w w w . s b f g n o s i a . o r g . b r / r e v i s t a
Original
Article
Phytochemical
analysis
of
Vernonanthura
tweedieana
and
a
validated
UPLC-PDA
method
for
the
quantification
of
eriodictyol
Layzon
Antonio
Lemos
da
Silva
a,
Larissa
Gabriela
Faqueti
a,
Flávio
Henrique
Reginatto
a,
Alan
Diego
Conceic¸
ão
dos
Santos
b,
Andersson
Barison
b,
Maique
Weber
Biavatti
a,∗aLaboratóriodeFarmacognosia,DepartamentodeCiênciasFarmacêuticas,CentrodeCiênciasdaSaúde,UniversidadeFederaldeSantaCatarina,Florianópolis,SC,Brazil bDepartamentodeQuímica,UniversidadeFederaldoParaná,Curitiba,PR,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received29May2015 Accepted12July2015 Availableonline8August2015
Keywords:
Asteraceae Eriodictyol UPLC-PDA
Vernonanthuratweedieana
a
b
s
t
r
a
c
t
Vernonanthuratweedieana(Baker)H.Rob.,Asteraceae,isusedintheBrazilianfolkmedicineforthe
treat-mentofrespiratorydiseases.Inthisworkthephytochemicalinvestigationofitsethanolextractsaswell
asthedevelopmentandvalidationofanUPLC-PDAmethodforthequantificationoftheeriodictyolfrom
theleaveswereperformed.Thephytochemicalstudyforthisspeciesleadtotheidentificationofethyl
caffeate,naringeninandchrysoeriolinmixture,eriodictyolfromleaves,andthemixtureof
3-hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)-propan-1-oneandevofolinB,apigenin,themixtureofcaffeicand
protocatechuicacidandluteolinfromstemswithroots,beingreportedforthefirsttimeforV.tweedieana,
exceptforeriodictyol.Thestructuralelucidationofallisolatedcompoundswasachievedby1Hand2D
NMRspectroscopy,andincomparisonwithpublisheddata.AnUPLC-PDAmethodforquantificationof
theeriodictyolinleavesofV.tweedieanawasdevelopedandvalidatedforspecificity,linearity,precision
(repeatabilityandintermediateprecision),limitofdetection(LOD)andlimitofquantification(LOQ),
accuracyandrobustness.Inthisstudy,anexcellentlinearitywasobtained(r2=0.9999),goodprecision
(repeatabilityRSD=2%andintermediateprecisionRSD=8%)andaccuracy(averagerecoveryfrom98.6%
to99.7%).ThecontentoferiodictyolintheextractofleavesofV.tweedieanawas41.40±0.13mg/g.Thus,
thisstudyallowedtheoptimizationofasimple,fastandvalidatedUPLC-PDAmethodwhichcanbeused
tosupportthequalityassessmentofthisherbalmaterial.
©2015SociedadeBrasileiradeFarmacognosia.PublishedbyElsevierEditoraLtda.Allrightsreserved.
Introduction
Naturalproducts,aswellasmedicinalplants,areconsideredas
agoodsourceforthedevelopmentofmedicinesanddrugsandas
atoolforthediscoveryofinnovativemolecularpatterns(Alvim etal.,2006;BalunasandKinghorn,2005;Craggetal.,2009;Cragg andNewman,2013).
BelongingtotheAsteraceaefamily,Vernonanthuratweedieana
(Baker)H.Rob.isknowninBrazilas“assapeixe”and“mata-pasto”
and theirleavesare popularlyused for thetreatmentof
respi-ratory diseases(Cabrera and Klein, 1980; Trevisanetal., 2012; Zanon, 2006; Zanon et al., 2008).Extracts of this species have
demonstrated antibacterial (Díaz et al., 2008),
immunomodula-tory(Petrietal.,2008),antioxidant(Zanon,2006),antinociceptive andantiedematogenic(Trevisanetal.,2012),antifungal(Portillo et al., 2005) and allelopathic (Olguin et al., 2005) activities.
∗ Correspondingauthor.
E-mail:[email protected](M.W.Biavatti).
AmongthesecondarymetabolitesalreadydescribedforV.
tweed-ieanaarelupeol,␣-and-amyrin,-sitosterol,stigmasterol(Díaz et al., 2008; Portillo et al., 2005; Zanon, 2006; Zanon et al., 2008), ␣-spinasterol (Trevisanet al.,2012; Zanon etal., 2008), eriodictyol(Rossatoetal.,2011;Zanon,2006), 6-cinnamoyloxy-1-hydroxyeudesm-4-en-3-one(Portilloetal.,2005)andpalmitic acid(Díazetal.,2008).
For the evaluation of quality of raw materials and
phy-tomedicinesisfundamentaltoknowandtostandardizethecontent oftheirmaincomponents(Baraetal.,2006).Sinceherbalextracts
contain many different chemical components, techniques with
highsensibilityand selectivityaremandatorytotheiradequate
qualitycontrol.Inthecontext,ultraperformanceliquid
chromatog-raphy (UPLC) has shorter run time and higher sensitivity and
resolutionwhencompared toHPLCandisaneleganttechnique
fordetectionandscreeningofcomplexanalytesinalargenumber
ofsamples(SteinmannandGanzera,2011;WalterandAndrews,
2014).
InordertobetterexplorethechemicalcompositionofV. tweed-ieana, ethanol extracts fromstems with roots and from leaves
http://dx.doi.org/10.1016/j.bjp.2015.07.009
(thelatterbeingpreparedaccordingtothepopularuse)were pre-paredandchemicallyfractioned,conductingtotheidentification
ofitsmaincompounds.Additionally,consideringtheimportance
ofqualitycontrolofV.tweedieanaleavesduetoitspopularuse,a
newandsimpleUPLC-PDAmethodforquantificationofitsmajor
compound(eriodictyol)wasdevelopedandvalidatedinthiswork.
Materialsandmethods
Chemicals
The solvents used in chromatographic procedures were
p.a. grade: n-hexane, dichloromethane, ethyl acetate, acetone,
methanol(Vetec®QuímicaFina,RiodeJaneiro,RJ,Brazil).For UPLC-PDAanalyseswereusedacetonitrileHPLCgrade(Tedia®Brazil,Rio deJaneiro,RJ,Brazil),formicacid85%p.a.grade(Vetec®Química
Fina,Rio de Janeiro, RJ, Brazil) and thewater was obtainedby
Millipore® MilliQ(Bedford,MA,USA)waterpurificationsystem. AllthesolutionspreparedforUPLCanalyseswerefilteredthrough
a0.22mmembraneandsonicatedbeforeuse.ForTLCanalyses
plates of silicagel 60 (SiliCycle®, Quebec,QC, Canada) and
sil-icaRPC18(Macherey-Nagel®,Düren,NRW,Germany)wereused.
ForNMRanalysesmethanol-d4andacetona-d6(CambridgeIsotope
Laboratories,Inc.,Andover,MA,USA)wereused.
Plantmaterial
Vernonanthura tweedieana (Baker) H. Rob., Asteraceae, was collectedduringbloomingseasoninFlorianópolis (27◦33′54.8′′S
48◦25’48.0′′W and 27◦35′54.7′′S 48◦31′00.3′′W), Santa Catarina,
BrazilinApril2013,andidentifiedbyProfessorDr.AdemirReis.A
voucherspecimenwasdepositedintheHerbariumRB(RB612274),
attheJardimBotânicodoRiodeJaneiro,RiodeJaneiro/RJ,Brazil.
Extractionandpartition
Thefreshleaves (2.6kg) and stemswith roots(5.9kg) were
extractedbymacerationwithethanol75%atroomtemperature,
for56days.Theextractivesolutionswerefilteredthroughpaper;
thesolventwasremovedunderreducedpressureandusedto
re-extracttheplantmaterial.Theresultingcrudeextractsfromleaves (ELF,172g)andfromstemswithroots(ESR,291.6g)aftersolvent removalwerekeptinadesiccatorinvacuo.
Bothcrude extracts were partitioned withincreasing
polar-itysolvents,yieldingn-hexane(17.9g),dichloromethane(CH2Cl2,
3.6g), ethyl acetate (EtOAc, 10.4g) and the aqueous residual
(104.3g)fractionsfromELF,andn-hexane(10.9g),CH2Cl2(4.2g),
EtOAc(2.5g)andtheaqueousresidual(244.9g)fractionsfromESR.
Phytochemicalstudy
TheCH2Cl2(3.6g)fractionfrompartitionofELFwassubjected
tosilicagel(230–400mesh)liquidchromatographyundervacuum
(flash-LCV)andelutedinagradientsystemconsistingofincreasing concentrationsofCH2Cl2inn-hexane(0–70%),followedbyacetone
inCH2Cl2 (0–80%)andfollowedbymethanol(MeOH)inacetone
(0–100%),yieldingseventeensubfractions(LF1–LF17).Pooled sub-fractionsLF7andLF8(676.6mg)wasalsosubjectedtoflash-LCV, elutedinagradientsystemconsistingofincreasingconcentrations ofCH2Cl2 inn-hexane(40–70%),followedbyacetonein CH2Cl2
(0–70%)andfollowedbyMeOHinacetone(0–100%),yielding
six-teen subfractions, named LF7-8A to LF7-8P. Subfraction LF7-8F
(368.7mg)wassubjectedtosilicagelcolumnchromatography(CC) andelutedinagradientsystemconsistingofincreasing
concentra-tionsofacetoneinhexane(25–100%)followedbyMeOH(100%),
yieldingtensubfractions,namedLF7-8FItoLF7-8FX,inwhichthe fractionLF7-8FVIIIcorrespondstocompound1(37.3mg).
TheEtOAc(10.4g)fractionfrompartitionofELFwassubjectedto flash-LCV,elutedinagradientsystemconsistingofincreasing con-centrationsofCH2Cl2inhexane(30–70%),followedbyacetonein
CH2Cl2(0–80%)andfollowedbyMeOHinacetone(0–100%),
yield-ingeighteensubfractions(LF18–LF35).SubfractionLF25(319mg)
wassubjectedtosilicagelCCelutedinagradientsystem consist-ingof increasingconcentrations of CH2Cl2 inhexane (80–95%),
followed byacetonein CH2Cl2 (0–70%)and followedby MeOH
inacetone(0–100%),resultingelevensubfractions(LF25A–LF25K).
ThesubfractionLF25G(22mg)wasfractionatedthroughSephadex
LH-20withacetoneaseluent,yieldingsevensubfractions,named
LF25GItoLF25GVII,whichfractionLF25GVcorrespondstothe mix-ture(3.4mg)ofcompounds2and3.SubfractionLF26(2079.3mg)
wassubjectedtosilica gelCCeluted ina gradient system
con-sisting of increasing concentrations of CH2Cl2 in hexane (80%),
followedbyacetoneinCH2Cl2(0–30%)andfollowedbyMeOHin
acetone(0–50%),untilwaterinMeOH(0–10%),resulting
seven-teensubfractions(LF26A-LF26Q).PooledsubfractionsLF26Kand
LF26L(262.1mg)wassubjectedtosilicagelCC,elutedina
gradi-entsystemconsistingofincreasingconcentrationsofacetonein
CH2Cl2 (15–70%)followedbyMeOHinacetone(0–100%),
yield-ingninesubfractions,namedLF26K-LItoLF26K-LIX,whichfraction
LF26K-LIVcorrespondstocompound4(104.7mg).
The CH2Cl2 (4.2g) fraction from partition of ESR was
sub-jectedtoflash-LCVandelutedinagradientsystemconsistingof increasingconcentrationsofCH2Cl2 inhexane(0–70%),followed
by EtOAc in CH2Cl2 (0–80%) and followed by MeOH in EtOAc
(0–70%),untilwaterinMeOH(0–50%),yieldingeighteen
subfrac-tions(SR1–SR18).ThesubfractionSR10(112mg)wassubjectedto
silicagelCC,elutedinagradientsystemconsistingofincreasing concentrationsofacetoneinCH2Cl2(3–70%)followedbyMeOHin
acetone(0–100%),yieldingtwelvesubfractions,namedSR10Ato
SR10L.ThesubfractionSR10E(19.7mg)wasfractionatedthrough
SephadexLH-20withacetoneaseluent,yieldingsixsubfractions, namedSR10EItoSR10EVI,whichfractionSR10EVIcorrespondsto themixture(0.8mg)ofcompounds5and6.
TheEtOAc(2.5g)fractionfrompartitionofESRwassubjected toflash-LCV,elutedinagradientsystemconsistingofincreasing concentrationsofEtOAcinCH2Cl2(0–70%)andfollowedbyMeOH
inEtOAc(0–100%),yieldingsevensubfractions(SR19-SR25).The
subfractionSR21(135.9mg)wassubjectedtosilicagelCC,eluted inagradientsystemconsistingofincreasingconcentrationsof
ace-toneinCH2Cl2 (3–70%)followedbyMeOHinacetone(0–100%),
yieldingfifteensubfractions,namedSR21AtoSR21O.The subfrac-tionSR21H(16.7mg)wasalsosubjectedtosilicagelCC,elutedin agradientsystemconsistingofincreasingconcentrationsofMeOH inCH2Cl2 (5–100%),resultingsixsubfractions,namedSR21HIto
SR21HVI.ThesubfractionSR21HV(9.6mg)wassubjectedto
prepar-ativeRPC18 TLC,elutedwithwater-MeOH(30:70,v/v)allowing
theisolationofcompounds4(4.9mg)and7(0.7mg).The
subfrac-tionSR21K(21.3mg)wasfractionatedthroughSephadexLH-20
withacetoneaseluent,yieldingeightsubfractions,namedSR21KI toSR21KVIII,whichfractionSR21KVcorrespondstothemixture
(11mg)ofcompounds8and9,andthefractionSR21KVII
corre-spondstothecompound10(1mg).
The elucidation of thecompounds was achieved by 1H and
2D(HSQCandHMBC)NMR spectroscopy,in Bruker® Ascend600
(600MHzforH,125MHzforC),andincomparisonwithpublished
data.
Ethylcaffeate(1):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)
J(Hz),7.16(1H,d,J=2.0,H-2),6.87(1H,d,J=8.2,H-5),7.04(1H, dd,J=8.2;2.0,H-6),7.54(1H,d,J=15.9,H-7),6.27(1H,d,J=15.9, H-8),4.18(2H,q,J=7.2,H-1′),1.27(3H,t,J=7.2,H-2′),8.17(1H,
br,OH-3)and8.41(1H,br,OH-4).13CNMR(acetone-d
TMS)ıC(ppm),127.5(C-1),115.2(C-2),146.4(C-3),148.7(C-4),
116.3(C-5),122.3(C-6),145.2(C-7),115.8(C-8),167.4(C-9),60.5 (C-1′)and14.7(C-2′).
Naringenin(2):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)
J(Hz),5.45(1H,dd,J=12.9;3.0,H-2),3.17(1H,dd,J=17.1;12.9, H-3a),2.74(1H,dd,J=17.1;3.0,H-3b),5.95(1H,d,J=2.1,H-6),5.96 (1H,d,J=2.1,H-8),7.39(2H,d,J=8.5,H-2′,H-6′),6.90(2H,d,J=8.5,
H-3′,H-5′)and12.17(1H,s,OH-5).13CNMR(acetone-d
6,150MHz,
TMS)ıC(ppm),80.0(C-2),43.5(C-3),197.2(C-4),166.8(C-5),96.4
(C-6),161.8(C-7),95.9(C-8),164.5(C-9),103.2(C-10),130.7(C-1′),
129.0(C-2′,C-6′),116.4(C-3′,C-5′)and158.7(C-4′).Thiscompound
wasidentifiedinmixturewithcompound3. Chrysoeriol(3):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)
J(Hz),6.69(1H,s,H-3),6.26(1H,d,J=2.0,H-6),6.55(1H,d,J=2.0, H-8),7.63(1H,d,J=2.1,H-2′),7.01(1H,d,J=8.3,H-5′),7.60(1H,dd, J=8.3;2.1,H-6′),4.00(3H,s,OCH
3-1′′)and13.00(1H,s,OH-5).13C
NMR(acetone-d6,150MHz,TMS)ıC(ppm),164.9(C-2),104.5
(C-3),182.9(C-4),163.3(C-5),99.7(C-6),165.0(C-7),94.7(C-8),158.7 (C-9),105.2(C-10),123.6(C-1′),110.7(C-2′),148.8(C-3′),151.3
(C-4′),116.4(C-5′),121.4(C-6′)and56.6(C-1′′).Thiscompoundwas
identifiedinmixturewithcompound2. Eriodictyol(4):1HNMR(methanol-d
4,600MHz,TMS),ıH(ppm)
J(Hz),5.28(1H,dd,J=12.8;3.0,H-2),3.06(1H,dd,J=17.2;12.8, H-3a),2.69(1H,dd,J=17.2;3.0,H-3b),5.88(1H,d,J=2.2,H-6),5.90 (1H,d,J=2.2,H-8),6.91(1H,m,H-2′),6.78(1H,m,H-5′)and6.79
(1H,m,H-6′).13CNMR (methanol-d
4,150MHz,TMS)ıC (ppm),
80.7(C-2),44.2(C-3),197.8(C-4),165.6(C-5),97.2(C-6),168.6 (C-7),96.2(C-8),164.9(C-9),103.5(C-10),131.8(C-1′),114.7(C-2′),
146.1(C-3′),147.1(C-4′),116.6(C-5′)and119.2(C-6′).
3-Hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)-propan-1-one (5):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)J(Hz),7.34(2H,
s,H-2,H-6),3.17(2H,d,J=6.2,H-8),3.91(2H,m,H-9)and3.91 (6H,s,OCH3-3,OCH3-5).13CNMR(acetone-d6,150MHz,TMS)ıC
(ppm),129.3(C-1),107.0(C-2,C-6),148.5(C-3,C-5),142.0(C-4), 198.2(C-7),41.8(C-8),58.6(C-9)and56.9(OCH3-3,OCH3-5).This
compoundwasidentifiedinmixturewithcompound6.
EvofolinB(6):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)
J(Hz),7.60(1H,d,J=2.0,H-2),6.84(1H,d,J=8.3,H-5),7.66(1H, dd,J=8.3;2.0,H-6),4.80(1H,dd,J=8.5;5.2,H-8),4.23(1H,m, H-9a),3.71(1H,m,H-9b),6.99(1H,d,J=2.0,H-2′),6.74(1H,d,J=8.1,
H-5′),6.80(1H,dd,J=8.1;2.0,H-6′),3.88(3H,s,OCH
3-3)and3.82
(3H,s,OCH3-3′).13CNMR(acetone-d6,150MHz,TMS)ıC(ppm),
130.5(C-1),112.3(C-2),148.2(C-3),152.1(C-4),115.3(C-5),124.5 (C-6),198.2(C-7),55.9(C-8),65.5(C-9),129.1(C-1′),112.8(C-2′),
148.5(C-3′),146.6(C-4′),115.9(C-5′),121.9(C-6′),56.3(OCH
3-3)
and56.4(OCH3-3′).Thiscompoundwasidentifiedinmixturewith
compound5.
Apigenin(7):1HNMR(methanol-d
4,600MHz,TMS),ıH(ppm)J
(Hz),6.60(1H,s,H-3),6.21(1H,d,J=2.1,H-6),6.46(1H,d,J=2.1, H-8),7.85(2H,d,J=8.9,H-2′,H-6′)and6.93(2H,d,J=8.9,H-3′,
H-5′).13CNMR(methanol-d
4,150MHz,TMS)ıC(ppm),166.3(C-2),
102.9(C-3),183.9(C-4),163.2(C-5),100.2(C-6),166.1(C-7),95.0 (C-8),159.5(C-9),105.3(C-10),123.3(C-1′),129.6(C-2′,C-6′),117.1
(C-3′,C-5′)and162.7(C-4′).
Caffeicacid(8):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)J
(Hz),7.16(1H,d,J=2.0,H-2),6.87(1H,d,J=8.2,H-5),7.03(1H,dd, J=8.2;2.0,H-6),7.55(1H,d,J=15.9,H-7)and6.27(1H,d,J=15.9, H-8).13CNMR(acetone-d
6,150MHz,TMS)ıC(ppm),127.6(C-1),
115.2(C-2),146.3(C-3),148.7(C-4),116.4(C-5),122.5(C-6),146.1 (C-7),115.9(C-8)and168.3(C-9).Thiscompoundwasidentifiedin
mixturewithcompound9.
Protocatechuicacid(9):1HNMR(acetone-d
6,600MHz,TMS),ıH
(ppm)J(Hz),7.53(1H,d,J=2.0,H-2),6.90(1H,d,J=8.3,H-5)and 7.48(1H,dd,J=8.3;2.0,H-6).13CNMR(acetone-d
6,150MHz,TMS)
ıC(ppm),123.2(C-1),117.6(C-2),145.5(C-3),150.6(C-4),115.8
(C-5),123.7(C-6)and167.7(C-7).Thiscompoundwasidentifiedin
mixturewithcompound8.
Luteolin(10):1HNMR(acetone-d
6,600MHz,TMS),ıH(ppm)J
(Hz),6.57(1H,s,H-3),6.25(1H,d,J=2.1,H-6),6.53(1H,d,J=2.1, H-8),7.50(1H,d,J=2.3,H-2′),7.00(1H,d,J=8.4,H-5′),7.46(1H,
dd,J=8.4;2.3,H-6′)and13.00(1H,s,OH-5).13CNMR(acetone-d
6,
150MHz,TMS)ıC(ppm),165.3(C-2),104.2(C-3),182.9(C-4),163.3
(C-5),99.6(C-6),164.9(C-7),94.7(C-8),158.9(C-9),105.3(C-10), 123.7(C-1′),114.1(C-2′),146.6(C-3′),150.2(C-4′),116.6(C-5′)and
120.1(C-6′).
Ultraperformanceliquidchromatographyoptimizedconditions
TheUPLCanalyseswerecarriedoutinaWaters®AcquityUPLC
systemequippedwithphotodiodearray(PDA)detection,anauto
samplerandcolumnoven.Thechromatographicseparationwas
achievedusingaWaters®AcquityUPLCBEHC
18(2.1mm×50mm
i.d.,1.7m)at40◦C(±2◦C).Theinjectionvolumewas2land
theflowratewaskeptconstantat0.4mlmin−1.Themobilephase
consisted of a 7min gradient system combining 0.1% aqueous
formicacidadjustedtopH3.0 (solventA)andacetonitrile (sol-ventB)asfollows:0–1mingradientof90–80%A(curve7);1–5min
80–55%A(curve7);5–6min55–35%A(curve9);6–7min35–90%A
(curve9).DatawereprocessedusingtheEmpower3(Waters®)
soft-ware.Thechromatogramswererecordedatwavelengthof287nm
whiletheUVspectraweremonitoredoverarangeof400–200nm.
PeakswereidentifiedbycomparingretentiontimesandUVspectra withreferencestandards.
Stocksolutionspreparation
Thequantificationoferiodictyolinthesampleswascarriedout
bytheexternalstandardmethod.Eriodictyol(purity≥98%,
veri-fiedbyPeakPuritytoolinEmpower3software)usedinthisworkas analyticalstandardwasobtainedfromphytochemicalinvestigation ofEtOAcfractionfrompartitionofELFofV.tweedieana.Thestock standardsolutionsoferiodictyol(1000and500g/gofsolution)
were freshly preparedin 0.1% aqueousformic acid–acetonitrile
(1:1,v/v) mixture.Thestockstandard solutionwasthen
appro-priatelydilutedin0.1%aqueousformicacid–acetonitrile(1:1,v/v) toobtainworkingstandardsolutions.
The sample solutions analyzed by UPLC-PDA were obtained
fromthecrudeextractsELFandESR,resultingofmacerationfrom leavesand fromstemswithrootsofV.tweedieana,respectively.
Bothcrudeextracts,ELFandESR,wereusedtopreparethestock
samplesolutions(1000g/gofsolution)in0.1%aqueousformic
acid–acetonitrile(1:1,v/v).Thestocksamplesolutionswerealso appropriatelydilutedin0.1%aqueousformicacid–acetonitrile(1:1, v/v)toobtainworkingsamplesolutions(about200g/gof solu-tion).
Thesestocksolutionswerefilteredthrougha0.22m
mem-braneandstoredat4◦C.
ValidationofUPLC-PDAmethod
TheUPLC-PDAmethodwasvalidatedintermsofspecificity, lin-earity,precision(repeatabilityandintermediateprecision),limit ofdetection(LOD)andlimitofquantification(LOQ),accuracyand robustness(Araujo,2009;ICH,2005;Shabir,2003;USP,2007).
Inordertocheckthespecificityofthemethod,thepurityof eriodictyolintheextractofV.tweedieanawasverifiedusingaPDA
detectorandthesoftwareEmpower3(Waters®
).
Linearitywasdeterminedwithcalibrationcurves,whichwere
constructed by plotting themeasurements of areapeak versus
resultswere analyzed by linear regression and the correlation coefficient(r2)wascalculated.One-wayANOVAwascalculatedto
comparethereplicatesofthecalibrationcurve(GraphPadPrism® version6.03).
Theprecisionofthemethodwasassessedinrepeatability
(intra-day) and intermediate precision (inter-day). Repeatability was
establishedbyanalyzingtherelativestandarddeviation(RSD%)of 6independentsamplesolutionsdeterminationsat100%ofthetest
concentration(200g/g).Intermediateprecisionwasdetermined
consideringtheRSD%bytheanalysisinsextuplicatebytwo
ana-lystsinthreedifferentdays(analystAperformedtheanalysisin
daysoneandtwo,whileanalystBperformedtheanalysisinday
three).
TheLODandLOQwereassessedatsignal-to-noiseof3and10,
respectively,byinjectingaseriesofdilutesolutionswithknown concentration.
Theaccuracywasevaluatedbytherecoverytest.Threedifferent concentrationsofstockstandardsolution(500g/g),
correspond-ingtheadditionof10, 14and 18g/gofsolution,werespiked
intoaknownsamplesolution.Thespikedsample,unspikedsample
andcorrespondingaddedstandard solutionsweremeasured, in
triplicate,underthesameconditions.Therecoverywascalculated bythereasonbetweentheconcentrationofspikedsampleanalyzed
andthetheoreticalspikedconcentration(establishedbythesum
ofmeasuredconcentrationsofunspikedsampleandthestandard
added).
Therobustnessofthemethodwasdeterminedrelativetothe
variation of retention time, resolution and tailingfactor of the peak(USP, 2007), and concentrationof eriodictyolfor
determi-nationsin nominalconditionsand againstvariationsofmethod
conditions.Experimentalvariablesweretheflowratevariedby(±) 0.1mlmin−1,columnoventemperaturevariedby(±)10◦Candthe
percentageoforganicmodifiervariedby(±)0.05%.Theresultswere
evaluatedbyone-wayANOVAfollowedbyBonferroniposthoctest,
withaconfidencelevelof95%(GraphPadPrism®version6.03).
Resultsanddiscussion
Phytochemicalstudy
Thephytochemicalinvestigationallowedcharacterizingten
dif-ferent known phenolic compounds from ethanol extractsof V.
tweedieana.Thesecompoundswereidentifiedby1Hand2DNMR
experimentsincomparisonwithpublisheddata.Consideringthat
themajorityofthepreviouslypublishedNMRdatapresentedsome
inconsistenciesregardingchemicalshifts(ppm)orwerenotinthe samesolvent,thenewdataarepresentedinMaterialsandMethods
section.Forcompound4differentchemicalshiftsofH-3aandH-3b
werepreviouslydescribedin2.69ppmforboth,andnowassigned
in3.06and2.69ppmrespectively.Forcompound5,thechemical
shiftsforcarbonsC-2,C-4,C-5andC-6werepreviouslypublishedas
115.5,152.9,112.4and124.6ppm,andnowassignedrespectively
in107.0,142.0,148.5and107.0ppm.Forcompound7,thechemical
shiftsH-6andH-8previouslypublishedin6.45and6.16ppmare
nowassignedin6.21and6.46ppm,respectively.Thecompounds
8and9werepublishedbeforeinmethanol-d4andnowacetone-d6
wasused.FromCH2Cl2 fractionofleaveswasisolatedethyl
caf-feate(1)(Uwaietal.,2008),andfromEtOAcfractionwasobtained naringenin(2)(Fatope etal.,2003)andchrysoeriol(3)(Lin and Kong,2006)inmixture,anderiodictyol(4)(Huangetal.,2014).
Fromstemswithrootsextract,theCH2Cl2fractionfurnished
3-hydroxy-1-(4-hydroxy-3.5-dimethoxyphenyl)-propan-1-one (5)
(Jonesetal.,2000)andevofolinB(6)(Wuetal.,1995)inmixture, andfromEtOAcfractionwereobtainedagaintheeriodictyol(4), apigenin(7)(Kimetal.,2014), caffeic(8)(Leeetal.,2012)and protocatechuic(9)(Liaoetal.,2014)acidsinmixtureandluteolin (10)(LinandKong,2006).
Exceptforeriodictyolthatwaspreviouslyreportedfortheleaves ofV.tweedieana(Rossatoetal.,2011;Zanon,2006),allsubstances aredescribedforthefirsttimeinthisspecies.Theseresultsallow increasingtheknowledgeforthespeciesV.tweedieana.
UPLC-PDAanalysesandmethodvalidation
Bothcrudeethanolextracts,fromleavesandfromcombination
ofstemswithrootsofV.tweedieana,wereanalyzedbyUPLC-PDA
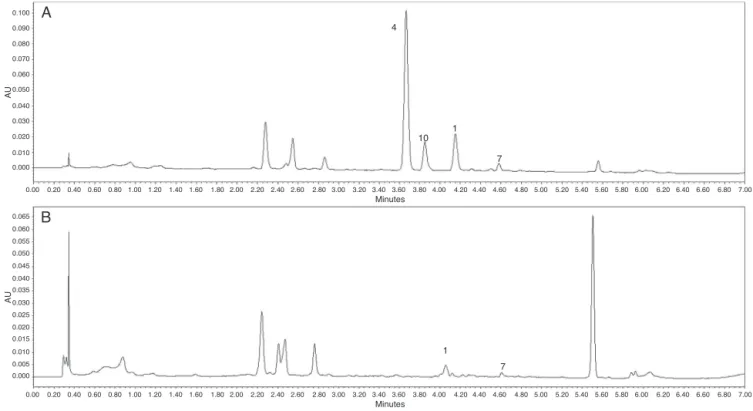
andthechromatogramsareshowedinFig.1A,B.
The comparative analysis of the crude extracts with all
isolatedcompoundsallowedcharacterizingfourpeaksinthe chro-matograms: ethyl caffeate (1), eriodictyol(4), apigenin (7) and luteolin(10).
The UPLC-PDA analyses of the two crude ethanol extracts
showeddistinctchromatographicprofiles.Eriodictyol(4)wasthe majorcompoundinELF,whileitwasnotobservedintheESR
chro-matogram,probablybecauseitscontentwasnotdetectable,since
theamountoferiodictyolisolatedfromESRwasverylow(4.9mg, 0.047%ofthecrudeextract).Flavonoidcontentvarieswithin indi-vidualleaves, stems or roots (Julkunen-Tiittoet al., 2015).The
presence of flavonoids within different cells and cellular
com-partmentscanberelatedtotheirfunctionsinplantenvironment
0.00 0.000 0.010 0.020 0.030 0.040 AU0.050 0.060 0.070 0.080 0.090 0.100
0.20 0.40 0.60 0.80 1.00 1.20 1.40 1.60 1.802.00 2.20 2.40 2.60 2.80 3.00 3.20 3.40 Minutes
4
A
10 1
7
3.60 3.804.00 4.20 4.40 4.60 4.80 5.00 5.20 5.40 5.60 5.80 6.00 6.20 6.40 6.60 6.80 7.00
0.00 0.000 0.005 0.010 0.015 0.020 0.025 0.030 0.035 0.040 0.045 0.050 0.055 0.060 0.065
AU
0.20 0.40 0.60 0.80 1.00 1.20 1.40 1.60 1.802.00 2.20 2.40 2.60 2.80 3.00 3.20 3.40 Minutes
B
1
7
3.60 3.804.00 4.20 4.40 4.60 4.80 5.00 5.20 5.40 5.60 5.80 6.00 6.20 6.40 6.60 6.80 7.00
Fig.1. UPLCchromatogramsofcrudeethanolextracts(200g/g)fromleaves(A)andfromcombinationofstemswithroots(B)ofV.tweedieana,withdiodearraydetection at287nm.1.ethylcaffeate;4.eriodictyol;7.apigenin;10.luteolin.Forchromatographicconditions,seeMaterialsandmethodssection.
Table1
Linearityandsensitivitydata.
Compound UVmax(nm) Rangeoflinearitya(g/g) Linearregressionequation r2 LOD(g/g) LOQ(g/g)
Eriodictyol 287.6 1.5–30.0 y=16,211x−277.7 0.9999 0.02344 0.06214
r2,correlationcoefficient;LOD,limitofdetection;LOQ,limitofquantification. aSevenlevelsconcentrations(n=3).
Consideringtheuseofleavesinpopularmedicinalpreparations ofV.tweedieanainBrazil(Trevisanetal.,2012),thecrudeextractof leaveswasanalyzedbyUPLC-PDA,inordertoverifythepotential oferiodictyolaschemicalmarkerforthisplantextract.
Thecalibration curves werefoundto belinear inthe range
1.5–30g/gofsolutionforeriodictyol.
CharacteristicparametersforlinearityaregiveninTable1.The
one-wayANOVAcalculatedtocomparethereplicatesofthe
cal-ibrationcurveshowednosignificantdifferencebetweengroups
(F=0.003254;p=0.9968).Thelinearregressionequationand cor-relationcoefficient(r2)of0.9999revealedagoodlinearityresponse
forthemethoddeveloped,sinceaccordingtotheliteraturea
coef-ficient ofdetermination for standard curves greater than0.999
indicatesanevidenceofacceptablefitofthedatatotheregression line(Shabir,2003).
TheLODand LOQvalues(Table1), respectively0.02344and
0.06214g/g,weresimilartothosereportedintheliteraturefora HPLCmethodforquantificationoferiodictyolinSorghumbicolour (Taleonetal.,2014).Thepeak purity,obtainedbyPDAdetector analysisoferiodictyolwasinagreementwithitspurityspectral profile.
Theresultsofrepeatability(intra-dayvariation)and
intermedi-ateprecision(inter-dayvariation)showedRSD%valuesof2%and
8%,respectively(Table2).Analyticalproceduresforbiologicaland biotechnologicalproductscanpresentahigherinherentvariation.
Naturalproductshavehighchemicalcomplexity,andconsidering
thatallreplicatesperformedinthisworkinvolvedthepreparation ofnewstockandworksamplessolutions,itcanrepresentasource ofvariation.However,thedataobtainedforprecisionwas accept-ableandisinaccordancewithotheranalyticalstudieswithsame
Table2
Repeatability,intermediateprecisionandaccuracydata.
Compound Repeatability(n=6) Intermediateprecision(n=18) Accuracy
AnalystA AnalystA AnalystB Standardadded(g) Recoveryb(%) RSD
Day1a RSD Day2a Day3a RSD
Eriodictyol 56.15±1.06 2% 46.66±0.34 53.74±0.57 8%
10 99.7±2.6 3%
14 98.6±1.6 2%
18 99.5±1.4 1%
RSD,relativestandarddeviation.
Table3 Robustnessdata.
Variables Levels Parameters
Concentrationa(mg/g) tRa(min) RSa T
fa
Nominalconditions 42.56±0.04 3.634±0.002 2.937±0.087 1.075±0.007
Flow 0.3mlmin−1 56.62±0.20 4.376±0.015 2.225±0.021 1.143±0.010
0.5mlmin−1 33.93±0.05 3.106±0.003 3.230±0.068 1.076±0.009
Columntemperature 30◦C 42.50±0.02 4.000±0.013 2.262±0.024 1.105±0.007
50◦C 42.22±0.14 3.249±0.017 3.180±0.035 1.092±0.000
Modifiercontent 0.05% 42.06±0.06 3.583±0.018 2.609±0.046 1.087±0.007
0.15% 42.34±0.01 3.546±0.015 2.653±0.038 1.070±0.008
Nominalconditionsareflowat0.4mlmin−1,columntemperatureat40◦Candmodifiercontent(formicacid)at0.1%.tR,retentiontime;RS,resolution(calculatedbetween peaks4and10);Tf,tailingfactor;ThetR,RSandTfvalueswerecalculatedaccordingUSPharmacopeia(2007).
aAverage±standarddeviation(n=3).
naturalsamples(Limetal.,2014;Motilvaetal.,2014;Oketal., 2014;Stincoetal.,2014).
TheaccuracydataaregiveninTable2.Theaveragerecovery
ofthethree amountaddedof eriodictyolrangedfrom98.6%to
99.7%,andtheirRSDvalueswerelessthan3.0%,characterizinggood reliabilityandaccuracyofthemethod.
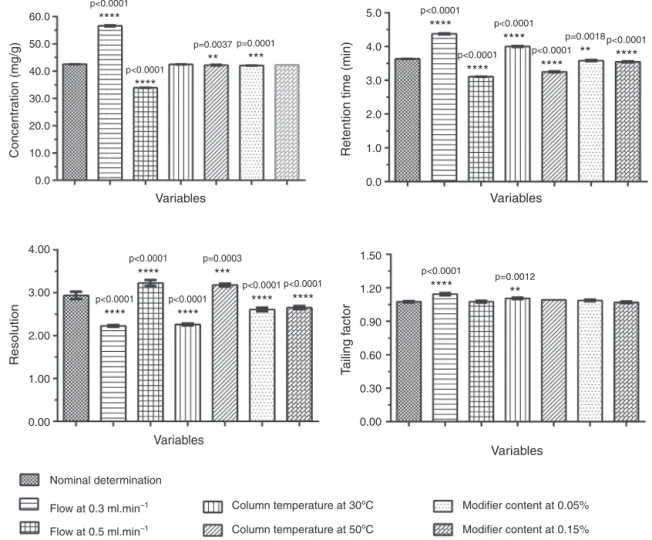
Thedeliberatevariationsanalyzedforrobustnessofthemethod areshowedinTable3.
The robustness was estimated by one-way ANOVA analysis
between the nominal determination and the different
lev-els of each variable (flow, column temperature and modifier
content),andassessedreferenttodifferentparameters
(concen-tration,retentiontime,resolutionandtailingfactorofthepeak) (Fig.2).
Amongrobustness evaluatedparameters,were notobserved
significantdifferencesintheeriodictyolconcentrationforcolumn temperatureat30◦Candmodifiercontentat0.15%variables.The
parametertailingfactorwastheleastaffectedbythedeliberate variations,toleratingchangesofmodifiercontent(lowerandhigher level),higherlevelofflow(0.5mlmin−1)andhigherlevelof
col-umntemperature(50◦C),withnostatisticalsignificantdifferences.
Theflowvariationwasthemostimpactingvariableinthemethod, exceptforthetailingfactor(Table3,Fig.2).Resultsshowthatall variablesarecriticalforthemethodandshouldbecarefully con-trolledduringtheanalysis.
Thequantitativeanalysisoferiodictyolinleavescrudeextract from of V. tweedieana showed a content of 41.40±0.13mg/g
(RDS=0.31%). Eriodictyolhasbeendescribed and quantifiedfor
60.0
p<0.0001
****
p=0.0001
***
p<0.0001****
p=0.0037
**
50.040.0
30.0
20.0
Concentr
ation (mg/g)
10.0
0.0
Variables
5.0 p<0.0001
****
p<0.0001****
p<0.0001
****
p<0.0001
****
p=0.0018**
p<0.0001****
4.04.00
3.0
3.00
2.0
2.00
1.0
1.00
Retention time (min)
0.0
0.00
Nominal determination
Flow at 0.3 ml.min–1 Column temperature at 30ºC Modifier content at 0.05%
Modifier content at 0.15% Column temperature at 50ºC
Flow at 0.5 ml.min–1
Variables
p<0.0001
****
p<0.0001
****
p<0.0001
****
p<0.0001
****
p<0.0001
****
p=0.0003
***
Resolution
Variables
p<0.0001
****
p=0.0012**
T
ailing f
actor
0.00 0.30 0.60 0.90 1.20 1.50
Variables
someotherplantextracts,butthecontentfoundintheleavesof V.tweedieanaseemstobesuperiortopreviouspublishedstudies (Borosetal.,2010;Linetal.,2007;Renetal.,2008).
Insummary,thephytochemicalinvestigationallowsto
char-acterizeten differentcompounds,ethyl caffeate(1), naringenin (2),chrysoeriol(3),eriodictyol(4), 3-hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)-propan-1-one(5),evofolinB(6),apigenin(7), caffeicacid(8),protocatechuicacid(9)andluteolin(10),reported forthefirsttimeforV.tweedieanaexceptforeriodictyol.Further,a simpleandfastUPLC-PDAmethodforquantificationoferiodictyol
incrudeextractfromleavesofV.tweedieanawasdevelopedand
validated,whichcanbeusedtothequalitycontrolpreparations
thatcontainsV.tweedieana.
Authors’contributions
LALS (MSc student) contributed in collecting plant sample,
phytochemicalinvestigation,elucidationofcompounds,
chromato-graphic and validation analyses, data analysesand draftedthe
paper.LGF(MScstudent)contributed inrunningthelaboratory
work,validationanalysesanddraftedthepaper.FHRcontributed
todesignandinterpretationofthevalidatedanalyticalconditions.
ADCS(PhDstudent)andABcontributedtoNMRanalysesand
elu-cidationofcompounds.MWBdesignedthestudy,supervisedthe
laboratorywork,dataanalysesandcontributedtocriticalreading anddraftofthemanuscript.
Conflictofinterest
Theauthorsdeclarenoconflictofinterest.
Acknowledgments
TheauthorsthankCNPqandCAPESforthefinancialsupportand thescholarships.TheauthorsalsothankProfessorDr.AdemirReis fortheidentificationofplantmaterial.
References
Agati,G.,Azzarello,E.,Pollastri,S.,Tattini,M.,2012.Flavonoidsasantioxidantsin plants:Locationandfunctionalsignificance.PlantSci.196,67–76.
Alvim,N.A.T.,Ferreira,M.A.,Cabral,I.E.,AlmeidaFilho,A.J.,2006.Theuseof medic-inalplantsasatherapeuticalresource:fromtheinfluencesoftheprofessional formationtotheethicalandlegalimplicationsofitsapplicabilityasanextension ofnursingcarepractice.Rev.Lat.Am.Enferm.14,316–323.
Araujo,P.,2009.Keyaspectsofanalyticalmethodvalidationandlinearityevaluation. J.Chromatogr.BAnalyt.Technol.Biomed.LifeSci.877,2224–2234.
Balunas,M.J.,Kinghorn,A.D.,2005.Drugdiscoveryfrommedicinalplants.LifeSci. 78,431–441.
Bara, M.T.F., Ribeiro, P.A.M., Arantes, M.C.B., Amorim, L., Paula, J.R., 2006.
Determinac¸ãodoteordeprincípiosativosemmatérias-primasvegetais.Braz.J. Pharmacogn.16,211–215.
Boros,B.,Jakabova,S.,Dornyei,A.,Horvath,G.,Pluhar,Z.,Kilar,F.,Felinger,A., 2010.Determinationofpolyphenoliccompoundsbyliquid chromatography-massspectrometryinThymusspecies.J.Chromatogr.A1217,7972–7980.
Cabrera,A.L.,Klein,R.M.,1980.FascículoCompostas:3.TriboVernonieae.In:Reitz, R.(Ed.),FloraIlustradaCatarinense.HerbárioBarbosaRodrigues,Itajaí/SC,Brasil, pp.324–327.
Cragg,G.M.,Grothaus,P.G.,Newman,D.J.,2009.Impactofnaturalproductson devel-opingnewanti-canceragents.Chem.Rev.109,3012–3043.
Cragg,G.M.,Newman,D.J.,2013.Naturalproducts:acontinuingsourceofnoveldrug leads.Biochim.Biophys.Acta1830,3670–3695.
Díaz,G.,Nogueira,M.A.,Olguín,C.F.A.,Somensi,A.,Vidotti,G.J.,2008.Estudo fito-químicoebiológicodeVernoniatweedianaBaker(Asteraceae).Lat.Am.J.Pharm. 27,56–61.
Fang,J.,Reichelt,M.,Hidalgo,W.,Agnolet,S.,Schneider,B.,2012.Tissue-specific distributionofsecondarymetabolitesinrapeseed(BrassicanapusL.).PLoSONE 7,e48006.
Fatope,M.O.,Al-Burtomani,S.K.S.,Ochei,J.O.,Abdulnour,A.O.,Al-Kindy,S.M.Z., Takeda,Y.,2003.Muscanone:a3-O-(1′′,8′′,14′′-trimethylhexadecanyl) narin-geninfromCommiphorawightii.Phytochemistry62,1251–1255.
Huang,Y.-H.,Zeng,W.-M.,Li,G.-Y.,Liu,G.-Q.,Zhao,D.-D.,Wang,J.,Zhang,Y.-L.,2014.
Characterizationofanewsesquiterpeneandantifungalactivitiesofchemical constituentsfromDryopterisfragrans(L.)Schott.Molecules19,507–513.
ICH,H.T.G.,2005.Validationofanalyticalprocedures:textandmethodology,Q2 (R1).CurrentStep4Version,ParentGuidelinesonMethodologyDated Novem-ber61996.IncorporatedinNovember2005.In:InternationalConferenceon HarmonisationofTechnicalRequirementsforRegistrationofPharmaceuticals forHumanUse,Geneva,Switzerland.
Jones,L.,Bartholomew,B.,Latif,Z.,Sarker,S.D.,Nash,R.J.,2000.ConstituentsofCassia laevigata.Fitoterapia71,580–583.
Julkunen-Tiitto,R.,Nenadis,N.,Neugart,S.,Robson,M.,Agati,G.,Vepsäläinen,J., Zipoli,G.,Nybakken,L.,Winkler,B.,Jansen,M.A.K.,2015.Assessingtheresponse ofplantflavonoidstoUVradiation:anoverviewofappropriatetechniques. Phytochem.Rev.14,273–297.
Kim,A.R.,Jin,Q.,Jin,H.G.,Ko,H.J.,Woo,E.R.,2014.PhenoliccompoundswithIL-6 inhibitoryactivityfromAsteryomena.Arch.Pharm.Res.37,845–851.
Laitinen,M.L.,Julkunen-Tiitto,R.,Rousi,M.,2002.Foliarphenoliccompositionof Europeanwhitebirchduringbudunfoldingandleafdevelopment.Physiol.Plant. 114,450–460.
Lee,S.Y.,Kim,K.H.,Lee,I.K.,Lee,K.H.,Choi,S.U.,Lee,K.R.,2012.Anewflavonol glycosidefromHylomeconvernalis.Arch.Pharm.Res.35,415–421.
Liao,C.-R.,Kuo,Y.-H.,Ho,Y.-L.,Wang,C.-Y.,Yang,C.-S.,Lin,C.-W.,Chang,Y.-S.,2014.
StudiesoncytotoxicconstituentsfromtheleavesofElaeagnusoldhamiiMaxim. innon-smallcelllungcancerA549cells.Molecules19,9515–9534.
Lim,S.J.,Jeong,D.Y.,Choi,G.H.,Park,B.J.,Kim,J.H.,2014.Quantitativeanalysisof matrineandoxymatrineinSophoraflavescensextractanditsbiopesticidesby UPLC.J.Agric.Chem.Environ.3,64–73.
Lin,L-Z.,Mukhopadhyay,S.,Robbins,R.J.,Harnly,J.M.,2007.Identificationand quantificationofflavonoidsofMexicanoregano(Lippiagraveolens)by LC-DAD-ESI/MSanalysis.J.FoodCompos.Anal.20,361–369.
Lin,Y.,Kong,L.,2006.StudiesonthechemicalconstituentsofDesmodium styraci-folium(Osbeck)Merr.AsianJ.Tradit.Med.1,34–37.
Motilva,M.J.,Macia,A.,Romero,M.P.,Labrador,A.,Dominguez,A.,Peiro,L.,2014.
Optimisationandvalidationofanalyticalmethodsforthesimultaneous extrac-tionofantioxidants:applicationtotheanalysisoftomatosauces.FoodChem. 163,234–243.
Ok,H.E.,Choi,S.W.,Kim,M.,Chun,H.S.,2014.HPLCandUPLCmethodsforthe determinationofzearalenoneinnoodles,cerealsnacksandinfantformula.Food Chem.163,252–257.
Olguin,C.d.F.A.,Hamerski,L.,Percio,M.F.,Somensi,A.,2005.Avaliac¸ãodopotencial biológicoalelopáticodosextratosfracionadosdaraizdaVernoniatweediana Baker.Rev.VariaSci.5,137–143.
Petri,R.D., Pletsch,M.U., Zeifert,M.,Schweigert,I.D.,2008. Efeito deextratos hidroetanólicosdeVernoniatweedieanaeVernoniacognatasobreimunidadede camundongos.Rev.Bras.Farmacogn.89,139–141.
Portillo,A.,Vila,R.,Freixa,B.,Ferro,E.,Parella,T.,Casanova, J.,Canigueral, S., 2005.AntifungalsesquiterpenefromtherootofVernonanthuratweedieana.J. Ethnopharmacol.97,49–52.
Ren,D.M.,Qu,Z.,Wang,X.N.,Shi,J.,Lou,H.X.,2008.Simultaneousdetermination ofninemajoractivecompoundsinDracocephalumrupestrebyHPLC.J.Pharm. Biomed.Anal.48,1441–1445.
Rossato,M.F.,Trevisan,G.,Walker,C.I.,Klafke,J.Z.,deOliveira,A.P.,Villarinho,J.G., Zanon,R.B.,Royes,L.F.,Athayde,M.L.,Gomez,M.V.,Ferreira,J.,2011.Eriodictyol: aflavonoidantagonistoftheTRPV1receptorwithantioxidantactivity.Biochem. Pharmacol.81,544–551.
Shabir,G.A.,2003.Validationofhigh-performanceliquidchromatography meth-odsforpharmaceuticalanalysis:Understandingthedifferencesandsimilarities betweenvalidationrequirementsoftheUSFoodandDrugAdministration,the USPharmacopeiaandtheInternationalConferenceonHarmonization.J. Chro-matogr.A987,57–66.
Steinmann,D.,Ganzera,M.,2011.RecentadvancesonHPLC/MSinmedicinalplant analysis.J.Pharm.Biomed.Anal.55,744–757.
Stinco,C.M.,Benítez-González,A.M.,Hernanz,D.,Vicario,I.M.,Meléndez-Martínez, A.J.,2014.Developmentandvalidationofarapidresolutionliquid chromatog-raphymethodforthescreening ofdietaryplantisoprenoids:carotenoids, tocopherolsandchlorophylls.J.Chromatogr.A1370,162–170.
Taleon,V.,Dykes,L.,Rooney,W.L.,Rooney,L.W.,2014.Environmentaleffecton flavonoidconcentrationsandprofilesofredandlemon-yellowsorghumgrains. J.FoodCompos.Anal.34,178–185.
Trevisan,G.,Rossato,M.F.,Walker,C.I.B.,Klafke,J.Z.,Rosa,F.,Oliveira,S.M.,Tonello, R.,Guerra,G.P.,Boligon,A.A.,Zanon,R.B.,2012.Identificationoftheplantsteroid ␣-spinasterolasanoveltransientreceptorpotentialvanilloid1antagonistwith antinociceptiveproperties.J.Pharmacol.Exp.Ther.343,258–269.
USP,2007.621Chromatography.In:UnitedStatesPharmacopeia.USPharmacopeial Convention,Rockville,MD.
Uwai, K., Osanai, Y., Imaizumi, T., Kanno, S., Takeshita, M., Ishikawa, M., 2008.Inhibitoryeffectofthealkylsidechainofcaffeicacidanalogueson lipopolysaccharide-inducednitricoxideproductioninRAW264.7macrophages. Bioorg.Med.Chem.16,7795–7803.
Walter,T.H.,Andrews,R.W.,2014.RecentinnovationsinUHPLCcolumnsand instru-mentation.TrAC–TrendsAnal.Chem.63,14–20.
Wu,T.-S.,Yeh,J.-H.,Wu,P.-L.,1995.TheheartwoodconstituentsofTetradium glabri-folium.Phytochemistry40,121–124.
Zanon,R.B.,2006.MetabólitossecundáriosemVernoniatweedianaBaker.In:Centro deCiênciasdaSaúde,ProgramadePós-Graduac¸ãoemCiênciasFarmacêuticas. UniversidadeFederaldeSantaMaria,SantaMaria/RS,Brasil.