D zal een astronaut zijn die lelijk zijn enkel verzwikt in de krater, die hij als eerste te laat ontdekte.
specific contributions by colleagues and collaborators are acknowledged in the Author Contributions section and by co-authorship.
Declaro que esta dissertação é da minha autoria e que os dados aqui incluídos são o resultado de trabalho original por mim desenvolvido entre 2010 e 2014 no laboratório do Dr. Lars Jansen no Instituto Gulbenkian de Ciência em Oeiras, Portugal. Sempre que apropriado, contribuições específicas dos colegas e colaboradores são reconhecidos na seção Author Contributions e por co-autoria.
Financial support was granted by Fundação para a Ciência e a Tecnologia, doctoral fellowhip SFRH/BD/74284/2010. Apoio financeiro da FCT e do FSE no âmbito do Quadro Comunitário de apoio, BD nº SFRH/BD/74284/2010.
To be defended at the Instituto Gulbenkian de Ciência in Oeiras, Portugal on the 8th of June 2015, before a jury
composed of:
Prof. Bill Earnshaw (Wellcome Centre for Cell Biology, Edinburgh, UK); Prof. Kerry Bloom (UNC, Chapel Hill, NC, USA);
Dr. Reto Gassmann (IBMC, Porto, PT); Dr. Jorge Carneiro (IGC, Oeiras, PT);
Dr. Lars Jansen (IGC, Oeiras, PT);
and presided over by a yet to be determined representative of ITQB
i
Summary — p.ii; Resumo em Português — p.iii; Acknowledgements — p.iv; List of Publications — p.ix
1. General Introduction: Epigenetics, Centromeres, and
Quantitative Biology P.1
Epigenetics — p.3; Centromeres — p.19; Quantitative Biology — p.34; References — p.46
2. Analysis of Protein Turnover by Quantitative SNAP-Based
Pulse-Chase Imaging P.71
Introduction — p.73; Pulse-Chase — p.77; Quench-Chase-Pulse — p.81; Combining SNAP Experiments with Cell Synchronization and RNAi — p.85; Live Imaging of Pulse Labeled Cells — p.91; Automated Quantification of SNAP-Tagged Protein Turnover at Centromeres — p.95; Supporting Protocols — p.102; Background Information — p.108; References — p.117; Appendix: Maps of SNAP- and SNAPf-tags — p.120
3. Assembly in G1 phase and Long-Term Stability are Unique Intrinsic Features of CENP-A Nucleosomes P.125
Introduction — p.127; Results — p.129; Discussion— p.144; Material and Methods — p.147; References — p.151; Supplementary Figures — p.155; Appendix: The Role of CENP-C in CENP-A Dynamics— p.158
4. The Quantitative Architecture of Centromeric Chromatin P.163 Introduction — p.165; Results — p.166; Discussion— p.184; Material and Methods — p.190; References — p.199; Figure Supplements — p.207
5. General Discussion; Or, What I’ve Learned and What I Have to
Say about It P.215
Non-Centromeric CENP-A — p.217; The Ultrastability of CENP-A — p.221; Mass Action vs. Ultrastability — p.228; The Critical Amount of CENP-A — p.232; Concluding Remark — p.237; References — p.238
ii
A PhD is like a box of chocolates, …… and in this thesis I will present what I got. My work has been focused on a cellular structure that is essential for accurate genome inheritance: the centromere. Centromeres are chromosomal domains that do not rely on the presence of any specific DNA sequence. Rather, they are determined by the presence of a histone variant called CENP-A. Stable transmission of CENP-A containing chromatin is accomplished through 1) an unusually high level of protein stability, 2) self-directed recruitment of nascent CENP-A near existing molecules, and 3) strict cell cycle regulation of assembly. Together, these features lead to a self-sustaining loop that allows for epigenetic maintenance of centromeres.
My own contributions to the understanding of epigenetic centromere inheritance are of a quantitative nature. To put my work in context, I will start with an extensive INTRODUCTION of epigenetics, centromeres, and
quantitative biology. Next, in CHAPTER 2, I will detail two of the main
methodologies that have allowed for the quantitative analysis of centromere inheritance in subsequent chapters. These are, firstly, fluorescent SNAP-based pulse-labeling, used to distinguish between old and new protein pools; and secondly, a macro for ImageJ that I have developed, allowing for the accurate and unbiased quantification of fluorescence signals at centromeres. In CHAPTER 3, the cis requirements for assembly and extreme
stability of centromeric nucleosomes are analyzed. I demonstrate that both G1 phase loading and long-term centromeric retention are unique features of the (CENP-A/H4)2
subnucleosomal core, and are self-directed through
a CENP-A encoded targeting domain. CHAPTER 4 provides a quantitative
analysis of centromeric chromatin. The absolute number of CENP-A molecules at centromeres has been determined in addition to its quantitative regulatory mechanism and distribution. Finally, an overarching DISCUSSION of my results is presented, providing an outlook on how my
iii
Um doutoramento é como uma caixa de chocolates, ... e nesta tese vou apresentar o que eu consegui. O meu trabalho focou-se numa estrutura celular essencial para fidelidade do processo de herança do genoma: o centrómero. Centrómeros são regiões cromossômicas que não dependem da presença de nenhuma sequencia de ADN específica. Invés, são determinados pela presença de uma histona chamada CENP-A. A transmissão estável de cromatina contendo CENP-A é possível graças 1) a uma inusual alta estabi-lidade da proteina, 2) o auto recrutamento da CENP-A nascente com base na presença da proteína antiga, 3) e um alto nível de regulação da sua incor-poração durante o ciclo celular. Em conjunto, estas princípios asseguram um ciclo auto sustentável de manutenção epigenética dos centrómeros.
A minha contribuição para a compreensão da herança epigenética do centrómero é de natureza quantitativa. Para contextualizar o meu trabalho, começo com uma INTRODUÇÃO extensa da epigenética, dos centrómeros, e da
biologia quantitativa. No CAPÍTULO 2, detalho duas das metodologias que foram usados nos capítulos seguintes para a análise da herança centromé-rico. Estas são, primeiro, marcação fluroescente baseada em SNAP-tagging, usada para distinguir as populações de proteinas antigas e novas; e segundo, uma macro de ImageJ desenvolvida por mim, que permite a quantificação dos sinais fluorescentes do centrómero de uma maneira precisa e imparcial. No CAPÍTULO 3 são analizados os requerimentos em cis da incorporação e
estabilidade extrema dos nucleossomas CENP-A. Demonstro que, ambas incorporação na fase G1 e retenção centromérica a longo prazo, são pro-priedades únicas da estrutura sub nucleossomal (CENP-A/H4)2, e definidas
por um domínio intrínseco de CENP-A. O CAPÍTULO 4 fornece uma análise
quantitativa da cromatina centromérica. O número absoluto de moléculas de CENP-A nos centrómeros foi determinado, assim como o aspecto quantita-tivo do mecanismo da sua regulação e distribuição. Por último é apresentada uma DISCUSSÃO abrangente dos meus resultados e do impacto que as minhas
iv
Honestly, I don’t really know where to begin. So many people have been helpful and supportive in so many ways. I guess maybe I should start by acknowledging those that I’m sure to forget further on: you deserve my fullest gratitude as well as my most humble apology. Also, I do apologize for this utterly unsophisticated and extensive acknowledgements section, if my (ab)use of the English language bothers you (which it probably should), please skip it; I promise that the rest of the thesis is much more eloquent.
OK, moving on...
Lars, I am really happy with the relationship that we’ve built up over the past 6 years. I think that from the first moment we were on a very similar wavelength regarding many things and have become even more in phase over the years. I am also very happy with the type of ‘supervision’ that I received from you: lots of hands-on support initially when I needed it; lots of independence later on when I appreciated that; always supportive to my random whims —whether to take an extra day off for yet another frisbee tournament or apply to a $10.000 course with a deadline in 2 days; you were always ok with it. I also very much appreciate the personal connection that I think we had from the beginning. I have enjoyed immensely working with and for you and couldn’t have asked for a better PI.
Yet, everyone in the EpiLab has been an amazing and fruitful collaborator over the years. Ana, it’s awesome to have a great buddy like you in the lab. I love our (many many) coffee breaks with random jumps from tedious boring discussions of antibody dilutions to tales of last weekend’s drinking bouts and bitching sessions about [....CONFIDENTIAL INFORMATION...]. Filipa, it has been an absolute pleasure working with you. I could not have asked for a better student and if you have learned even half as much from me as I have from you, then I would be as proud of myself as I am of you. Mariana, thanks for welcoming me to the lab and to the country from the very beginning. I very much appreciated the heated arguments and
v
having you around for a while. Mariluz, Maxi, Dragan, Nuno, it has been great having worked with all of you; Ruben, Sreyoshi, Wojtek, I wish you all the luck in the EpiLab and am sorry we have only barely had an opportunity to work together.
Still missing one EpiLabber, right...: João, I know that you always say that you’re just doing your job —and you probably actually really feel that you do— but you do so much more. Whenever needed, whatever’s the matter, you are always ready to be as helpful as humanly possible! Whether it is to drop me off at the airport, fix my computer over the weekend, lend me your car for random errands, or discuss for a few hours a single sentence of some random translation I need for some obscure reason, I know I can always count on you. And then I won’t even mention the immense help you are in the lab, which one could potentially argue (although I personally wouldn’t) is indeed part of your job. Please know that all this, as well as your friendship over the last years, is and always has been very much appreciated. Hangout-clan, thanks a whole frickin’ bundle for sharing the joys (not many) and pains of thesis writing. The countless screens we’ve shared as well as the p*** that we didn’t was instrumental in pulling me through and I hope it’s been as useful for you too. Ewa, thanks for your patience, advice, and help about the tedious details of putting together a representable thesis. Also thanks to the theses of Babs, Ewa, Ines, Mariana, Mariluz, and Matilde for being great examples of what my boekje should look like.
I would also like to thank the IGC for having been a great host institution. The open-lab philosophy and highly interactive atmosphere created here has been extremely stimulating and productive for both work and social purposes. A special thanks goes to everyone that has passed through the Zheng-Ho wing and to the cell cycle club and chromatin club communities. Nuno, the first sentence you said to me when you saw me — “what do you think you’re doing” — and the resulting collaboration has been one of the most influential events of my entire PhD, although one of the few
vi
taught me to think like a microscope. Alekos, Mónica, Jorge, Raquel, thanks for the many fruitful discussions we’ve had about my projects. I am also very much indebted to everyone at the 2012 Physiology course for having reshaped my way of thinking about scientific problems and solutions. Thanks also to Élio for getting me out of a pickle: I was really reluctant to sit it out and your help was probably the one thing that could’ve and did rescue me. Indeed, my mind reels with appreciation of what it means to have been able to do a PhD at ITQB.
Tons of thanks also go to all the people that made my time in Portugal and at IGC soooooo much fun for such a long time. An incomplete list could be (in alphabetical order): Ana, Babs, Cláudia, Ewa, Filipa, Inês, Jess, João, João Beer, Jordi, Jorge, Krzys, Laura, Lars, Luís, Mada, Marc, Mariana, Mihailo, Nicole, Nuno, Pol, Roksana, s, Stefan, Tiago. Also lots of thanks to everyone who has kept on throwing discs at me to keep me sane all this time, especially Sof, Trick, Patrão, Carla, Cons, Rui, Fred, Rui, Pifre, Inês, Seb, Morris, and of course Filipa who introduced me to this all.
ZZ, KJAJBDTK!
Natuurlijk gaat er ook onwijze dikke dank naar al mijn lieve vrienden, ex-collega’s en mentoren thuis, die mij na al die lange jaren hopelijk nog niet vergeten zijn. Sander, Adri, Matilde (en alle anderen waarmee ik in Sander’s lab gewerkt heb); Paulien, Stan en Veronica; jullie hebben stuk voor stuk op een onmiskenbare manier bijgedragen aan de vorming van de wetenschapper die ik vandaag ben, en ik herken in mezelf nog steeds de specifieke invloed van ieder van jullie. Piet, Petertje, Matthia, it has been a joy and honor om samen met jullie biologisch grootgebracht te worden: onze tijden van SPI___RAAL, ik spreek Oebli-Oebli en in je broek waren onmisbaar om mij de biloloog te maken die ik vandaag ben. Sanne, Ditte, Banafsheh (waarschijnlijk de enige 3 buiten mijn familie waarvan ik ervoor zorg dat ik elke keer dat ik in Nederland ben minstens een klein beetje tijd vind om bij te kletsen): hoera!
vii
sentence, which I was told probably around age 9, instantaneously transformed me into a scientist. Peter, you probably don’t even remember saying this to me, but I will never forget (at least, well, I haven’t forgotten it yet).
(MaPaDaNo(SaToMi)); Worte fehlen mir... ausser: Danke für alles! Kommt noch eine Person die ich noch nicht gennant habe: Papa, ich widme dir diese Dissertation. Ich glaube es gibt niemanden auf der Welt der einen grösseren Einfluss auf meine Bildung, in jeder möglichen Hinsicht, gehabt hat. Papa, es tut mir schrecklisch leid das du nicht hast sehen können was aus mir geworden ist.
ix In chronological order:
Silva MCC, Bodor DL, Stellfox ME, Martins NMC, Hochegger H, Foltz DR & Jansen LET (2012) Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev. Cell 22: 52–63
Bodor DL, Rodríguez MG, Moreno N & Jansen LET (2012) Analysis of
Protein Turnover by Quantitative SNAP-Based Pulse-Chase Imaging. Curr. Protoc. Cell Biol. Chapter 8: Unit8.8
Bodor DL, Valente LP, Mata JF, Black BE & Jansen LET (2013)
Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol. Biol. Cell 24: 923–932
Bodor DL & Jansen LET (2013) How two become one: HJURP
dimerization drives CENP-A assembly. EMBO J. 32: 2090–2092
Bodor DL, Mata JF, Sergeev M, David AF, Salimian KJ, Panchenko T,
Cleveland DW, Black BE, Shah JV & Jansen LET (2014) The quantitative architecture of centromeric chromatin. eLife Sciences 3: e02137
General Introduction:
Epigenetics, Centromeres, and Quantitative Biology
Dani L. Bodor
3
E
PIGENETICSInheritance systems
Inheritance from a biological perspective is the transfer of information from one (cell) generation to th e next. In order for a system of inheritance to persist, a number of criteria need to be fulfilled. The bare minimal requirement is that there is a carrier (or carriers) of information that can be propagated through generations. In addition, to allow for sustained passage of information into subsequent generations, the carrier needs to be replicated in each generation. Moreover, in many cases it is important that there is careful regulation to ensure that the correct number of heritable units is passed on. Temporal regulation can play a role in quantitative control so that e.g. one new unit is formed for each pre-existing one exactly once per cell generation. In summary, the basic properties of a successful inheritance system include: 1) propagation, 2) replication, and 3) copy-number regulation.
Up to the early 1940s, there was a heated debate on the molecular nature of heritability. Two opposing ideas were that either protein or nucleic acids would be the carriers of genetic information (Deichmann, 2004). Among other factors, the low apparent complexity of DNA led to the common notion that genes were more likely composed of proteins. However, In the 1940s and ‘50s a number of breakthrough discoveries were made that irrevocably showed that, in fact, DNA was responsible for genetic inheritance. Instrumental were experiments showing that DNA is the agent that is responsible for the transformation of non-virulent into virulent pneumococcus (Griffith, 1928; Avery et al, 1944), as well the famous Hershey-Chase experiment, showing that viral DNA, but not protein, enters the host upon bacteriophage infection (Hershey & Chase, 1952). Soon afterwards, Watson and Crick published their breakthrough model of the double-helical structure of DNA, including the now famous statement “it has not escaped our notice that the specific pairing we have postulated
4
immediately suggests a possible copying mechanism for the genetic material” (Watson & Crick, 1953a). Indeed, the semi-conservative ‘copying mechanism’ that was intended, where each of the two existing strands of DNA form the template for a nascent strand (Figure 1.1A), was later confirmed by Meselson & Stahl (1958) in what is often called ‘the most beautiful experiment in biology.’ Much later, and over the course of decades, the regulation mechanisms were elucidated, which ensure that the entire genetic complement is replicated exactly once per cell division, such that there is no under- or overduplication of the genetic material (Sclafani & Holzen, 2007). In short, once per cell division cycle, a defined number of replication origins are licensed with an initiation complex that is consumed when DNA replication begins at this site, thus ensuring that the same stretch of DNA is not replicated more than once. In addition, progression of cell division is halted until a complex machinery, called a checkpoint, has ensured that DNA replication is complete. In conclusion, although some details may still need to be resolved, a fairly good understanding of the mechanism of genetic inheritance has emerged.
As is clear from the section above, DNA perfectly fits all criteria given above for the carrier of heritable information. This molecule is stably propagated when cells divide, it is replicated after each cell division, and regulated such that each molecule gives rise to only one new molecule exactly once per division. Thus, genetic inheritance is a showcase model of an effective inheritance system.
Non-genetic inheritance
Ever since the discovery that DNA was the carrier of genetic information, the study of inheritance from a biological perspective has been dominated by DNA and its nucleotide sequence. This system is perfectly able to account for Mendel’s laws of inheritance (Mendel, 1866) as well as some more complex variations of these principles, which together govern inheritance of the majority of traits in sexually reproducing organisms. However, certain
5
heritable features do not strictly dependent on the genetic code of a cell. This is most apparent from the fact that within a single multicellular organism there can be many different cell types with the exact same genetic material. Generally, when cells that have acquired a certain developmental status divide, they give rise to the same cell type, e.g. a dividing skin cell will not suddenly give rise to a heart muscle cell, and vice versa. In addition to such non-genetic inheritance that is contained within a single organism, a number of transgenerationally inherited traits have been described that do not seem to follow the typical laws of inheritance. One famous example is ‘helmet’ size in the waterflea Daphnia cucullata: if exposed to a predator, the size of this protective structure is altered throughout multiple generations (Agrawal et al, 1999), even in the absence of a predatory cue in the offspring. Another well-known case is the toadflax Linaria vulgaris that exists in two distinct heritable morphological states, but can spontaneously switch between generations without any apparent mutations in the responsible gene (Cubas et al, 1999). Thus, there must be other structures present in cells that are able to carry information from mother to daughter cells, or even through organismal generations. Below, some typical examples of alternative inheritance systems, and their method of transferring information, are discussed.
Self-sustaining loops
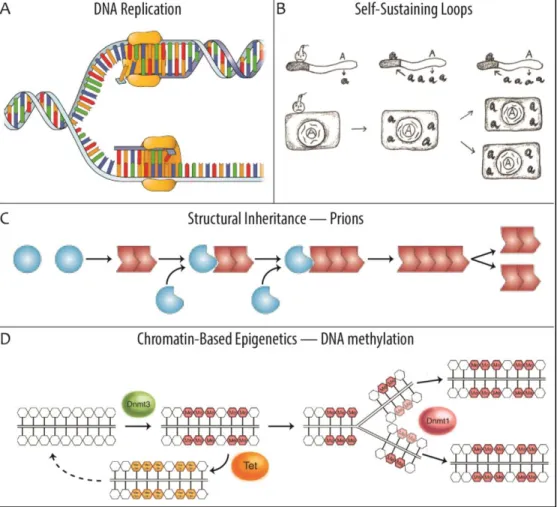
Perhaps the simplest possible form of (non-genetic) inheritance is a self-sustaining loop (Figure 1.1B). If the expression of a gene is driven by its own product (protein or RNA), then the cytoplasmic inheritance of this factor during cell division will ensure that the active state of the gene will also be inherited (Rosenfeld, 2011). Gene products can either drive such feed-forward loops directly (e.g. a transcription factor that activates the gene by which it is produced), or, more commonly, indirectly (e.g. a protein that initiates a genetic cascade, ultimately leading to its own expression). In either case, gene-activity will effectively be maintained throughout generations until it is actively (or spontaneously) interrupted. This type of
6
self-sustaining loop is common in bacteria and other unicellular organisms (Santillan et al, 2007; Jablonka & Raz, 2009), and likely contributes to the maintenance of cell identity in multi-cellular organisms as well (Hobert, 2011; Holmberg & Perlmann, 2012; Ptashne, 2013).
Figure 1.1 Examples of inheritance systems. (A) DNA is replicated in a semiconservative fashion. During replication, a single DNA duplex untwines and individual nucleotides on each strand form the template for production of a new strand of DNA (image adapted from: The Nucleus and DNA Replication, 2015). (B) Once initiated by an external cue (indicated by a bomb), gene products that maintain their own expression through a self-sustaining loop can be inherited through the cytoplasm during cell division. In this way, they maintain their activity in the next cellular generation, even in the absence of the original initiating signal (image adapted from: Jablonka & Lamb, 2006). (C) Prion transmission is an example of structural inheritance. The amyloid protein conformer (red) catalyzes conversion of native protein isoforms of identical amino acid sequence (blue balls) into its own conformation (image adapted from: Shorter & Lindquist, 2005). (D) DNA methylation is the best understood form of chromatin-based epigenetics. DNMT3 is a de novo methyltransferase that is capable of adding methyl groups (red hexagons) to cytosines on unmethylated DNA. During DNA replication, the maintenance methyltransferase DNMT1 associates with the core replication machinery and specifically methylates hemimethylated DNA, thus retaining the pre-replication methyl-pattern in the next generation. Conversely, TET enzymes can oxidize methylated cytosine into hydroxymethylcytosine (orange hexagons), which can initiate a pathway that restores unmethylated DNA (image adapted from: Li & Zhang, 2014).
7
In this inheritance system, the carrier of heritable information is the gene product (let’s call it Factor X). Factor X is propagated through the cytoplasm of a dividing cell, oftentimes by random segregation of the total pool of existing molecules (Rosenfeld et al, 2005). Replication in the next generation is achieved by activating the gene that is responsible for producing Factor X. Although in this case there is no absolute requirement for copy number regulation with a high degree of accuracy, sufficient molecules are required to ensure that each daughter sustains and perpetuates gene activity. In summary, self-sustaining loops represent a very basic example of a stable inheritance system.
Structural inheritance
In some cases, a given three dimensional structure propagates itself by forming the template for assembly of the same structure. Perhaps the most elegant (and best understood) structural inheritance system is in fact genetic inheritance, where nascent strands of DNA are templated onto existing molecules (Watson & Crick, 1953a, 1953b; Meselson & Stahl, 1958). However, many additional structural inheritance systems have been described. A clear example are prions (Figure 1.1C), proteins of identical amino-acid sequence that can exist in multiple conformational states, at least one of which drives conversion of the other(s) into itself (Prusiner, 1982, 1998; Halfmann et al, 2010). Although prions are generally considered detrimental or pathogenic, it has been shown that they can have a physiological role by conferring advantageous traits in certain environments (Halfmann et al, 2010, 2012). Prion inheritance is in many ways analogous to the self-sustaining loops described above (it is itself a type of feed forward loop), as it is inherited through the cytoplasm where it will replicate by mediating a nascent protein isoform into its own conformational state.
An interesting case is presented by the centrosome, the primary microtubule organizing center (MTOC) in most animal cells. A single centrosome contains two centrioles, cylindrical structures composed mainly
8
of tubulin, each of which nucleate a nascent daughter centriole exactly once per cell division cycle (Bettencourt-Dias & Glover, 2007; Nigg & Stearns, 2011). Conversely, centrioles can also form de novo under certain conditions, although this is strongly suppressed be the presence of pre-existing centrosomes (Marshall et al, 2001; Terra et al, 2005; Rodrigues-Martins et al, 2007). However, this inheritance mechanism differs from true structural inheritance, as there is no evidence for actual templating of one centrosome against another. Rather, centrosomes are more likely sites where enzymes, regulatory, and structural proteins accumulate to regulate the biogenesis of nascent structures (Rodrigues-Martins et al, 2007, 2008), allowing for a semi-conservative replication mechanism that is carefully regulated by the cell cycle (Bettencourt-Dias & Glover, 2007; Nigg & Stearns, 2011). In this system, the carrier of heritable information are the centrioles, although it is not completely clear what the information is that they carry. Nevertheless, their replication is strictly regulated in time, space, and number to ensure the propagation of the correct number of structures to the following generation.
Other examples of structural (or structural-like) inheritance systems include the organization of ciliary rows on the cell cortex of certain ciliates (Sonneborn, 1964), cellular membranes (Cavalier-Smith, 2004), certain organelles (Warren & Wickner, 1996), or even the cell as a complete entity. In summary, structural inheritance is a common mechanism to pass information from one generation to the next.
Chromatin-based epigenetics
The term epigenetics was originally coined by Conrad Waddington in 1942 to indicate “the mechanism by which the genes of the genotype bring about phenotypic effects” (Waddington, 1942). In this definition, epigenetics does not refer to any heritable features, but is more similar to what today is considered gene regulation or developmental biology. However, throughout the last 70-odd years, the word epigenetics has been used and redefined in
9
many different ways (Jablonka & Lamb, 2002; Bird, 2007; Marris et al, 2008). One very broad definition of an epigenetic phenomenon is: “a change in phenotype that is heritable but does not involve DNA mutation” (Gottschling, 2004). However, if taken literally, this definition encompasses certain heritable features that are usually not intended, such as traits acquired through social learning (Jablonka & Lamb, 2005; Shea, 2009) or vertically transmitted infections and symbionts (Ford-Jones & Kellner, 1995; Moran et al, 2008). Nevertheless, more recently, during a conference on chromatin-based epigenetics at Cold Spring Harbor, a consensus definition was formulated as: “a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” (Berger et al, 2009). Perhaps unsurprisingly, this consensus definition only includes what the main topic of the conference was, namely chromatin-based epigenetics (see below), while excluding all other potential forms of epigenetics, including self-sustaining loops and structural inheritance. In my own opinion, the most useful definition of epigenetic inheritance goes along the lines of: information that cells can pass to their progeny without changing their DNA sequence (paraphrased from Jablonka & Lamb, 2005, p. 113). In this case, heritable features at the cellular molecular scale (e.g. self-sustaining loops and structural inheritance) are included, while features heritable at the organismal scale (e.g. symbiosis and learning) are not. Deceptively, yet more definitions exist outside of biology, e.g. epigenetic robotics, which is related to machine learning (Prince & Demiris, 2003), and the epigenetic theory of human development, a psychological theory of transitions in human development through psycho-social crises (Erikson, 1950). Therefore, although I only partially agree, Adrian Bird makes a reasonable point when he says: “epigenetics is a useful word if you don't know what's going on — if you do, you use something else” (Marris et al, 2008).
Despite the ongoing controversy on the exact meaning of epigenetics, practically speaking, chromatin-based epigenetics is the most actively
10
studied form of non-genetic inheritance. The structure and organization of chromatin allows for a plethora of modifications, many of which can either be inherited or participate in a pathway that drives inheritance. In addition, this complex nature of chromatin allows for tight control of the transmission of the epigenetic signal. I will first proceed with a brief introduction on chromatin and then delve deeper into its role in epigenetic inheritance.
Chromatin structure
Generally, the existence of chromatin is attributed to the necessity of fitting a large (eukaryotic) genome into a much smaller nucleus. If we take human cells as an example, the total length of the 46 chromosomes, together comprising over six billion base pairs of DNA, would exceed two meters if placed head-to-tail (Flicek et al, 2014). However, in analogy to packing a suitcase, it does not make much sense to lay all ones clothes in a neat line next to other and then wonder how this will ever fit into a small carry-on bag (Morse, 2013). Similarly, chromosomes are not linearly extended molecules, but are folded and packaged into three-dimensional structures. In fact, the paradox of fitting 2 meters worth of DNA into an average sized nucleus of ~7 μm in diameter is easily resolved by the fact that the volume of this nucleus is almost 30 times as big as that of the total DNA (respective volumes ~180 μm3 and ~6.3 μm3). Thus, chromatinization is a means of proper folding of
the DNA, and has additional roles in organizing and regulating the genome. The primary organizational unit of chromatin is the nucleosome (Kornberg, 1974; Olins & Olins, 1974). A single nucleosome consists of ~145 bp of DNA tightly wrapped around an octamer consisting of two copies of each of the histone proteins H2A, H2B, H3, and H4 (Luger et al, 1997). The octamer itself is composed of a central (H3/H4)2 tetramer, flanked by two
H2A/H2B dimers. These core histones are among the most highly conserved eukaryotic proteins (Sullivan et al, 2000, 2002; Malik & Henikoff, 2003), arguing that little structural variability is tolerated for their function. This is especially true in their histone fold domain (HFD), which form the major
11
interactions between the separate histones as well as with the DNA (Luger et al, 1997) and are 100% identical between human and certain plants and fungi (Sullivan et al, 2002). Histone H1 serves as a linker-histone, which binds DNA between neighboring nucleosomes, thereby helping to stabilize the chromatin structure (Thoma et al, 1979). Further organization is likely achieved by multiple forms of higher order structures, the precise in vivo nature of which has proven to be very challenging to determine (Woodcock & Ghosh, 2010). Despite the high level of conservation and strong interaction of the histone-DNA binding, chromatin is both a heterogeneous and a dynamic structure (Gasser, 2002; Flaus & Owen-Hughes, 2004; Chakravarthy et al, 2005). Indeed, both replication and transcription machineries displace, reorganize, and remodel the nucleosomes as DNA and RNA polymerases, respectively, plough through the chromatin (Mousson et al, 2007; Groth et al, 2007). In addition, certain regions of the chromosome can be highly compacted, while flanking regions remain accessible to external factors, such as transcription factors or other DNA binding proteins (Wu et al, 1979; Larsen & Weintraub, 1982; Song et al, 2011). Furthermore, major rearrangements of this chromatin organization commonly occur, e.g. throughout the cell cycle (Reeves, 1992; Aragon et al, 2013; Raynaud et al, 2014) and during cell differentiation (Meshorer & Misteli, 2006; Kobayakawa et al, 2007; Probst & Almouzni, 2008). In summary, while composed of fairly simple units, chromatin is a highly complex structure that is regulated at the level of configuration, organization, and dynamics.
Consistent with its complexity, a large variety of processes exist that help effectively regulating chromatin homeostasis and dynamics in cells. The close association of chromatin and its modifications to the genome of the cells makes it an excellent candidate for driving epigenetic inheritance, e.g. of gene activities. Three of the major mechanisms are DNA methylation, incorporation of histone variants, and modification of histone proteins. Each of these processes has the potential, supported at least by some evidence, to drive epigenetic inheritance, and will be briefly discussed below.
12 DNA methylation
DNA methylation, the covalent addition of a methyl group to the DNA backbone, is found throughout the tree of life (Colot & Rossignol, 1999; Jeltsch, 2002; Ponger & Li, 2005). However, this modification was lost multiple times in evolution, and is absent from a wide variety of species including D. discoideum, S. cerevisiae, S. pombe, and C. elegans (Ponger & Li, 2005). Methylation of DNA can affect many cellular processes, including gene-regulation, transposon silencing, heterochromatin formation, and susceptibility to restriction enzymes, depending to some extent on the species (Colot & Rossignol, 1999). In eukaryotes, methylation at carbon 5 in the pyrimidine ring of cytosine, thus creating 5-methylcytosine (meC), is the only known form of methylated DNA (Jeltsch, 2002). In plants, any cytosine in the genome has the potential to be methylated, although separate enzymes are responsible for the methylation of CG-dinucleotides (CpG), CHG-sites (where H is any non-guanine nucleotide), and CHH-sites (Law & Jacobsen, 2010). In mammalian cells, however, DNA methylation is largely restricted to CpGs (Sinsheimer, 1955), although low levels of meC can be observed on other sites, especially in germ and stem cells (Ramsahoye et al, 2000; Ichiyanagi et al, 2013). Importantly, not every potential site is methylated, e.g. ~14% of cytosines are methylated in Arabidopsis thaliana leaf tissue (Capuano et al, 2014), while ~70–80% of CpGs are methylated in somatic human tissues (Ehrlich et al, 1982; Bird, 2002). Furthermore, the pattern of methylation can differ between different cell types of the same organism and change during differentiation (Reik et al, 2001). Thus, sequence determinants are not sufficient to explain the existing pattern of DNA methylation.
The vast majority of meC sites in the mammalian genome are symmetrically methylated. In other words, either both strands of a minipalindromic CpG site are methylated, or neither is (Bird, 1978). However, the process of DNA replication inevitably leads to the formation of hemimethylated DNA, where a nascent strand of unmethylated DNA is
13
templated against a methylated pre-existing strand. The DNA methyltransferases DNMT1 has been shown to have a high preference for hemimethylated DNA (Bestor & Ingram, 1983) and associate with the core DNA replication machinery protein PCNA (Chuang et al, 1997) as well as with NP95, which specifically recognizes hemimethylated DNA (Sharif et al, 2007). In this way, DNMT1 is accurately targeted to hemimethylated DNA during its formation and can restore the pre-existing pattern of methylation. This shows that DNA methylation is a semiconservatively inherited epigenetic feature and intrinsically coupled to cell cycle regulation (Figure 1.1D).
Although DNA methylation is generally considered a stable epigenetic modification, its genomic pattern is largely reset in each generation. Demethylation can potentially occur in two fundamentally different ways. One is the passive dilution of meC during successive rounds of DNA replication in the absence of maintenance methylation. The other is by active removal of methylated cytosines, although claims of finding such mechanisms have a history of being highly controversial (Ooi & Bestor, 2008). Only recently has a bona fide active demethylation pathway been described, where meC is iteratively oxidized into hydroxymethylcytosine (Tahiliani et al, 2009), formylcytosine, and carboxylcytosine (Ito et al, 2011; He et al, 2011), the latter two of which can be converted back to unmodified cytosine through base-excision repair (He et al, 2011; Maiti & Drohat, 2011). This pathway may explain how, in the absence of replication, methylated DNA is rapidly lost from the mouse paternal pronucleus after fertilization (Mayer et al, 2000; Oswald et al, 2000). Embryonic stem cells re-initiate a nascent pattern of DNA methylation (Jähner et al, 1982; Stewart et al, 1982) using the de novo DNA methyltransferases DNMT3a and DNMT3b (Okano et al, 1998, 1999). However, a recent analysis on the genome-wide methylation patterns of three great apes, including human, argues that methylation patterns can gradually change over generations and may ultimately even contribute to variability between species (Martin et al, 2011;
14
Boffelli & Martin, 2012). Nevertheless, generally speaking, it appears that DNA methylation in mammals is mainly involved in epigenetic inheritance through mitotic divisions, and has a relatively minor role in transgenerational inheritance.
Histone variants
As mentioned above, canonical nucleosomes contain a histone octamer consisting of four different types of histone proteins: H2A, H2B, H3, and H4. Multiple different variants exist for each of these histone proteins in most species analyzed (Talbert et al, 2012), with the exception of H4, for which a sole known non-canonical variant exists in Trypanosoma (Siegel et al, 2009). In humans, up to 47 non-allelic variants, i.e. proteins with different amino acid sequence, have been described in total for the four nucleosomal histones (Wiedemann et al, 2010; Khare et al, 2011). However, it remains unclear whether each variant actually has distinct properties, especially in cases with only one or few residues divergence. Nevertheless, one example where this is indeed the case is histone H3.3, which differs from its canonical H3.1 counterpart by a mere 5 amino acids, yet its dynamics and regulation are drastically different. H3.1 is assembled throughout the genome by the CAF complex in a strictly DNA replication-coupled manner, while H3.3 assembly occurs preferentially at specific loci by the histone chaperones HIRA, DAXX, and DEK and is independent of the cell cycle (Smith & Stillman, 1989; Ray-Gallet et al, 2002; Ahmad & Henikoff, 2002; Tagami et al, 2004; Drané et al, 2010; Goldberg et al, 2010; Sawatsubashi et al, 2010). Therefore, altered histone variant compositions of the nucleosome are good candidates as carrier of epigenetic information.
The process of DNA replication forms an inherent challenge to the local heritability of histones. In order for a megadalton sized replication complex to pass through the chromatin, nucleosomes are disassembled prior to the denaturation and replication of DNA (Groth et al, 2007; Alabert & Groth, 2012). However, pre-existing subnucleosomal (H3/H4)2 tetramers are
15
recycled behind the replication fork, possibly through their association with the histone chaperone Asf1 (Groth et al, 2007; Mousson et al, 2007; Alabert & Groth, 2012). Conversely, it appears that histones H2A and H2B are more dynamic than H3 and H4 (Jackson, 1987; Kimura & Cook, 2001; Bodor et al, 2013) and thus less likely to carry epigenetic information. Consistently, evidence exists that at least two variants of histone H3 are carriers of epigenetic information. The role of the centromeric variant CENP-A is described in detail in part 2 of the introduction. The replacement variant H3.3 is enriched at sites of high gene activity (Ahmad & Henikoff, 2002; Mito et al, 2005; Goldberg et al, 2010), and is enriched in post-translational modifications associated with active transcription (McKittrick et al, 2004; Hake et al, 2006). Importantly, it has been shown that H3.3 is involved in the resistance to reprogram an active gene expression profile in Xenopus after transplantation of somatic cell nuclei into oocytes (Ng & Gurdon, 2008). Interestingly, a similar role for macroH2A was found by maintaining a repressed state on the X-chromosome (Pasque et al, 2011) and on pluripotency genes (Gaspar-Maia et al, 2013). Although the precise mode of action of these histone variants remains unclear, it appears that they are somehow involved in the transmission of an epigenetic state.
Histone modifications
In addition to modifying the histone variant composition of nucleosomes, each of the histones can undergo a large number of post-translational modifications (PTMs). Common modifications on histones include acetylation, methylation, phosphorylation, ubiquitylation, citrullination, biotinylation, and ADP-ribosylation (Khare et al, 2011). Most PTMs exist in the protruding N-terminal histone tails, while only few are found within the HFD (Khare et al, 2011). In some cases, a single residue is known to exist in multiple different modified forms; e.g., lysine 9 of Histone H3 (H3K9) can be mono-, di-, or trimethylated, acetylated, or biotinylated. Indeed, on histone H3 alone, there are at least 44 separate known modifications, spread over 21 individual sites, resulting in over three billion
16
potential combinatorial states of modification on each molecule (Khare et al, 2011). Interestingly, many modifications are shown to correlate with specific (functional) states, such as high or low gene-activity, splicing, DNA repair, and DNA replication (Bannister & Kouzarides, 2011). These findings have spurred the hypothesis of a ‘histone code’ that can be read by downstream effector proteins or have a function in epigenetic memory (Strahl & Allis, 2000; Jenuwein & Allis, 2001; Turner, 2002; Rothbart & Strahl, 2014). However, because most PTMs are not exclusively associated with any one particular state (Barski et al, 2007), such a histone code can at best be seen as a highly complex combinatorial code or language (Lee et al, 2010; Rothbart & Strahl, 2014), unlike e.g. the linear genetic code (1 codon => 1 amino acid). Nevertheless, similar to histone variants, PTMs on histone tails have the potential to propagate epigenetic information.
PTMs are often equated to epigenetic marks, even in the scientific literature (e.g. Turner, 2002). However, in many cases there is clear evidence that the PTMs are not inherited at all, but are transient structures that mediate e.g. cell cycle progression (Van Hooser et al, 1998), DNA replication (Benson et al, 2006), or DNA repair (Rogakou et al, 1999; Hunt et al, 2013). In addition, for many modifications that are associated to gene-activity, it remains unclear whether they are the cause or consequence of the transcriptional state (Ng et al, 2003; Soshnikova & Duboule, 2009; Muramoto et al, 2010). Nevertheless, while, at least to my knowledge, there is no direct evidence that PTMs carry and transmit epigenetic information, they remain strong candidates at least for certain modifications.
Epigenetics in evolution
Above, it has been thoroughly established that heritability is not exclusively mediated by the genome. Although most examples given refer mainly to inheritance of features through mitotic divisions, i.e. within the somatic cells of a single organism, more than 100 examples of trans-generational epigenetic inheritance from 40 different species have been
17
documented by Jablonka & Raz (2009). Given this wealth of epigenetic heritability, at least some of the traits must be adaptive and advantageous phenotypes to certain environments have been observed for variable methylomes in the plant species Arabidopsis thaliana (Johannes et al, 2009) and Mimulus guttatus (Scoville et al, 2011), as well as prions in S. cerevisiae (Halfmann et al, 2012), heritable antiviral RNA molecules in C. elegans (Rechavi et al, 2011), and gene silencing in D. melanogaster (Stern et al, 2012). Together, these observations lead to the interesting possibility that non-genetic inheritance can contribute to evolutionary dynamics.
To illustrate that evolution can be driven by epigenetic inheritance, Jablonka and Lamb (2005) used an interesting thought-experiment approximately along the following lines:
Imagine a faraway planet that is as rich and dynamic a world as our own, featuring many different environments and climates; let’s call it CB (for Complex Biosphere). This world is inhabited by a population of creatures that does not tolerate any divergence in its genome whatsoever; let’s call them SAM (for Species in the Absence of Mutation). Given the richness of the environment, there is a great potential for SAM to adapt to many different niches. Therefore, as time goes by, SAM plays it (again) in a way that does not require any genetic change. Rather, SAM differentially produces epigenetic traits, e.g. through altering the gene methylation states, generating novel prion-like protein con-formations, or activating self-sustaining loops. If advantageous in a given milieu, adapted individuals will prosper, compete more successfully for the available resources, and produce a higher number of offspring. Thus, by means of natural selection, the epigenetic diversification of SAM in different environments will ultimately be the origin of species.
Given that imagination is the only weapon in the war against reality, we do not want to argue here that actual evolution is driven solely by epigenetic changes. Nevertheless, this story does clearly make the point that adaptation, and thus evolution, can in principle occur through inheritance of variable, non-genetic traits. Accepting that “variations, however slight and from whatever cause proceeding, if they be in any degree profitable to the individuals of a species [...], will tend to the preservation of such individuals” (Darwin, 1859: p.61; emphasis mine), it is difficult to imagine that natural selection would not work on epigenetically inherited traits.
18
The influence of epigenetic mechanisms on evolution could be very different from genetic inheritance. Importantly, reproduction of epigenetic states in the next generation is generally much less accurate than genetic inheritance. For example, while DNA replication occurs at an error rate in the range of ~10-6–10-8 (Kunkel, 2004), errors in copying DNA methylation
occur as frequently as ~0.3–4% (Laird et al, 2004; Goyal et al, 2006). Although a higher error rate likely makes epigenetic traits less stable, it may also lead to a more rapid acquisition in response to changing environments (Cubas et al, 1999; Pryde & Louis, 1999). These and other epigenetic specific effects (Jablonka, 2012) make that the classical models of evolution and population dynamics need to be reevaluated. However, only recently have different aspects of epigenetics started to be integrated in such models (Tal et al, 2010; Day & Bonduriansky, 2011; Geoghegan & Spencer, 2012). In addition, epigenetic mechanisms have been proposed to have a role in speciation, macroevolution, and even the major transitions in evolution (Jablonka & Lamb, 2006; Jablonka & Raz, 2009; Boffelli & Martin, 2012; Jablonka, 2012).
19
C
ENTROMERESThe function of centromeres
Centromeres were originally defined cytologically by Walther Flemming in the late 19th century, as the site of a ‘primary constriction’ in mitotic
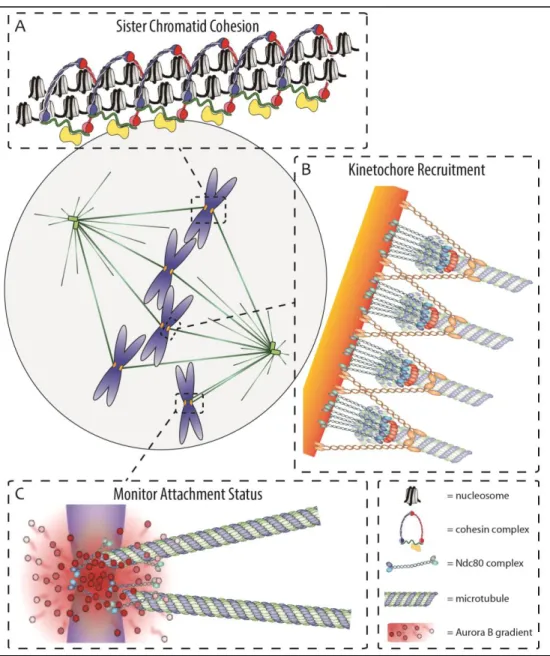
chromosomes (Flemming, 1880). Today, we have a fairly good understanding of what brings about this particular structure. During DNA replication, nascent sister chromatids are held together by a protein complex called cohesin (Figure 1.2A), thus preventing precocious separation and chromosome missegregation (Michaelis et al, 1997; Uhlmann & Nasmyth, 1998). Upon entry into mitosis (or meiosis), the chromosomes condense (Koshland & Strunnikov, 1996) and the majority of cohesin is removed from the chromosomes (Losada et al, 1998). However, cohesin is preferentially retained at a single site on each sister chromatid pair, the centromere (Losada et al, 2000; Waizenegger et al, 2000), where it is protected by Shugoshin proteins (Kerrebrock et al, 1995; Salic et al, 2004). Only when cells are ready to exit mitosis and segregate sister chromatids to the daughter cells is the remaining centromeric cohesin cleaved by a protein called separase (Uhlmann et al, 1999, 2000). Thus, centromere specific cohesion is responsible for the X-shaped conformation of mitotic chromosomes and Flemming’s primary constriction (Haarhuis et al, 2014).
Centromeres are also the chromosomal loci that form the point of contact between the DNA and the mitotic spindle (Figure 1.2B). A large group of proteins, the constitutive centromere associated network (CCAN), are present at the centromere throughout the cell cycle (Foltz et al, 2006; Izuta et al, 2006; Cheeseman & Desai, 2008). During mitosis, the CCAN recruits a secondary protein complex known as the kinetochore, which includes the conserved microtubule-binding KMN network, consisting of the protein KNL1 as well as the Mis12 and Ndc80 complexes (Cheeseman et al, 2004, 2006; DeLuca et al, 2006). Poleward directed pulling forces are exerted on centromeres by stable binding of depolymerizing microtubules at
20
kinetochores, which drag the sister chromatids in opposite directions during anaphase (Brinkley & Cartwright, 1975; Salmon et al, 1976; Mitchison et al, 1986; Inoué & Salmon, 1995). Thus, the centromere is the primary structure responsible for recruiting the entire chromosome segregation machinery.
Figure 1.2 Centromeres control chromosome segregation. (A) Sister chromatid cohesion is maintained specifically at centromeres during mitosis to prevent precocious chromosome separation (image adapted from: Nasmyth & Haering, 2009). (B) During mitosis, centromeres form a recruitment hub for kinetochores, including the microtubule binding Ndc80 complex, which drive chromosome segregation during anaphase (image adapted from: Cheeseman & Desai, 2008). (C) An Aurora B gradient emanating from the inner centromere destabilizes proximal kinetochore-microtubule interactions to prevent asymmetric chromosome segregation (image adapted from: Lampson & Cheeseman, 2011).
21
Finally, centromeres have an integral role in monitoring proper kinetochore-microtubule interactions. The formation of amphitelic attachments, where sister centromeres are attached to microtubules of opposing spindle poles, guarantees that chromosomes are pulled in opposite directions during anaphase (Cimini et al, 2001). The spindle assembly checkpoint (SAC), aka the mitotic checkpoint, is recruited to centromeres at the onset of mitosis (Chen et al, 1996; Li & Benezra, 1996) and monitors the attachment status of centromeres (Sacristan & Kops, 2015). Attachment of microtubules to the kinetochore allows for the active removal of SAC proteins from the centromere (Waters et al, 1998). However, kinetochore-microtubule interactions are destabilized by the Aurora B kinase (Figure 1.2C)., localized in between the sister centromeres, in a distance dependent manner often called the Aurora B gradient (Pinsky et al, 2006; Liu et al, 2009). Only upon formation of amphitelic attachments are kinetochores sufficiently distant from Aurora B to allow for stable microtubule attachments. The SAC is silenced once amphitely has been accomplished on all chromosomes, leading to the activation of APC/C, an E3 ubiquitin ligase that marks target proteins for destruction (Hardwick & Shah, 2010). Important targets include Cyclin B (Amon et al, 1994; Irniger et al, 1995; King et al, 1995; Sudakin et al, 1995), which activates the mitotic master regulator Cdk1, and securin (Zur & Brandeis, 2001), which inhibits separase from cleaving cohesin. Thus cells are inhibited from exiting mitosis until proper amphitelic attachments are made on all chromosomes and accurate chromosome segregation is ensured.
In summary, centromeres play a key role in the regulation of mitotic progression. Centromeres are responsible for maintenance of sister chromatid cohesion, recruitment of the microtubule binding kinetochore complex, and monitoring proper kinetochore-microtubule attachments. Together, the concerted action of these processes allows for dividing cells to accurately segregate their chromosomes to the two nascent daughters.
22
Specification of centromere identity
Centromeric DNA
Because centromeres are chromosomal loci, the simplest possible mechanism to specify them is by a particular nucleotide sequence. Indeed, in the budding yeast S. cerevisiae, centromeric sequences consist of three elements, called CDEI, CDEII, and CDEIII (for centromeric DNA element 1– 3). CDEI (8 bp) and CDEIII (25 bp) are both highly conserved between the sixteen S. cerevisiae centromeres, and CDEII (~80–85 bp), although not well conserved, systematically has an AT-richness of >90% (Hieter et al, 1985; Niedenthal et al, 1991; Hegemann & Fleig, 1993). Mutations in any of these elements can cause a dramatic increase in chromosome loss, indicative of failure to form functional centromeres (Gaudet & Fitzgerald-Hayes, 1989; McGrew et al, 1989; Niedenthal et al, 1991; Hegemann & Fleig, 1993; Meluh & Koshland, 1995), with the most severe effects in CDEIII, where specific single point mutations can completely abolish centromere function (McGrew et al, 1986). Conversely, a naïve 125 bp sequence encompassing the three centromere elements is sufficient to operate as a functional centromere (Cottarel et al, 1989). In summary, specific DNA sequences are both sufficient and required for centromere function in budding yeast.
Based on the budding yeast model system, it was originally thought that centromeres in other species would also be critically dependent on specific DNA sequences or motifs (Willard, 1990). However, unlike budding yeast, centromeres in most other species contain highly repetitive tandem repeat sequences, making them muchly much much more difficult to study. In fission yeast, for example, centromeres consist of a small complex (i.e. non-repetitive) central core (~4–7 kbp) flanked by ~40–100 kbp of repeat sequences (Fishel et al, 1988; Chikashige et al, 1989), while centromeric DNA of Drosophila is characterized by 5 bp repeats, interspersed with transposable elements (Sun et al, 1997). Human centromeres are formed by megabase-sized stretches of so-called alpha-satellite DNA, which consists of
23
imperfect repeats of a 171 bp AT-rich sequence (Manuelidis & Wu, 1978; Manuelidis, 1978; Willard & Waye, 1987). Surprisingly, conservation of centromeric sequences is quite poor, even between closely related species (Haaf & Willard, 1997; Csink & Henikoff, 1998; Malik & Henikoff, 2002; Lee et al, 2005, 2011). In addition, it has been observed in multiple lineages that the position of centromeres along the chromosomes can change independently of the surrounding sequences or structural rearrangements (Montefalcone et al, 1999; Rocchi et al, 2012). Interestingly, as was first described in the long bug Protenor belfragei (Schrader, 1935), centromeres are not necessarily restricted to any one locus, but can instead be diffusely spread along the length of the chromosome in what is called a holocentric arrangement. C. elegans is probably the best known example (Albertson & Thomson, 1982), but holocentricity has been observed in many species and has evolved multiple independent times in both animals and plants (Melters et al, 2012). Given all these observations, centromeres are considered among the fastest evolving chromosomal regions in eukaryotes (Henikoff et al, 2001), which conflicts with the hypothesis that centromere identity is driven by a specific sequence context.
Positive evidence against DNA sequences being essential for human centromere specification came with the discovery of centromeres on atypical loci. So-called neocentromeres were first identified in 1993 on a stably segregating fragment of chromosome 10 that lacked typical α-satellite or other centromeric sequences (Voullaire et al, 1993). Although centromere repositioning appears to be a rare event, over 130 unique human neocentromeres, spanning all chromosomes except 22, have been found to date (Marshall et al, 2008; Liehr, 2014). In the majority of cases analyzed, virtually all cells (within one lineage) contained the same neocentromere, arguing in favor of stable inheritance of the neocentric locus through mitotic divisions (Marshall et al, 2008). Moreover, at least seven independent neocentromeres have been described, which are inherited through human generations (Wandall et al, 1998; Tyler-Smith et al, 1999; Knegt et al, 2003;
24
Amor et al, 2004; Ventura et al, 2004; Capozzi et al, 2009; Hasson et al, 2011), arguing that they are stable in meiosis as well. Importantly, large arrays of α-satellite sequences that did not display any centromeric function can be retained neocentric chromosomes, including meiotically stable ones (Bukvic et al, 1996; Tyler-Smith et al, 1999; Amor et al, 2004; Ventura et al, 2004; Capozzi et al, 2009; Liehr et al, 2010; Hasson et al, 2011). In summary, observations on neocentromeres argue that centromeric sequences are neither required nor sufficient for centromere specification in human cells.
Although not strictly required for centromere identity, specific sequences cannot be excluded to have a function. Indeed, one well known feature of mammalian centromeric DNA is the recruitment of CENP-B, a sequence specific DNA binding protein that recognizes a 17 bp site found within a proportion of α-satellite monomers (Masumoto et al, 1989). Although CENP-B is non-essential (Hudson et al, 1998), it may play a role in organizing centromeric chromatin (Pluta et al, 1992; Hasson et al, 2011) and it has recently been suggested to contribute to centromere function (Fachinetti et al, 2013). Moreover, in an effort to create centromeres de novo on human artificial chromosomes, it was found that both α-satellite DNA and centromeric CENP-B binding sites are essential (Ohzeki et al, 2002). Another interesting observation is that a surprisingly high number of human neocentromeres have been found at regions that correlate with centromere positions in other primates (Ventura et al, 2003, 2004; Cardone et al, 2006; Capozzi et al, 2008, 2009). Moreover, it was found that orthologous loci have been used in multiple species for evolutionary centromere repositioning events that have become fixed in the population (Ventura et al, 2004). Together, these observations suggest that while specific sequences are dispensable for centromere function and maintenance, they appear to have at least some influence on de novo centromere formation.
25 CENP-A
Because DNA sequences are not responsible for centromere identity, another defining factor must exist. Using auto-immune sera from human scleroderma patients, centromere protein A (CENP-A) was among the first proteins (together with CENP-B and CENP-C) to be identified at human centromeres (Earnshaw & Rothfield, 1985). Soon after its discovery, it was found that CENP-A has many histone-like properties and copurifies with core histone proteins (Palmer et al, 1987). In addition, it shares sequence homology to histone H3, which strongly suggested that CENP-A can replace this histone in centromeric nucleosomes (Palmer et al, 1987, 1991), which was confirmed by in vitro reconstitution studies some 10 years later (Yoda et al, 2000). The first piece of evidence indicating that CENP-A may be the defining feature for centromere identity came from the discovery that it is absent from inactive centromeres in dicentric chromosomes, but readily detected on neocentromeres (Earnshaw & Migeon, 1985; Warburton et al, 1997). In addition, clear centromere specific CENP-A homologues exist in nearly all species analyzed (Malik & Henikoff, 2003; Talbert et al, 2012), with the notable exception of kinetoplastids (Akiyoshi & Gull, 2013). Surprisingly, it was recently found that multiple holocentric insects appear to have lost CENP-A (Drinnenberg et al, 2014), although the presence of centromere specific H3 variants not matching their criteria was not excluded. Furthermore, loss of CENP-A is lethal and results in severe defects of chromosome segregation in all species analyzed (Stoler et al, 1995; Buchwitz et al, 1999; Henikoff et al, 2000; Howman et al, 2000; Blower & Karpen, 2001; Talbert et al, 2002; Régnier et al, 2005; Black et al, 2007b). Conversely, CENP-A is sufficient for the recruitment of virtually all known centromere and kinetochore proteins (Foltz et al, 2006; Heun et al, 2006; Liu et al, 2006; Okada et al, 2006; Carroll et al, 2009, 2010; Barnhart et al, 2011; Guse et al, 2011; Mendiburo et al, 2011), with the exception of the sequence specific DNA binding protein CENP-B (Pluta et al, 1992; Voullaire et al, 1993). Importantly, CENP-A nucleosomes are stably transmitted
26
through both mitotic (Jansen et al, 2007) and meiotic (Palmer et al, 1990; Raychaudhuri et al, 2012; Dunleavy et al, 2012) cell divisions. Together, these observations have for many years spurred the hypothesis that CENP-A is primarily responsible for specifying centromeric identity.
Despite these indications, direct evidence that CENP-A defines centromere identity was lacking until very recently. In a seminal study, Mendiburo et al (2011) used cultured Drosophila S2 cells in which they expressed a fusion protein of CENP-ACID and LacI that can be targeted to a
chromosomally integrated LacO array. Using this cell line, the authors were able to show that ectopically targeted CENP-ACID is assembled into
nucleosomes, recruits virtually all known Drosophila centromere and kinetochore proteins, stably binds kinetochore microtubules, and behaves as a functional centromere (Mendiburo et al, 2011). Most importantly, it was shown that a substantial pool of naïve CENP-ACID, which has no intrinsic
affinity for LacO sequences, is present on the array up to 7 days after pulse-expression of targeted CENP-ACID-LacI (Mendiburo et al, 2011). More
recently, it was shown that LacO-tethering of the CENP-A loading factor HJURP is not only sufficient to induce neocentromere formation, but it is also able to rescue chromosome stability and cell viability after deletion of the endogenous centromere in chicken DT40 cells (Hori et al, 2013). Intriguingly, this same study found similar results after tethering of CCAN components CENP-C or CENP-I. Thus, almost 15 years after the original suggestion by Warburton et al (1997), these beautiful experiments were finally able to provide compelling evidence that CENP-A is sufficient for the initiation of a feedback loop allowing for the stable inheritance of a centromere structure.
The question that arises next is how CENP-A is able to specify a centromere. One controversial hypothesis is that it is integrated into a particle with a radically different conformation than canonical nucleosomes. Indeed, a number of different conformational models have been proposed (reviewed in Black & Cleveland, 2011), including heterotypic CENP-A/H3
27
nucleosomes (Lochmann & Ivanov, 2012), a stable (CENP-A/H4)2 tetramer
lacking H2A and H2B (Williams et al, 2009) and the replacement of H2A and H2B by a non-histone protein (Mizuguchi et al, 2007). However, these models are supported by a very limited amount and oftentimes ambiguous evidence (Black & Cleveland, 2011). Nevertheless, the hemisome model, where particles are composed of a single copy of CENP-A, H4, H2A, and H2B, continually makes its way into high impact publications. The main argument used in favor of the existence of hemisomes is that CENP-A containing particles measured by atomic force microscopy (AFM) have a reduced height of approximately 50% as compared to canonical nucleosomes (Dalal et al, 2007; Dimitriadis et al, 2010; Bui et al, 2012). However, a recent study suggested that AFM measurements of in vitro reconstituted octameric CENP-A nucleosomes are in fact only half the size of their H3 counterparts (Miell et al, 2013), perhaps due to a more flexible packaging of DNA around the histone octamer (Palmer et al, 1987; Conde e Silva et al, 2007; Tachiwana et al, 2011; Hasson et al, 2013). However, these results have almost immediately been refuted by the Dalal and Henikoff labs, practically the exclusive proponents of the hemisome model, after measuring in vitro assembled octameric CENP-A nucleosomes at canonical size ranges (Codomo et al, 2014; Walkiewicz et al, 2014), and it thus remains unclear what the true height is of CENP-A nucleosomes (Miell et al, 2014). Additional observations used in favor of the existence of hemisomes comes from: 1) a nucleosome-crosslinking assay indicating the presence of a single copy of each histone (Dalal et al, 2007), although this could easily be the result of a missing crosslinkable lysine in CENP-ACID (Black & Bassett, 2008;
Zhang et al, 2012) as cysteine-crosslinking readily produced CENP-ACID
dimers (Zhang et al, 2012); 2) an apparent reversed directionality of DNA supercoiling around the CENP-ACse4 particle, which would be most
consistent with a hemisomal conformation (Furuyama & Henikoff, 2009), although alternative, energetically more favorable explanations for the specific observations of the assay have been proposed (Black & Cleveland,