DIPEA N,N-Diisopropiletilamina
DMAP 4-dimetilaminopiridina
DMF N,N-dimetilformamida
DMSO Dimetilsulfóxido
eq. Equivalente (s)
f Forte
fr Fraco
Hex Hexano
HMQC Heteronuclear Multiple-Bond Correlation = Correlação heteronuclear de múltiplas
ligações
xxii
J Constante de acoplamentem Multipleto (em RMN) ou média (em IV)
n.d. Não descrito
MeOH Metanol
ppm Partes por milhão
rf Factor de retenção
RMN Ressonância
s Singuleto
t Tripleto
TBDMS tert-bultildimetilsilil
THF Tetrahidrofurano
TMS Tetrametilsilano
xxiii
Lista de compostos sintetizados
1 1,2,3,4,6-penta-D-glucopiranose O
-acetil-2 1-bromo-2,3,4,6-tetra-acetil-α-D-glucopiranose O
-3 1-O-(1-bromoetil)-2,3,4,6-tetra-O-acetil-β
-D-glucopiranose
4 1-2,3,4,6-tetra-O-polietilenoglicol 2000-O-acetil-α-D-
glucopiranose
5 1-deoxi-1-azido-2,3,4,6-tetra-O-acetil-β
-D-glucopiranose
6 benzil-1-O-metil-2,3,4,6-tetra-O
-α-D- glucopiranose
7 benzil-1-hidroxi-2,3,4,6-tetra-α-D-glucopiranose O
xxiv
9 (4,8-dimetil-7-3-cumarinil)-3-propanoato O -propargil-de etilo
10
Ácido (4,8-dimetil-7-O
- propargil-3-cumarinil)-3-propanóico
11 Tosilato de PEG2000
12 tosilPEG2000 Succinato de
13 PropagilPEG2000
14
5’-O-tert
xxv
15 3’butildimetilsilil-timidina -O-propargil-5’-O-tert
-16 3’-O-(pent-4-inoíl)-5’-O
-tert-butildimetilsilil-timidina
17 3’butildimetilsilil-timidina -O-succinoíl-5’-O-tert
-18
1-O-(succinoíl
polietilenoglicolil2000)-2,3,4,5-tetra-O-acetil-α
-D-glucopiranose
19
5’’-O-tert
-butildimetilsililtimidina-3’’
-O (1-O
-[polietilenoglicol2000- 2,3,4,6-tetra-O-acetil-α
xxvi
20
3-[triazolil-4-(3’ -O-propanoato-5’ -O-tert-
butildimetilsilil-timidina)]7-hidroxicumarina
21
1-[triazolil-2’’-(ácido(4’,8’ -dimetil-7’-O-etil-3’
-cumarinil)-3’ -propanóico)]-2,3,4,6-treta-O-acetil-β
1
Capítulo I – Introdução
Nanomedicina é uma das áreas científicas em permanente evolução, sendo um domínio interdisciplinar muito promissor, com tecnologias revolucionárias, capazes de oferecer acentuados benefícios, quer a nível de diagnóstico precoce, quer num tratamento inteligente e rentável de várias doenças como sejam, cancro, problemas cardiovasculares, diabetes e doenças de Alzheimer e de Parkinson.1
As principais vantagens inerentes ao uso de nanocompostos são essencialmente as seguintes: maior estabilidade, libertação controlada, diminuição significativa da toxicidade, bem como melhorar o efeito terapêutico devido à maior probabilidade de o medicamento alcançar o alvo pretendido de uma forma mais dirigida. Neste contexto, vários materiais têm sido usados na preparação de nanopartículas, como sejam macromoléculas naturais e polímeros aceites farmacologicamente.2
No processo de distribuição do medicamento (“drug delivery”) existem duas propriedades
essenciais que deverão estar presentes nos polímeros usados: biodegradabilidade e biocompatibilidade.
Ao longo do trabalho desenvolvido ao abrigo da tese de mestrado subordinada ao título
”Síntese de novas glico-macromoléculas poliméricas com potencial atividade biológica“
procurou-se desenvolver a síntese de macromoléculas potencialmente úteis na preparação de novas nanopartículas poliméricas. É expectável que conjugados contruídos com base em blocos naturais (açúcares) sejam biocompatíveis e biodegradáveis. Por outro lado. combinando unidades moleculares com funcionalidades e propriedades distintas (cumarina, triazole, polietilenoglicol e timidina), deverão ser obtidos novos conjugados que possam intervir como um somatório das suas diferentes potencialidades, sendo algumas delas posteriormente comentadas.
Figura 1.1 – Representação esquemática do tipo de macromolécula pretendido.
I.1. Carboidratos
2
Figura 1.2 – Estrutura de vancomicina.
Os carboidratos podem existir na forma de monómeros, polímeros ou oligómeros.
Originalmente, carboidratos eram todos os compostos que possuíssem a fórmula empírica Cn(H2O)n. Atualmente, considera-se em geral, que carboidratos (glicídios, glucídios, glúcidos ou
hidratos de carbono), são compostos com uma função mista do tipo poliálcool-aldeído ou poliálcool-cetona, bem como outros compostos que, por hidrólise, dão poliálcoois-aldeídos e/ou poliálcoois-cetonas. É cada vez mais evidente que os carboidratos são moléculas chave para a comunicação biológica, controlando vários tipos de processos como infeções microbiais, inflamações, entre outros, participando no reconhecimento molecular e na função intracelular. Por exemplo, a glucose participa em vários processos metabólicos, para além de estar na base de moléculas mais complexas, também com elevada importância. Assim, assume um papel fundamental no corpo humano. O açúcar que circula na corrente sanguínea está, essencialmente, na forma de glucose. A glucose ao ser quebrada em unidades mais pequenas, liberta energia que é utilizada num grande número de órgãos do corpo e é a única que o cérebro é capaz de utilizar.4
Carboidratos são, portanto, compostos que apresentam uma vasta funcionalidade, o que os torna numa fonte explorada na obtenção de bibliotecas de novos compostos úteis, com particular interesse no desenvolvimento de novos medicamentos.2 Um dos aspetos fulcrais
inerente às moléculas de açúcares resulta do facto de possuírem vários centros quirais e com estereoquímica bem definida. Com efeito, os carboidratos podem ser usados na sua forma opticamente pura (enantiómeros) e sendo ricos em centros assimétricos, são substratos promissores na síntese de novas moléculas complexas, com eventual interesse biológico.
Nesse contexto, os carboidratos são substratos promissores dentro da química e da biologia, podendo ser usados na síntese de (macro)moléculas de grande importância química, farmacêutica e biológica. Atualmente, muitos produtos naturais são glicosados, constatando-se que os açúcares participam numa grande variedade de funções biológicas. Vários estudos de relação estrutura-atividade deste tipo de compostos têm sido alvo de atenção significativa por parte da sociedade científica.5
3
Figura 1.3 –Estrutura de alguns exemplos de D-monossacáridos.
I.1.1. Glucose
A glucose (Fig. 1.4) pertence ao grupo de carboidratos e é a molécula orgânica mais abundante no planeta. A glucose é formada por fotossíntese a partir de água e dióxido de carbono, utilizando a energia lumiosa proveniente do sol. É armazenada nas plantas sob a forma de um polímero de amido nos animais na forma de glicogénio. A glucose comercial é geralmente obtida por hidrólise enzimática de amido.6
Figura 1.4 –Representação da β-D-Glucose.
4
Esquema 1.1 – Configurações possíveis da glucose, em condições de equilíbrio.
A molécula de glucose possui 5 grupos hidroxilo. Ao longo do projeto procurou-se explorar o potencial da reatividade do carbono anomérico (C-1), uma vez que possui um comportamento químico diferente dos restantes carbonos presentes na molécula.5
A glucose em solução aquosa, estabelece um equilíbrio entre as formas alfa – e beta-glucopiranose (Esq. 1.1 e 1.2), sendo que a orientação equatorial é preferencial (mais estável). Existe ainda uma quantidade muito reduzida de glucose na forma furanose e cadeia aberta.4
Esquema 1.2 –Representação do anómero α e β da glucose.
Por outro lado, verifica-se que, em presença de determinados substituintes, nem sempre o estereoisómero beta é o mais estável, contrariando assim os efeitos estereoquímicos previstos. Esta estabilização do isómero alfa, resulta dum fator adicional, designado por efeito anomérico. Por exemplo, a presença de substituintes electronegativos num anel piranose tem preferência por uma orientação axial (Esq. 1.3).4
α-glucopiranose
5
Esquema 1.3 – Efeito da presença de substituintes electroatractores no carbono anomérico.
Como a glucose possui vários grupos hidroxilo livres, torna-se muitas vezes imperativo recorrer a métodos que permitam distingui-los. Uma via frequentemente utilizada passa pelo uso de grupos protetores, os quais ao reagirem preferencialmente em determinados grupos funcionais, vão essencialmente omitir (esconder) temporariamente a sua reatividade. O grupo protetor escolhido é muito importante, pois pode influenciar a reatividade da molécula, como por exemplo, a estereoquímica do composto pretendido.7 Os acetatos e benzoatos são excelentes
grupos de proteção, pois podem ser introduzidos e removidos facilmente com rendimentos elevados (Esq. 1.4).
Esquema 1.4 – Reação de acetilação da D-Glucose.
I.2. Cumarina
O sistema dicíclico designado por cumarina (benzopirano-2-ona ou cromen-2-ona), encontra-se presente em vários produtos naturais, como feijão de tonka (Fig. 1.5), varfarina e folha de trevo, apresentando propriedades farmacológicas importantes.8 A molécula original foi
isolada pela primeira vez por Vogel, a partir do feijão tonka.9,10 O nome vem de um termo francês
para o feijão tonka, ‘’coumarou’’, uma das fontes das quais a cumarina foi isolada como produto
natural em 1820.9,10
6
A cumarina (Fig. 1.6) foi sintetizada pela primeira vez em 186811 e tem sido utilizada na
indústria farmacêutica como reagente precursor na síntese de uma série de produtos farmacêuticos anticoagulantes.
Figura 1.6 – Estrutura base da cumarina.
Derivados da cumarina geralmente ocorrem como metabolitos secundários presentes em sementes, raízes e folhas de muitas espécies de plantas. Mais de 300 cumarinas diferentes (derivados da estrutura base) foram identificadas a partir de fontes naturais, possuindo diferentes aplicações farmacológicas, bioquímicas e terapêuticas.8
Figura 1.7 – Exemplos de cumarinas com atividade biológica.
O sistema dicíclico de cumarina pode ser obtido a partir do ácido cinâmico, via orto-hidroxilação (a), seguida de trans-cis isomerização da dupla ligação da cadeia lateral (b), e posterior lactonização (c). Como a forma trans é estável, não podendo ocorrer ciclização, torna-se necessário provocar a isomerização, através da intervenção da enzima isomeratorna-se (Esq. 1.5).12
7
Alternativamente, cumarina e seus derivados (genericamente designados por cumarinas) podem ser preparados através de uma série de reações (Esq. 1.6), como sejam: a) reação de Perkin, entre salicilaldeído e anidrido acético; b) por condensação tipo Pechmann, entre fenol e acetoacetato de etilo; c) por condensação de Knoevenagel.
Esquema 1.6 – Possíveis métodos de síntese de cumarinas.13
Por outro lado, atendendo a que a espectroscopia de fluorescência é uma das técnicas analíticas mais informativas e sensíveis, a qual tem dado um forte contributo em métodos de pesquisa, o facto das cumarinas serem fluorescentes faz com que sejam ótimos candidatos neste domínio científico. Com efeito, o estudo do funcionamento interno de biomoléculas, células e organismos partiu do desenvolvimento de ferramentas baseadas em fluorescência.14
Figura 1.8 – Exemplos do efeito de substituintes na fluorescência.
8
descritos na literatura evidenciam o uso da cumarina como fluoróforo, por ser uma molécula pequena, biocompatível e com aplicação em processos de síntese.16
I.3. Polietileno glicol (PEG)
Polietileno glicol (PEG, Fig. 1.9) é um polímero de monómeros de óxido de etileno que apresenta diversas utilidades, desde o nível industrial até ao medicinal.
Figura 1.9 – Molécula de polietilenoglicol.
O tamanho da sua cadeia define as suas potenciais aplicações. Normalmente são designados por PEG os compostos com uma massa molecular menor que 20.000g/mol e por PEO os polímeros com massa molecular superior a 20.000g/mol.17 O número que por norma
está associado ao nome PEG indica qual a massa molecular média do composto.
Apesar do tamanho da cadeia variar, as possíveis aplicações, propriedades físicas e as propriedades químicas não são muito diferentes. O estado físico dos polímeros varia conforme a massa molecular, pelo que, quando esta é abaixo de 200g/mol o polímero é um líquido viscoso, enquanto que se for entre 200g/mol e 2000g/mol tem um aspeto tipo cera e se a massa molecular for superior a 2000g/mol apresenta-se na forma de um cristal sólido branco opaco (Fig. 1.10).17
São utilizados comercialmente em diversas aplicações, como sejam como surfactante, nas indústrias alimentar, cosmética, farmacêutica, entre outras. São polímeros solúveis em água e composto orgânicos como metanol, etanol, acetonitrilo, diclorometano e benzeno.18
Fig. 1.10 – Composto PEG com diferentes massas.
9
Esquema 1.7 - Processo químico de obtenção de uma poliamida (''nylon'').
Polietilenoglicol é geralmente considerado biologicamente seguro e inerte, apesar de estarem descritos na literatura alguns casos (uma minoria) de pessoas que apresentam alergias.20 Este composto tem grande potencial para aplicações farmacêuticas por ser
metabolizado pelo fígado ou pelos rins quando no interior do corpo humano.21 Com efeito, o PEG,
ao ser considerado seguro e não tóxico, foi aprovado pela FDA para administração humana intravenosa, dermal e oral.22 Neste sentido, é considerado um composto muito promissor a nível
medicinal devido à sua baixa toxicidade, excelente biocompatibilidade e biodegradabilidade23,
pelo que tem sido usado como uma ferramenta complementar aos métodos tradicionais de libertação de fármacos.
A otimização do potencial deste polímero passa pela diferenciação dos grupos hidroxilos terminais, de modo a permitir reatividades diferenciadas e consequentemente poderem ser ligadas unidades diferentes.24
I.4. Timidina
O AZT (3’-azido-3’-desoxitimidina, Fig.1.11) é um exemplo de fármaco mundialmente conhecido e cuja estrutura deriva de uma unidade de açúcar, tendo sido o primeiro antirretroviral usado contra o HIV (vírus da imunodeficiência humana).25 Posteriormente, novos derivados têm
sido testados, constatando-se a presença de atividade ainda mais acentuada.
Figura 1.11 – Estrutura do AZT.
A timidina (desoxitimidina, Fig. 1.12) é um derivado da pirimidina, sendo constituída por uma timina (base azotada) ligada a um anel de desoxirribose (açúcar, pentose) através de uma
10
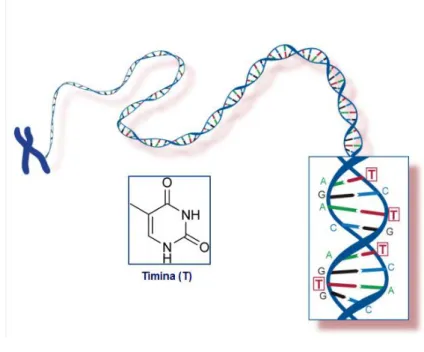
Figura 1.12 – Estrutura da timidina.
A timidina é o nucleosídeo T do DNA, que emparelha com a adenosina (A) na dupla hélice do DNA. A timidina existe na forma de cristais ou pó brancos. É um composto não tóxico, que ocorre naturalmente e existente em todos os seres vivos, fazendo dele um composto biológico chave. Com efeito, muitos estudos no campo da biologia molecular estão dependentes do uso da timidina ou derivados.
Figura 1.13 – Representação da molécula de DNA, em que se observa o emparelhamento entre
nucleosídeos.
Desde há 50 anos que os cientistas têm mostrado um interesse contínuo pela química de nucleosídeos, principalmente no campo da síntese de fármacos.26 A reatividade dos
nucleosídeos é complexa devido à presença de vários centros nucleofílicos, pelo que para gerar derivados de nucleosídeos é necessário recorrer a processos de proteção e desproteção de alguns grupos funcionais.
11
I.5. Triazole
O composto designado por triazole, também conhecido como pirrodiazole, é um composto orgânico heterocíclico, formado por um anel de cinco membros, di-insaturado, possuindo três átomos de azoto e dois átomos de carbono. Ocorre normalmente como um par de isómeros (1,2,3-triazole e 1,2,4-triazole, Fig. 1.14).28
Figura 1.14 – Estrutura do 1,2,3-triazole e 1,2,4-triazole, respetivamente.
A síntese de sistemas heterocíclicos, com alto teor de azoto, tem sido, ao longo do tempo, de crescente interesse para indústrias farmacêuticas e agroquímicas, sendo a unidade triazole uma das mais exploradas.28 Com efeito, anéis heterocíclicos estão amplamente presentes em
diversos tipos de fármacos. Indicam-se na Fig. 1.15 alguns exemplos mundialmente conhecidos. Com efeito, alguns estudos efetuados sobre a relação estrutura-atividade mostram o seu envolvimento em diversas atividades farmacológicas.28
Figura 1.15 – Exemplos de anéis de heterocíclicos presentes em fármacos.
O triazole e os seus derivados são compostos relativamente estáveis e versáteis que possuem uma vasta gama de atividades farmacológicas, tais como antimicrobianas, analgésicas, anti-inflamatórias, antiepilética, antidepressivos, anti-histamínicos, anti tuberculares, etc., bem como atividades biológicas, tais como antivirais, anti cancro e antibióticos.28
Neste contexto, a inserção de um anel triazole, como espaçador (união) entre dois compostos, pode ser considerada uma vertente promissora, porque não atua exclusivamente como um ligante passivo, mas também pode contribuir para potenciais alvos biológicos.29
Aparentemente, os anéis de triazole são exclusivamente de origem sintética37 e
normalmente a sua metodologia faz parte de um conjunto de reações designadas por “Click Chemistry”. Esta terminologia foi introduzida por K.B. Sharpless em 2001, para descrever
12
O desenvolvimento de reações de cicloadição azida-alcino catalizada por Cu(I) (CuAAC) foi impulsionado por Meldal e Sharpless, posteriormente à descoberta pioneira das cicloadições 1,3-dipolar de Huisgen.30 Infelizmente, a cicloadição térmica de Huisgen 1,3-dipolar de alcinos e
azidas requer temperaturas elevadas, o que conduz, na presença de alcinos assimétricos, a misturas de regioisómeros (Esq. 1.8). Neste sentido, a clássica cicloadição 1,3-dipolar de Huisgen não pode ser considerada uma reação ‘click’.38 A reação catalisada por cobre permite a
síntese dos regioisómeros 1,4-dissubstituídos especificamente, apresentando grandes vantagens operacionais comparativamente à mesma reação de cicloadição não catalisada.31
Esquema 1.8 – Comparação entre as reações de cicloadição azida-alcino de Huisgen e a catalisada por
cobre.
Com efeito, uma vez que os alcinos são pouco reativos com azidas (Esq. 1.9), a eficiência de uma reação CuAAC está fortemente dependente da presença dum catalisador metálico, como o cobre no estado de oxidação +1 [(Cu(I)]. Existem várias fontes de cobre e reagentes redutores disponíveis. O catalisador ativo de Cu (I) pode ser gerado a partir de sais de Cu (I) ou sais de Cu (II), mas o uso do sal de cobre(II) (CuSO4) como fonte de cobre juntamente com ascorbato (que
funciona como agente redutor)em solução aquosa de tert-butanol, tem sido a técnica mais
13
Esquema 1.9 – Mecanismo possível para o ciclo catalítico CuAAC.34
A elevada seletividade conseguida nas reações de cicloadição catalisadas por CuAAC tem tido um acentuado impacto científico na preparação de triazoles.35
Esquema 1.10 – Representação de uma reação CuAAC. Uma molécula funcionalizada com azida reage
com uma molécula contendo um alcino terminal formando um conjugado via triazole.
I.6. Métodos de análise
I.6.1. Espectroscopia de Infravermelho
A radiação infravermelho refere-se sobretudo à parte do espectro electromagnético entre a região da luz visível e das microondas. Esta técnica de espectroscopia é muito útil e prática em química orgânica, em particular a região entre os 4000 e os 400 cm-1. A espectroscopia IV é uma
técnica não dispendiosa e relativamente rápida que permite detetar as vibrações das ligações de certos grupos funcionais através da absorção do comprimento de onda da radiação IV produzida através da interação com a matéria.
14
Os espectros de IV são utilizados em conjunto com outros dados espectrais para determinar a estrutural molecular. Serve para complementar a análise obtida por Ressonância Magnética Nuclear.
I.6.2. Espectrometria de Ressonância Magnética Nuclear
A ressonância magnética nuclear é uma técnica de análise estrutural que tem por base as propriedades magnéticas de certos núcleos presentes na molécula, como os núcleos dos isótopos de hidrogénio e carbono. Esta técnica é basicamente outra forma de espectroscopia de absorção que tem por base as transições entre os níveis energéticos incitadas pela absorção da radiação electromagnética na região das radiofrequências.
É uma ferramenta de análise muito benéfica por fornecer informações sobre o ambiente químico dos protões e carbonos presentes num determinado composto, dando origem a espectros mono e bidimensionais. É uma técnica não destrutiva, para uma análise necessita de pouca quantidade de composto (entre 5 a 20 mg) num solvente deuterado que consiga dissolver a amostra. O RMN permite-nos obter diferente informação estrutural do que o IV.
I.6.3. Polarimetria – Rotação Ótica Específica
A rotação ótica específica é uma propriedade característica de compostos opticamente ativos. A polarimetria tem sido bastante utilizada na identificação de compostos.
Esta técnica permite medir a rotação do plano da luz polarizada quando incide num determinado composto quiral, usando um polarímetro. É muito importante na química dos açúcares por possuírem centros quirais.
Os valores de rotação ótica específica podem ser positivos, quando a luz polarizada sofre um desvio para a direita, e negativos quando o desvio é para a esquerda, ou zero (quando se possui uma mistura racémica). O conhecimento da rotação ótica permite identificar substâncias e contribuir para a sua caracterização estrutural (configuração absoluta).
É uma técnica sensível, existem vários fatores que influenciam a sua leitura, como a temperatura, concentração, solvente, distância percorrida e comprimento de onda da luz polarizada.
Equação 1.1 – Equação para determinar a rotação ótica específica. T –temperatura; α – medida de
15
Capítulo II – Parte Experimental
II.1 Preâmbulo
Todos os reagentes comerciais utilizados na síntese dos compostos indicados foram fornecidos pela Sigma-Aldrich e Pronalab.
Todos os solventes utilizados foram previamente secos ou destilados seguindo os procedimentos descritos na literatura.36
- Dimetilformamida (DMF): Juntou-se BaO ou sílica ao DMF e deixou-se a agitar em atmosfera inerte de um dia para o outro. Filtrou-se a mistura e o DMF foi destilado a pressão reduzida. Armazenou-se o solvente em garrafa de vidro escuro, sob atmosfera de árgon e contendo peneiros moleculares de 3 Å previamente ativados a 300oC.
- Diclorometano (DCM): O diclorometano foi aquecido em refluxo na presença de hidreto de cálcio durante uma hora antes de cada utilização. Foi, de seguida, recolhido numa ampola e transferido para o balão reacional com uma seringa.
- Tetrahidrofurano (THF): Foram adicionados fios de sódio e benzofenona num balão com THF. A solução foi aquecida a refluxo sob atmosfera inerte durante algumas horas, até que o solvente ficou com uma cor profundamente azul, indicativa de ausência de água. Destilou-se o volume desejado, que foi recolhido numa ampola e transferido com uma seringa para o balão reacional.
Os solventes das reações foram evaporados a pressão reduzida, usando um evaporador rotativo Büchi R-114 acoplado a um banho de água B-480 e a uma bomba de vácuo V- 700 com módulo de V-801. Qualquer vestígio de solvente foi posteriormente eliminado utilizando uma bomba de vácuo da marca Edwards, modelo P2212.
Para pesagens de reagentes e produtos utilizou-se uma balança de marca Sartorius BL210S com 4 casas decimais e com uma precisão de ±0,1mg.
Todas as síntese foram realizadas sob agitação constante e atmosfera de árgon. As reações foram controladas através de cromatografia de camada fina (c.c.f.) utilizando placas de sílica gel 60g/UV254nm Macherey-Nagel com 0,20mm de espessura em suporte de alumínio. Após
a eluição as placas foram observadas sob luz ultravioleta num comprimento de onda de 365nm (Camag) e revelados em soluções como: ácido sulfúrico em metanol; permanganato de potássio;
e ácido fosfomolibdico em etanol.
A purificação dos compostos foi realizada por cromatografia em coluna (c.c.) utilizando sílica gel Carlo Erba (40-63 μm) ou cromatografia em camada preparativa (c.c.p.) usando-se a sílica gel com indicador UV254nm com espessura de 1 mm.
Os sistemas de solventes utilizados para o c.c.f., c.c. e c.c.p. são referidos em cada uma das sínteses no capítulo II.
Para medições de ponto de fusão dos compostos recorreu-se ao aparelho Electrothermal Melting Point Apparatus.
O pH foi medido com papel indicador universal da Merck.
Os espectros de infravermelho (IV) foram traçados num espectrómetro Bruker, modelo Tensor 27 e tratados no software OPUS 6,0. As análises foram efetuadas em células de cloreto de sódio (NaCl). Na descrição das sínteses são apresentadas as bandas mais intensas e características de cada espectro, os dados estão apresentados na seguinte ordem: suporte da amostra, frequência do máximo de absorção (טmax em cm-1), tipo de banda (f- forte, m- média,
fr- fraco) e a atribuição do grupo funcional na molécula (quando possível).
Os espectros de ressonância magnética nuclear (RMN) foram obtidos num espectrómetro Bruker ARX (400 MHz para 1H e 101 MHz para 13C). Os desvios químicos (δ) são expressos em
16
carbono são apresentados na seguinte forma: solvente deuterado, desvio químico e atribuição na molécula em estudo (quando possível). Como referência interna utilizou-se o tetrametilsilano (TMS) no caso do clorofórmio deuterado (CDCl3), a água no caso do óxido de deutério (D2O), e
17
II.2 Reações de síntese de compostos
Síntese de 1,2,3,4,6-penta-O-acetil-D-glucopiranose (1)
Dissolveu-se o composto comercialmente disponível D-glucose (5,00 g; 2,78x10-2 mol) em
75 mL de piridina, em atmosfera de árgon. Em seguida foi adicionado anidrido acético (20 mL; 2,08x10-1 mol; 7,5 eq.). A reação foi controlada através de c.c.f. usando como eluente acetato de
etilo/MeOH/H2O (5:2:1). A solução ficou sob agitação durante 5 horas. A piridina foi evaporada
na bomba de vácuo com o auxílio de tolueno. A mistura bruta obtida foi purificada através de cromatografia em coluna de sílica usando como eluente a mistura hexano/AcOEt (1:1).
Aspeto: sólido branco cristalino; Rendimento: 97%; P.f.: 130-132°C P.f. lit.: 130-133°C
IV (NaCl): טmax 2964 (fr, C-H), 1754 (f, C=O), 1434 (m, CH-CH), 1371 (f, CH3C), 1222 (f, CO-O)
cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 6,33 (d, J=3,3 Hz, H-1); 5,47 (t, J=9,9 Hz, H-4); 5,12 (dd, J=11,8, 8,4 Hz, H-2 H-3); 4,28 (m, H-5); 4,11 (t, J=8,0 Hz, H-6); 2,18 (s, CH3); 2,10 (s, CH3); 2,05
(s, CH3); 2,03 (s, CH3); 2,02 (s, CH3).
13C NMR (CDCl3, 400 MHz): δ ppm 170,63 – 168,75 (C=O OAc); 89,06 (C-1); 69,82 (C-5); 69,18
(C-2); 67,88 (C-3); 67,74 (C-4); 61,45 (C-6); 20,87 – 20,44 (CH3 OAc).
Síntese de 1-bromo-2,3,4,6-tetra-O-acetil-α-D-glucopiranose (2)
Dissolveu-se o composto (1) (0,50 g; 2,43x10-3 mol), em 15 mL de diclorometano seco, em
atmosfera inerte de árgon, e foi adicionado HBr em ácido acético 33% (2,25 mL; 1,28x10-2 mol;
10 eq.). A mistura reacional foi deixada sob agitação e controlada por c.c.f usando como eluente Hex/AcOEt (1:1). Após 2 horas observou-se o consumo total do açúcar inicial, formando-se um produto menos polar. Procedeu-se para o tratamento da reação que consistiu na eliminação do ácido bromídrico efetuando lavagens com água gelada e bicarbonato de sódio. A fase orgânica foi recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente obtendo-se um óleo incolor.
18
Síntese de 1-O-(1-bromoetil)-2,3,4,6-tetra-O-acetil-β-D-glucopiranose (3)Num balão com 3 tubuladuras, revestido com papel alumínio, dissolveu-se o composto (1) (0,99 g; 2,52x10-3 mol) em 2 mL de diclorometano, foi adicionado bromoetanol (0,22 mL; 3,10x10 -3 mol). A reação foi arrefecida a 0oC e em seguida, com o auxilio de um funil de droping,
juntou-se BF3.OEt2 (1,60 mL; 1,30x10-2 mol) em 2 mL de DCM gota a gota. A reação ficou sob agitação
durante a noite e depois foram adicionados 0,80 mL de DCM e 4 mL de água gelada. Foram lavagens com água e bicarbonato de sódio, a fase orgânica foi recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente.
Aspeto: óleo amarelo; Rendimento: 51%
IV (NaCl): טmax 3445 (m, OH); 2964 (fr, C-H); 1753 (s, C=O); 1643 (m, CH3); 1430 (fr, CH-CH);
1371 (m, CH3C); 1225 (f, CO-O); 1041 (f, C-C) cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 5,24 (t, J=9,5 Hz, H-3); 5,11 (t, J=9,7 Hz, H-4); 5,04 (dd, H-2);
4,60 (d, J=7,9 Hz, H-1); 4,28 (dd, J=12,3, 4,8 Hz, 6); 4,18 (m, 7); 3,84 (m, 6); 3,73 (m,
H-5); 3,48 (dd, J=9,3, 4,3 Hz, H-8); 2,12 – 2,03 (s, CH3 OAc).
13C NMR (CDCl3, 400 MHz): δ ppm 170,65 – 169,40 (C=O OAc); 101,03 (C-1); 72,61 (C-5); 71,94
(C-4); 71,03 (C-2); 69,79 (C-7); 68,33 (C-3); 61,85 (C-6); 29,85 (C-8); 20,74 – 20,59 (CH3 OAc).
Síntese de 1-O-polietilenoglicol 2000-2,3,4,6-tetra-O-acetil-α-D-glucopiranose (4)
Dissolveu-se o composto comercialmente disponível PEG 2000 (0,81 g; 4,03x10-4 mol)
em 1,50 mL de DCM seco, em atmosfera de árgon. O balão reacional foi protegido da luz e adicionou-se Ag2CO3 (0,22 g; 1,21x10-3 mol; 2 eq.). A mistura ficou sob agitação durante 15
minutos e juntou-se lentamente o composto (2) (0,20 g; 4,84x10-4 mol; 1,2 eq.) dissolvido em
1,50 mL de DCM seco. A reação ficou sob agitação durante 48 horas, sendo controlada por c.c.f. utilizando como eluente CHCl3/MeOH (9:1). A mistura foi filtrada através de um funil de poros
com celite. O composto foi purificado usando cromatografia em coluna de sílica usando como eluente a mistura CHCl3/MeOH (9:1).
Aspeto: composto tipo cera; Rendimento: 76%
IV (NaCl): טmax 3446 (banda, OH); 2918 (fr, C-H); 1748 (m, C=O); 1643 (m, C-CH3); 1467 (fr,
19
1H NMR (CDCl3, 400 MHz): δ ppm 5,55 (t, J=9,9 Hz, H-4); 5,48 (d, J=3,4 Hz, H-1); 5,10 (t, J=9,4
Hz, H-2); 4,93 (dd, J=10,2, 3,7 Hz, H-3); 4,47 (m, H-5); 4,14 (d, J=10,0 Hz, H-6); 3,60 – 3,75 (m,
PEG) 2,09 (m, CH3 OAc).
13C NMR (CDCl3, 400 MHz): δ ppm 170,75 – 170,09 (C=O OAc); 90,18 (C-1); 71,05 – 70,28 (CH2
PEG); 69,86 (C-5); 68,48 (C-2); 68,42, 68,28 (C-3); 67,24 (C-4); 61,96 (C-6); 20,73 – 20,60 (CH3
OAc).
Síntese de 1-azido-2,3,4,6-tetra-O-acetil-β-D-glucopiranose (5)
Dissolveu-se o composto (2) (0,25 g; 6,08x10-4 mol) em 8 mL de DMF seco, em atmosfera
de árgon, e adicionou-se NaN3 (0,18 g; 2,74x10-3 mol; 4,5 eq). A reação foi deixada sob agitação
durante a noite. Através do controlo da reação por c.c.f. usando como eluente Hex/AcOEt (1:1) observou-se, no dia seguinte, o consumo total do composto. Foi então procedido o tratamento da reação que consiste em evaporar o DMF, diluir o composto com diclorometano e lavar a fase orgânica com água. A fase orgânica foi recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente. A purificação foi efetuada através de c.c. de sílica utilizando como eluente a mistura Hex/AcOEt (1:1).
Aspeto: sólido cristalino branco; Rendimento: 86%; P.f.: 125-126°C; P.f. lit.: 124-125°C
IV (NaCl): טmax 2949 (fr, C-H); 2121 (m, N3); 1754 (f, C=O); 1434 (m, CH-CH); 1372 (m, CH3
C-(C=O)); 1231 (m, C-O-C) cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 5,24 (t, J=9,5 Hz, H-3); 5,12 (t, J=9,7 Hz, H-4); 4,98 (t, J=9,2
Hz, H-2); 4,67 (d, J=8,8 Hz, H-1); 4,29 (dd, J=12,4, 4,6 Hz, H-6); 4,18 (dd, H-6); 3,82 (m, H-5);
2,11 (d, J=10,1 Hz, 6H, CH3 OAc); 2,04 (d, J=8,8 Hz, 6H, CH3 OAc).
13C NMR (CDCl3, 400 MHz): δ ppm 170,62 – 169,22 (C=O OAc); 87,91 (C-1); 74,02 (C-5); 72,60
(C-4); 70,63 (C-2); 67,87 (C-3); 61,65 (C-6); 20,71 – 20,55 (CH3 OAc).
Síntese de 1-O-metil-2,3,4,6-tetra-O-benzil-α-D-glucopiranose (6)
Dissolveu-se o composto comercialmente disponível 1-O-metil-α-D-glucopiranose (0,98 g;
-20
2 mol; 8 eq.) em banho de gelo e a mistura ficou sob agitação durante 30 minutos, adicionou-selentamente brometo de benzilo (3,68 mL; 3,10x10-2 mol; 6 eq.). A reação manteve-se a
temperatura baixa durante 30 minutos sendo depois deixada atingir a temperatura ambiente, ficou sob agitação durante a noite. A evolução da reação foi controlada por c.c.f. usando como eluente AcOEt/MeOH/H2O (5:2:1). Quando se observou o consumo total do açúcar inicial
diluiu-se a solução em 150 mL de água gelada. Foram feitas extrações com CH2Cl2, sendo a fase
orgânica lavada com solução saturada de NaCl, recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente. O composto foi purificado usando cromatografia em coluna de sílica tendo como eluente a mistura Hex/AcOEt (5:1).
Aspeto: óleo; Rendimento: 61%
IV (NaCl): טmax 3060 (m, C=C-H); 3027 (f, C-H aromático); 2909 (f, C-H); 1604 (f, C=C); 1494 (m, C=C); 1452 (f, C-H2); 1359 (f, C=C-H); 1191 (f, C-O-C); 912 (m, C-C); 736 (f, C-H) cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 7,45 – 7,18 (m, 20H, OBn); 5,03 (d, J=10,9 Hz, H-1); 4,86 –
4,52 (m, 8H, CH2 OBn); 4,03 (t, J=9,2 Hz, H-2); 3,77 (dd, J=16,7, 7,0 Hz, H-3); 3,67 (t, J=16,5 Hz,
H-4); 3,61 (dd, J=9,6, 3,5 Hz, H-5); 3,43 (s, 3H).
13C NMR (CDCl3, 400 MHz): δ ppm 138,85 – 137,96 (4C, OBn); 128,49 – 127,62 (20C, OBn);
98,25 (C-1); 82,17 (C-3); 79,89 (C-2); 77,71 (C-4); 75,79 – 73,43 (4C, CH2 OBn); 70,10 (C-5);
55,20 (OCH3).
Tentativa de síntese de 1-hidroxi-2,3,4,6-tetra-O-benzil-α-D-glucopiranose (7)
Preparou-se uma solução de 4 mL de ácido acético glacial e 1 mL de H2SO4 0,1M e
aqueceu-se à temperatura de 105°C. O composto (6) (0,50 g; 8,98x10-4 mol) dissolvido em 0,60
21
Tentativa de síntese de 3-azido-7-hidroxicumarina (8)Num balão reacional foram dissolvidos os compostos 2,4-dihidroxibenzaldeído (1,40 g; 1,01x10-2 mol), N-acetilglicina (1,18 g; 1,01x10-2 mol) e acetato de sódio anidro (3,33 g ; 4,06x10 -2 mol) em 5 mL de Ac2O, em atmosfera de árgon. A mistura foi mantida sob agitação e em refluxo
a 160°C durante 4 horas. Filtrou-se o precipitado amarelo, sendo lavado com água e acetato de
etilo. O composto formado foi dissolvido em 10 mL de HCl 1M e 5 mL de EtOH, mantendo a mistura sob agitação e a 80°C durante 2 horas e em seguida foram adicionados 20 mL de água
gelada à solução. Baixando a temperatura da mistura com um banho de gelo, juntou-se lentamente NaNO2 (1,34 g; 1,94x10-2 mol; 2 eq.), deixando reagir durante 10 minutos.
Acrescentou-se NaN3 (1,89 g; 2,90x10-2 mol; 3 eq.) e deixou-se a reação atingir a temperatura
ambiente. A mistura ficou sob agitação durante a noite, com um tubo de secagem com cloreto de cálcio, e foi depois filtrada e lavada com água. Não se conseguiu isolar o composto pretendido.
Síntese de (4,8-dimetil-7-O-propargil-3-cumarinil)-3-propanoato de etilo (9)
Uma mistura do composto 3,8-dimetil-2-propanoato de etilo-7-hidroxi-cumarina (2,01 g; 6,92x10-3 mol), carbonato de potássio (1,91 g; 1,38x10-2 mol) e brometo de propargilo (0,95 mL;
1,01x10-2 mol) em 60 mL de acetona foi aquecida a refluxo durante 24 horas. O volume de
acetona foi reduzido para metade e em seguida a solução foi colocada em 100 mL de água. O precipitado foi filtrado, lavado com água e recolhido. O composto resultante ficou durante a noite a secar a 50°C.
Aspeto: pó beje; Rendimento: 72%; P.f.: 154-156°C; P.f. lit.: 153-154°C
IV (NaCl): טmax 3433 (banda, C-H metil); 3242 (m, C-H aromático); 1721 (f, C=O, éster); 1693 (m, C=C) cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 7,45 (d, J=8,9 Hz, H-5); 6,96 (d, J=8,9 Hz, H-6); 4,80 (d, J=2,1
Hz, H-1’); 4,12 (q, J=7,1 Hz, H-16); 2,97 (t, J=7,6 Hz, H-13); 2,60 (t, J=7,6 Hz, 14); 2,54 (s,
22
13C NMR (CDCl3, 400 MHz): δ ppm 172,92 (C-15); 161,74 (C-2); 157,40 (C-7); 151,39 (C-9);
147,38 4); 122,42 5); 121,77 3); 114,96 10); 114,69 8); 108,21 6); 78,18
(C-2’); 75,96 (C-3’); 60,49 (C-16); 56,41 (C-1’); 32,69 14); 23,17 13); 14,99 12); 14,21 (C-17); 8,30 (C-11).
Síntese de Ácido (4,8-dimetil-7-O-propargil-3-cumarinil)-3-propanóico (10)
Dissolveu-se o composto (9) (0,50 g; 1,53x10-3 mol) em 20 mL de EtOH e adicionou-se
NaOH 0,25M (6,67 mL; 3,55x10-1 mol), a solução ficou sob agitação e a 80°C durante 1 hora. A
reação arrefeceu até à temperatura ambiente, e adicionou-se 33 mL de água. O pH foi aferido para pH=7. Evaporou-se o composto até à secura. O término da reação foi confirmado por c.c.f. usando como eluente CHCl3/MeOH (95:5).
Aspeto: pó beje; Rendimento: 89%; P.f.: 220-223°C; P.f. lit.: 190-191°C
IV (NaCl): טmax 3858 (f, C-H, CH3); 3389 (banda, OH); 3055 (fr, C-H aromático); 2943 (m, C-H, CH3); 2831 (f, C-H, alílico); 2228 (fr, C-H alcino); 1763 (m, (C=O)-O); 1704 (f, C=O); 1641 (m, C=C); 1450 (m, COOH) cm-1.
1H NMR (D2O, 400 MHz): δ ppm 7,15 (d, J=8,9 Hz, H-5); 6,75 (d, J=8,9 Hz, H-6); 4,67 (s, H-1’);
2,56 (m, H-13); 2,15 (dd, J=17,1, 9,3 Hz, H-14); 2,10 (s, H-12); 1,85 (s, H-11).
13C NMR (D2O, 400 MHz): δ ppm 181,73 (C-15); 163,81 (C-2); 156,93 (C-7); 150,13 (C-9); 149,66
(C-4); 122,85 (C-5); 120,68 (C-3); 114,29 (C-10); 113,12 (C-8); 108,78 (C-6); 78,05 (C-2’); 77,08 (C-3’); 56,53 (C-1’); 35,83 (C-14); 24,07 (C-13); 14,30 (C-12); 7,31 (C-11).
Síntese de Tosilato de PEG2000(11)
Dissolveu-se o composto comercialmente disponível PEG 2000 (1,01 g; 5,04x10-4 mol) em
10 mL de DCM seco, em atmosfera de árgon. Adicionou-se TsCl (0,11 g; 5,98x10-4 mol; 1,2 eq.)
e Et3N (0,10 mL; 6,05x10-4 mol; 1,2 eq.), a reação foi mantida sob agitação durante dois dias,
sendo controlada por c.c.f. usando como eluente CH3Cl/MeOH (9:1). O solvente foi evaporado à
23
Aspeto: cera esbranquiçada; Rendimento: 74%; P.f.: 49-50°C; P.f. lit.: 48-53°C
IV (NaCl): טmax 3524 (f, O-H); 2871 (banda, C-H aromático); 1643 (m, C-CH3); 1351 (f, S(=O)2)
cm-1.
1H NMR (CDCl3, 400 MHz): δ ppm 7,80 (d, J=8,0 Hz, 2H aromático Ts); 7,35 (d, J=7,8 Hz, 2H
aromático Ts); 4,47 – 3,48 (m, PEG); 2,46 (s, CH3 Ts); 1,26 (s, OH).
13C NMR (CDCl3, 400 MHz): δ ppm 144,78 – 127,98 (C aromático); 72,54 – 61,70 (PEG); 21,64
(CH3).
Tentativa de síntese de Succinato de tosilPEG2000 (12)
A uma solução do composto (12) (0,80 g; 3,70x10-4 mol) em 10 mL de DCM juntou-se
DMAP (0,05 g; 3,70x10-4 mol), Et3N (0,04 mL; 3,70x10-4mol) e anidrido succínico (0,08 g; 7,40x10 -4 mol; 2 eq.). A reação ficou sob agitação durante 48 horas e foi controlada por c.c.f. usando
como eluente CH3Cl/MeOH (9:1). A mistura foi diluída em DCM, em seguida foi sujeita a
sucessivas lavagens com HCl 0.1M, solução saturada de NaCl e água. A fase orgânica foi recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente. Não foi possível isolar o composto pretendido.
Síntese de PropargilPEG2000 (13)
Dissolveu-se o composto comercialmente disponível PEG 2000 (1,05 g; 5,25x10-4 mol;
2,5 eq.) em 4 mL de THF seco, em atmosfera de árgon, foi adicionado NaH (0,01 g; 2,1x10-4 mol;
1 eq.) e a mistura ficou sob agitação durante 20 minutos, altura em que se adicionou brometo propargilo (0,02 mL; 2,1x10-4 mol; 1 eq.). A mistura ficou sob agitação durante 24 horas, foram
depois feitas lavagens com NH4Cl. A fase orgânica foi recolhida e seca com sulfato de sódio
anidro, posteriormente foi filtrada para em seguida evaporar o solvente.
Aspeto: composto tipo cera; Rendimento: 80%; P.f.: 50-52°C; P.f. lit.: 48-53°C
IV (NaCl): טmax 3448 (banda, OH); 3270 (m, C
≡
C-H); 2879 (f, C-H2); 1352 (m, C-H2); 1281 (m,24
1H NMR (CDCl3, 400 MHz): δ ppm 4,39 – 3,48 (m, CH2 PEG); 4,14 (s, H-1); 2,38 (s, H-3); 1,19
(s, OH).
13C NMR (CDCl3, 400 MHz): δ ppm 79,65 (C-2); 74,58 (C-3); 72,61 – 61,66 PEG); 58,18
(C-1).
Síntese de 5’-O-tert-butildimetilsilil-timidina(14)
Dissolveu-se o composto comercial timidina (1,00 g; 4,13x10-3 mol) em 4 mL de DMF seco
e adicionou-se TBDMSCl (0,69 g; 4,55x10-3 mol) e imidazol (0,62 g; 9,09x10-3 mol) na proporção
1.1:2.2. A solução foi mantida a 37°C durante uma hora. Foi então procedido o tratamento da reação que consiste em evaporar o DMF. Em seguida o composto foi purificado através de cromatografia em coluna de sílica usando como eluente CHCl3/MeOH (95:5).
Aspeto: sólido branco; Rendimento: 50%; P.f.: 195-197°C; P.f. lit.: 196-197°C
IV (NaCl): טmax 3555 (banda, OH); 3177 (fr, N-H); 2930 (fr, C-H aromático); 2855 (fr, C-H alilico); 1699 (f, C=O); 1471 (f, C=C) cm-1.
1H NMR(DMSO, 400 MHz): δ ppm 11,32 (s, NH); 7,47 (s, H-6); 6,17 (t, J=6,9 Hz, H-1’); 5,27 (d, J=4,1 Hz, H-3’); 4,20 (s, H-4’); 3,81 (d, J=5,0 Hz, H-5’); 2,07 (m, H-2’); 1,78 (s, H-7); 0,89 (s, 9H,
CH3-tert); 0,08 (s, 6H, CH3-Si).
13C NMR (DMSO, 400 MHz): δ ppm 164,13 (C-4); 150,84 (C-2); 135,96 (C-6); 109,84 (C-5); 87,25
(C-1’); 84,30 (C-4’); 70,97 (C-3’); 63,75 (C-5’); 39,64 (C-2’); 26,26 (CH3-tert); 18,49 (C-tert); 12,69
25
Síntese de 3’-O-propargil-5’-O-tert-butildimetilsilil-timidina(15)Dissolveu-se o composto (14) (0,05 g; 1,38x10-4 mol) em 1 mL de THF seco, adicionou-se
NaH (0,05 g; 2,01x10-3 mol) e manteve-se a mistura a 0°C durante 1 hora. Quando a reação
atingiu a temperatura ambiente foi adicionado brometo de propargilo (0,03 mL; 3,37x10-4 mol) e
a solução ficou sob agitação durante 24 horas, sendo controlada através de c.c.f. usando como eluente CH2Cl2/MeOH (95:5). Foram depois feitas lavagens com EtOAc, solução saturada de
NaCl e água. A fase orgânica foi recolhida e seca com sulfato de sódio anidro, posteriormente foi filtrada para em seguida evaporar o solvente. O composto foi purificado através de cromatografia em coluna de sílica usando como eluente CHCl3/MeOH (95:5).
Aspeto: sólido branco; Rendimento: 43%
IV (NaCl): טmax 3448 (f, NH); 2924 (fr, C-H); 1642 (f, C=C) cm-1.
1H NMR(DMSO, 400 MHz): δ ppm 11,37 (s, NH); 7,49 (s, H-6); 6,10 (dd, J=8,5, 5,7 Hz, H-1’);
4,24 (s, H-3’, H-1’’); 4,00 (s, H-4’); 3,76 (dd, J=10,7, 6,5 Hz, H-5’); 2,29 (dd, J=13,3, 4,8 Hz,
H-3’’); 2,13 (m, H-2’); 1,79 (s, H-7); 0,90 (s, 9H, CH3-tert); 0,10 (s, 6H, CH3-Si).
13C NMR (DMSO, 400 MHz): δ ppm 164,09 (C-4); 150,83 (C-2); 135,81 (C-6); 110,04 (C-5); 84,37
(C-1’); 80,50 (C-4’); 78,76 (C-2’’); 77,86 (C-3’’); 75,45 (C-3’); 63,69 (C-5’); 56,25 (C-1’’); 36,44
(C-2’); 26,25 (CH3-tert); 18,44 (C-tert); 12,68 (C-7); -4,98 (CH3-Si); -5,02 (CH3-Si).
Síntese de 3’-O-(pent-4-inoíl)-5’-O-tert-butildimetilsilil-timidina (16)
Dissolveu-se o composto (14) (0,50 g; 1,41x10-3 mol) em 10 mL de DCM seco e
adicionou-se N-N’-diciclohexilcarboxilato (0,31 g; 1,50x10-3 mol; 1,1 eq.) e 4-dimetilaminopiridina (0,02 g;