PROGRAMA DE PÓS-GRADUAÇÃO EM PATOLOGIA
LÍVIA MONTEIRO GALVÃO
ESTUDO DE HAPLÓTIPOS DO GENE DA BETA GLOBINA S, FATOR DE CRESCIMENTO ENDOTELIAL VASCULAR-A (VEGF-A), HEMOGLOBINA FETAL
E MANIFESTAÇÕES CLÍNICAS EM PACIENTES COM ANEMIA FALCIFORME
ESTUDO DE HAPLÓTIPOS DO GENE DA BETA GLOBINA S, FATOR DE CRESCIMENTO ENDOTELIAL VASCULAR-A (VEGF-A), HEMOGLOBINA FETAL E
MANIFESTAÇÕES CLÍNICAS EM PACIENTES COM ANEMIA FALCIFORME
Dissertação apresentada ao Programa de Pós-Graduação em Patologia da Universidade Federal do Ceará, como requisito parcial à obtenção do título de Mestre em Patologia. Área de concentração: Medicina II.
Orientador: Profa. Dra. Romélia Pinheiro Gonçalves Lemes.
Co-orientador: Prof. Dr. Max Victor Carioca Freitas
ESTUDO DE HAPLÓTIPOS DO GENE DA BETA GLOBINA S, FATOR DE CRESCIMENTO ENDOTELIAL VASCULAR-A (VEGF-A), HEMOGLOBINA FETAL E
MANIFESTAÇÕES CLÍNICAS EM PACIENTES COM ANEMIA FALCIFORME
Dissertação apresentada ao Programa de Pós-Graduação em Patologia da Universidade Federal do Ceará, como requisito parcial à obtenção do título de mestre em Patologia. Área de concentração: Medicina II.
Aprovada em: ___/___/______.
BANCA EXAMINADORA
________________________________________ Profa. Dra. Romélia Pinheiro Gonçalves Lemes (Orientadora)
Universidade Federal do Ceará (UFC)
_________________________________________ Profa. Dra. Arlândia Cristina Lima Nobre de Morais
Universidade de Fortaleza (UNIFOR)
_________________________________________ Prof. Dr. Roberto César Pereira Lima Junior
Universidade Federal do Ceará (UFC)
respectivamente.
Ao Profa. Dra. Romélia Pinheiro Gonçalves Lemes por acreditar no meu potencial e me oferecer a oportunidade, investimento e orientação.
Ao Prof. Dr. Max Victor Carioca Freitas pelas generosas contribuições e orientações. Aos professores Dr. Roberto César Pereira Lima Junior, Dra. Rosângela Pinheiro Gonçalves Machado, Dr. José Ajax Nogueira Queiroz e Dra. Arlândia Cristina Lima Nobre de Morais pelo tempo, pelas valiosas colaborações e sugestões.
Aos voluntários que participaram do estudo cedendo seu tempo e as amostras biológicas.
Aos funcionários do laboratório do HEMOCE e da UFC pela ajuda e o café mautino. A Valéria Cordeiro de Oliveira pelos conselhos, organização e ajuda nos momentos difíceis.
Aos colegas da turma de mestrado, pelas reflexões, críticas e sugestões recebidas. A Maritza Barbosa, Talytta Santos, Amanda Menezes, Marília Laurentino, Alano Pedrosa, Marilena, Tarcisio, Pedro Aurio, Renata Eleutério, André Jhonatan, Iran Davi e Monalisa Figueredo pelo suporte científico e emocional e pela alegria compartilhada no dia-a-dia..
Aos meus amigos, Anália Almeida, Anamaria Falcão, Bruna Vitoriano, Daisy Lima, Juliano Casemiro, Liana Sales, Luana David, Pablo Vitoriano e Tamiris Goebel, pelas valiosas horas de ócio produtivo, pelas risadas infinitas e por toda comida ofertada. A Océlia Monteiro e Fátima Sales por terem me adotado e zelado por mim quando precisei.
A meu avô, Wanderley Neves, e meus irmãos, Lílian Galvão, Wanderley Galvão, Larissa Adeodato e Mª Odete Rocha.
“Nada do que você veste, pensa ou diz te define tão bem quanto suas prioridades”
A anemia falciforme (AF) é uma doença hereditária decorrente de uma mutação pontual no gene da globina S gerando uma hemoglobina anormal denominada de hemoglobina S (HbS) , que quando desoxigenada sofre polimerização favorecendo o fenômeno de vaso-oclusão, dano endotelial vascular e a expressão de fatores angiogênicos. Fatores genéticos como os haplótipos da beta globina S são responsáveis pelas diferenças clínicas entre os pacientes. O objetivo deste trabalho foi verificar a associação dos haplótipos da globina s com o fator de crescimento endotelial vascular-A (VEGF-A), com hemoglobina fetal (HbF) e manifestações clínicas em pacientes com AF. O estudo foi do tipo transversal, analítico e observacional. A população em estudo foi constituída por 51 pacientes com diagnóstico molecular de AF, sendo 39 em uso hidroxiúreia (HU) (617 mg/dia) e 12 sem uso. No total de 61 indivíduos sem hemoglobinopatias, considerados sadios, participaram como grupo controle. A análise dos haplótipos foi realizada por PCR-RFLP e a dosagem de VEGF-A por ELISA. Os dados clínicos e laboratoriais foram obtidos por consulta em prontuários. A análise estatística foi realizada mediante a utilização do programa estatístico GraphPad Prism versão 5.0. Os pacientes com AF apresentaram parâmetros hematológicos característicos da doença e aumento significativo de VEGF-A em relação ao grupo controle, quando se estratificou os pacientes pelo uso de HU foi observada concentrações maiores entre os pacientes que não estavam em uso de HU. O haplótipo predominante no grupo estudado foi o Bantu, não houve diferença nas concentrações de VEGF-A entre os grupos de haplótipos da globina s e manifestações clínicas, excetuando-se as crises vaso-oclusivas (CVO), onde o uso de HU influenciou as concentrações de VEGF-A no grupo com maior quantidade de crises por ano. Observou-se uma correlação negativa entre as concentrações de VEGF-A e as concentrações de HbF e uma correlação positiva com a contagem de reticulócitos. Conclui-se que o aumento do VEGF-A nos pacientes sem uso de HU possa ser atribuído a fisiopatologia da doença e que provavelmente o uso da HU teve uma ação anti-angiogênica. No entanto mais estudos são necessários para avaliar o papel do VEGF-A e da angiogênese na AF.
ABSTRACT
Sickle cell anemia (SCA) is a hereditary disease caused by a point mutation in S globin gene causing an abnormal hemoglobin called hemoglobin S (HbS), when deoxygenated polymerizes favoring the vaso-occlusion phenomenon, vascular endothelial injury and expression of angiogenic factors. Genetic factors such as haplotypes beta globin S are responsible for the clinical differences among patients. The aim of this study was to
investigate the association of haplotypes of s globin with vascular endothelial growth factor -A (VEGF--A), with fetal hemoglobin (HbF) and clinical manifestations in patients with SCD. The study was cross, analytical and observational. The study population consisted of 51 patients with a molecular diagnosis of SCD, 39 in use hydroxyurea (HU) (617 mg/day) and 12 unused. A total of 61 individuals without hemoglobinopathies, considered healthy, participated as a control group. The haplotype analysis was performed by PCR-RFLP and the dosage of VEGF-A by ELISA. Clinical and laboratory data were obtained in consultation records. Statistical analysis was performed by using the statistical program GraphPad Prism version 5.0. Patients with SCD showed characteristic hematological disease and significant increase in VEGF-A in the control group when stratified patients by the use of HU was observed higher concentrations among patients who were not in use HU. The predominant haplotype in the group studied was the Bantu, there was no difference in VEGF-A concentrations between groups of haplotypes of S globin and clinical manifestations, except for the vaso-occlusive crises (CVO), where the use of HU influenced VEGF-a concentrations in the group with the largest number of attacks per year. There was a negative correlation between VEGF-A concentrations and concentrations of HbF and a positive correlation with the reticulocyte count. It is concluded that the increase of VEGF-A in patients without use of HU can be attributed to the pathophysiology of disease and probably the use of HU had an anti-angiogenic effect. However more studies are needed to evaluate the role of VEGF-A and angiogenesis in SCD.
LISTA DE FIGURAS
Figura 1 – Distribuição global da HbS ... 16
Figura 2 – Alteração da membrana do eritrócito por polímeros de hemoglobina S ... 19
Figura 3 – Interações dos eritrócitos falcizados com endotélio... 20
Figura 4 – Patogênese da vaso-oclusão na AF ... 22
Figura 5 – Isquemia-reperfusão na AF ... 23
Figura 6 – Distribuição das áreas de origem do gene da HbS ... 32
LISTA DE GRÁFICOS
LISTA DE TABELAS
Tabela 1 – Enzimas de restrição utilizadas para a detecção de haplótipos do cluster do gene
S
LISTA DE ABREVIATURAS E SIGLAS
AF Anemia falciforme
ANOVA Análise de variância AVC Acidente vascular cerebral
BCAM/LU Molécula de adesão basal/Lutheran
Ca++ Cálcio
CD36 Cluster of differentiation 36 CD47 Cluster of diferentiation 47 CD64 Cluster of diferentiation 64 CEC Células endotéliais circulantes
CHCM Concentração da hemoglobina corpuscular média
CVO Crises vaso-oclusivas
DNA Ácido desoxirribonucléico
EDTA Ácido etilenodiamino tetra-acético ELISA Enzyme-linked immunosorbent assay
EPO Eritropoietina
ET-1 Endotelina-1
FBS Fibronectina
FDA Food and Drug Administration
Fe3+ Ferro férrico
FS Fosfatidilserina
GLU Ácido glutâmico
H2O2 Peróxido de hidrogênio
Hb Hemoglobina
HbF Hemoglobina fetal
HbS Hemoglobina S
HbS/ Heterozigose para HbS e talassemia
HbSA Heterozigose da HbS
HbSC Heterozigose para HbS e C
HbSD Heterozigose para HbS e D
HbSS Homozigose para HbS
HIF-1α Fator induzido por hipóxia-1α
HU Hidroxiuréia
HUWC Hospital universitário Walter Cantídeo ICAM-1 Molécula de adesão intercelular - 1
IL-1 Interleucina-1
IL-6 Interleucina-6
IL-8 Interleucina-8
K+ Potássio
kDa Quilodalton
LACT Laboratório de análises clínicas e toxicológicas
LHGDH Laboratório de pesquisa em hemoglobinopatias e genética das doenças hematológicas
Masc Masculino
NF-κB Fator nuclear kappa B
NO Óxido nítrico
O2 Oxigênio
PAF Fator de ativação plaquetária
pb Pares de base
PCR-RLFP Restriction fragment length polymorphism
PDGF Fator de crescimento derivado das plaquetas
pH Potencial hidrogeniônico
PIGF Fator de crescimento placentário ROS Espécies reativas de oxigênio
STA Síndrome torácica aguda
TCLE Termo de conscentimento livre e esclarecido TNF-α Fator de necrose tumoral
TSP Trombospondina
UFC Universidade Federal do Ceará
VAL Valina
VCAM-1 Molécula de adesão celular e vascular
VCM Volume corpuscular médio
VEGF Fator de crescimento endotelial vascular
α4 1 ou VδA Antígeno de ativação tardia
LISTA DE SÍMBOLOS
% Porcentagem
® Marca registrada
dL Decilitro
fL Fentolitro
g Grama
mL Mililitro
nm Nanômetro
ºC Grau Celsius
pg Picograma
α Alfa
Beta Gama Delta
κ Kappa
SUMÁRIO
1. REVISÃO BIBLIOGRÁFICA ... 15
1.1. Histórico da Anemia Falciforme ... 15
1.2. Definição da AF ... 16
1.3. Fisiopatologia ... 17
1.3.1. Polimerização da HbS ... 17
1.3.2. Moléculas de adesão ... 19
1.3.3. Papel do Óxido nítrico (NO) na homeostase vascular ... 21
1.4. Inflamação na AF ... 22
1.5. Dano endotelial ... 24
1.6. Eventos clínicos na AF ... 25
1.7. Angiogênese ... 27
1.8. Papel do VEGF na AF ... 28
1.9. Moduladores genéticos da AF ... 31
1.10. Tratamento da AF ... 34
1.11. Justificativa ... 35
2. OBJETIVOS ... 36
2.1. Objetivo geral ... 36
2.2. Objetivos específicos ... 36
3. CASUÍSTICA E MÉTODOS ... 37
3.1. Desenho do estudo ... 37
3.2. Casuística ... 37
3.3. Local do Estudo ... 37
3.4. Seleção das amostras ... 38
3.4.1. Critérios de inclusão ... 38
3.4.2. Critérios de exclusão ... 38
3.4.3. Coleta das amostras biológicas... 38
3.4.4. Coleta dos dados... 38
3.4.5. Definição das variáveis avaliadas nos grupos ... 39
3.4.5.1. Variáveis demográficas ... 39
3.4.5.2. Variáveis laboratoriais ... 39
3.5. Análises laboratoriais ... 39
3.5.1. Análises moleculares ... 39
3.5.1.1. Extração do DNA genômico ... 39
3.5.1.2. PCR-RFLP ... 40
3.5.1.3. Análise dos haplótipos da mutação da globina ßS ... 40
3.5.2. Dosagem de VEGF-A ... 41
3.6. Descarte de material biológico ... 42
3.7. Critérios éticos ... 42
3.8. Análise estatística ... 42
4. RESULTADOS ... 43
5. DISCUSSÃO ... 49
6. CONCLUSÃO ... 53
7. CONSIDERAÇÕES FINAIS ... 54
REFERÊNCIAS ... 55
ANEXO A – PARECER COMITÊ DE ÉTICA EM PESQUISA ... 68
ANEXO B – PROSPECTO DO KIT DE VEGF-A ... 71
APÊNDICE A – TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO ... 88
1. REVISÃO BIBLIOGRÁFICA 1.1.Histórico da Anemia Falciforme
A anemia falciforme (AF) é uma hemoglobinopatia hereditária causada por uma mutação que afeta o DNA e leva à formação da hemoglobina S (HbS). O termo “anemia
falciforme” foi usado pela primeira vez por εaçom em 19ββ, quando foram relacionadas
algumas manifestações clínicas e hematológicas ao defeito genético: pacientes de etnia afrodescendente que apresentavam icterícia, fraqueza, úlceras de membros inferiores, anemia intensa e hemácias falcizadas no sangue periférico (GONÇALVES et al., 2003; ORLANDO
et al., 2000).
O primeiro relato científico da AF foi em 1910, por James B. Herrick, onde células em forma de “foice” foram encontradas no sangue de um jovem estudante negro que tinha anemia severa. A estrutura falciforme das hemácias, experimentalmente, foi revertida pelo fornecimento de oxigênio às células, por Hahn e Gillespie, em 1927, sugerindo que a anóxia causa a falcização. Estes pesquisadores também foram os primeiros a associarem a falcização das hemácias ao pH ácido do meio. Em 1930 foram registrados os primeiros dados clínicos da doença associados a população negra no Brasil (CAVALCANTI; MAIO, 2011). As pesquisas experimentais construídas com base na anomalia morfológica das hemácias revelaram que as células falciformes são menos flexíveis que os glóbulos vermelhos normais e, portanto, teriam dificuldade para passar em capilares (SHERMAN, 1940).
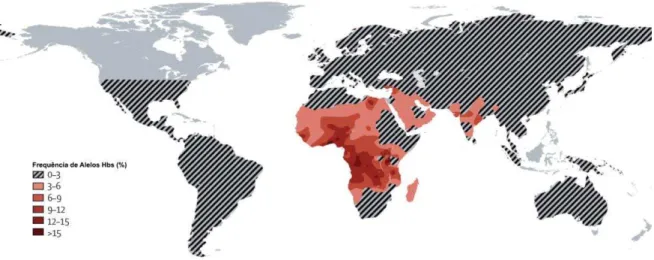
Apesar de ter origem desconhecida, a AF foi encontrada primeiramente na África, onde se verificou posteriormente sua alta frequência na região central e ocidental. Atualmente é uma doença de distribuição mundial (Figura 1), tendo importância significativa na população brasileira devido à miscigenação da população com grupos africanos trazidos para o Brasil nos séculos XV a XIX (BITOUNGUI et al., 2015; REES; WILLIAMS; GLADWIN, 2010; SALDANHA, 1957; ZAGO, M. A.; FIGUEIREDO; OGO, 1992)
Figura 1 – Distribuição global da HbS
Fonte: Adaptado de Rees e coloaboradores ( 2010).
Em 1948, Linus Pauling observou que a hemoglobina (Hb) das células falciformes tinha uma carga elétrica positiva maior que a Hb das células vermelhas normais e que estas poderiam ser separadas por eletroforese. A partir desta pesquisa foi introduzido o termo
“doença molecular” para caracterizar a doença falciforme. No mesmo ano, Janet Watson e colaboradores, correlacionaram os sintomas da doença a diminuição da concentração da hemoglobina fetal (HbF) observando que recém-nascidos com elevada concentração de HbF permaneciam assintomáticos. Somente em 1956, Ingram avaliou que o efeito molecular envolvido na AF é a troca de uma base nitrogenada no DNA codificante da molécula de hemoglobina.
1.2.Definição da AF
As hemoglobinopatias consistem em um conjunto de alterações na estrutura ou na síntese da hemoglobina, resultantes de defeitos genéticos, condicionando um aumento da morbidade em condições ambientais normais. A maior parte das alterações ocorre em cadeias
globínicas da hemoglobina, mas alterações de cadeias α, e são também relativamente comuns. As mutações nas cadeias globínicas são responsáveis pela produção de uma expressiva variedade de hemoglobinas anormais e entre as variantes mais frequentes encontram-se as hemoglobinas S, C, D e E (KIMURA et al., 2008; OLD, 2003).
A doença falciforme é um grupo representado pela presença da hemoglobina S (HbS). Estão inclusas neste grupo a AF (HbSS), interações com talassemias (HbS/ 0
talassemia, HbS/ +
HbSD) e a associação com a HbA (HbSA) denominado traço falcêmico (ORLANDO et al., 2000).
Quando a HbS está em heterozigoze (HbSA, HbSC, HbSD) o indivíduo apresenta uma forma mais branda da doença, com sintomas mais leves ou ausentes e uma expectativa de vida maior que a parcela da população que tem a mutação em homozigose (HbSS), onde as hemácias apresentam cerca de 80% de HbS, tornando estes pacientes portadores de anemia hemolítica crônica grave (ASHLEY-KOCH; YANG; OLNEY, 2000).
1.3.Fisiopatologia
1.3.1. Polimerização da HbS
A produção da HbS é resultante de uma alteração genética que causa a mutação pontual de uma base nitrogenada no DNA, na posição 6 do cromossomo 11, resultando na troca do códon do aminoácido ácido glutâmico pela valina (GACGTC) nas cadeias da globina. Esta substituição leva a modificações físico-químicas na molécula da hemoglobina e redução da solubilidade da molécula de deoxihemoglobina, fazendo com que os eritrócitos assumam uma forma falciforme (INGRAM, 1956; STUART; NAGEL, 2004; WAGENER et al., 2001).
A substituição de um aminoácido hidrofílico (GLU) por um hidrofóbico (VAL) causa uma modificação na carga elétrica da hemoglobina promovendo a polimerização da HbS quando esta se encontra em um meio com baixa concentração de O2. O polímero que se
forma possui 7 cadeias duplas de fibras que tem contato axial e lateral permitindo a interação entre os aminoácidos valina, fenilalanina e leucina. Esta alteração causa a mudança na forma discóide normal das hemácias para forma de foice, chamada drepanócito, e reduz o tempo de vida média da hemácia de 120 dias para 7 a 25 dias. Além da concentração de O2 outros
fatores interferem neste processo, como a concentração de HbS e HbF, a temperatura e o pH do meio (FERRONE, FRANK A.; HOFRICHTER; EATON, 1985; IQBAL et al., 2013). Quanto mais rígido o polimero formado, menor é a flexibilidade da célula, menor sua estabilidade e mais difícil a reversão da polimerização (CHRISTOPH; HOFRICHTER; EATON, 2005; FERRONE, FRANK.; HOFRICHTER; EATON, 1985; FERRONE, FRANK, 2004; YOSMANOVICH et al., 2016).
viscosidade sanguínea e a densidade no interior celular. Além destes eventos, a hemoglobina polimerizada se liga a proteínas de membrana o que pode causar perda do grupo heme e liberação do Fe3+ promovendo um microambiente oxidativo no interior da célula. Se ocorrer algum dano definitivo na estrutura dos eritrócitos durante o ciclo de polimerização as células tornam-se irreversivelmente falcizadas, perdendo a capacidade de transporte de oxigênio e sofrendo hemólise intravascular. A polimerização da hemoglobina é considerada por muitos o evento primário responsável pela fisiopatologia da doença (FRENETTE; ATWEH, 2007; LEW; BOOKCHIN, 2005; VANDORPE et al., 2010; ZAGO; PINTO, 2007). Os danos causados pela falcização ocorrem de maneira heterogênea no organismo, alguns órgãos têm condições mais propícias para o fenômeno que outros, como o baço onde comumente as hemácias falcizadas são sequestradas e causam múltiplos infartos podendo gerar esplenectomia (ZAGO; PINTO, 2007).
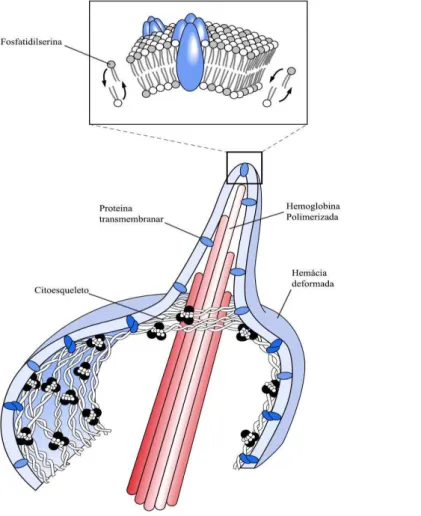
Figura 2 – Alteração da membrana do eritrócito por polímeros de hemoglobina S
Fonte: Adaptada de Statius Van Eps (1999).A formação de polímeros de HbS ocasiona a desorganização lipídica e a exposição da fosfatidilserina (FS) que altera as propriedades adesivas dos eritrócitos falciformes contribuindo para um estado pró-inflamatório.
1.3.2. Moléculas de adesão
Durante períodos em que o paciente apresenta anemia, a medula é estimulada a liberar eritrócitos imaturos, os reticulócitos, que apresentam em sua superfície níveis elevados
de moléculas de adesão, como o CDγ6 e a integrina α4 1 , que facilitam as interações com o
endotélio. Pacientes com AF apresentam número aumentado de reticulócitos no sangue periférico devido à anemia hemolítica crônica. Os eritrócitos deformados pela polimerização da HbS e reticulócitos são mais suscetíveis à adesão, pelo aumento da viscosidade sanguínea e aumento da expressão de moléculas na superfície celular. (ATAGA et al., 2008; CONRAN; FRANCO-PENTEADO; COSTA, 2009; HEBBEL, 2008).
Mais de uma dúzia de moléculas têm sido implicadas no processo de adesão das células ao endotélio: O antígeno de ativação tardia (VδA) ou integrina α4 1 do eritrócito se
interação com proteínas plasmáticas; O CD36, que é um receptor glicoprotéico, liga a trombospondina (TSP) e o fator de Von Willebrand às proteínas endoteliais (HEBBEL, 2008); Além destes também é conhecido a atuação do CD47, que é uma glicoproteína de membrana que associa a TSP ao endotélio via receptores de vibronectina αv 3, a proteína produzida pelo
grupo sanguíneo Lutheran e o BCAM/LU (molécula de adesão basal), que também se liga à lamina na matriz extracelular; A FS que está expressa na superfície dos drepanócitos e se liga
ao receptor da fibronectina (αv 3) do endotélio (Figura 3) (ELION et al., 2004; KAUL;
FINNEGAN; BARABINO, 2009; TELEN, 2016). Outra molécula envolvida na ligação dos
drepanócitos através do αv 3 é a molécula de adesão intercelular (ICAM-1). Estudos em
murinos mostraram que esta molécula esta envolvida nas crises vaso-oclusivas (CVO) e no aumento de ligação dos leucócitos ao endotélio (ZENNADI et al., 2008, 2007).
Figura 3 – Interações dos eritrócitos falcizados com endotélio
Fonte: Adaptado de Telen (2016). As moléculas de adesão dos eritrócitos se ligam as proteínas plasmáticas, matrix extracelular e ao endotélio.
(GUTSAEVA et al., 2011). CD64, CD36, L-selectinas e P-selectina são também encontrados na superfície de neutrófilos ativados e favorecem a adesão ao endotélio, recrutam plaquetas e outros neutrófilos para o sítio inflamatório e secretam H2O2 que juntamente com os ROS
lesionam o endotélio (OKPALA, 2004).
1.3.3. Papel do Óxido nítrico (NO) na homeostase vascular
A vaso-oclusão e a hemólise intravascular são os eventos responsáveis pelas manifestações clínicas evidenciadas na AF como a anemia crônica, icterícia, sequestro esplênico, crises de dor, priapismo, síndrome torácica aguda, acidente vascular cerebral, hipertensão pulmonar, úlceras de perna, necrose asséptica do fêmur e maior suscetibilidade a infecções (REES; WILLIAMS; GLADWIN, 2010).
O endotélio regula a homeostase vascular através do controle do tônus vasomotor, do fluxo sanguíneo, do crescimento de células do musculo liso e da inflamação local (HUANG; VITA, 2006). As células endoteliais produzem vasoconstritores como endotelinas e um potente vasodilatador, chamado NO, responsáveis pela regulação do tônus vascular (GALLEY; WEBSTER, 2004; WAGENER et al., 2001). A AF é caracterizada por níveis baixos de NO principalmente por causa da hemólise intravascular que libera hemoglobina livre, heme e arginase no meio extracelular. A arginase tem como substrato a L-arginina, precursora do NO, quando consumida reduz as quantidades de NO que deixa de agir no musculo liso dos vasos, diminuindo a vasodilatação. Reforçando este efeito as células endoteliais e o músculo liso de pacientes falciformes produzem níveis elevados de endotelina-1 (ET-endotelina-1), um potente vasoconstritor produzido em resposta ao estímulo inflamatório, hipóxia ou estresse (ERGUL et al., 2004). A vasoconstrição desempenha papel importante na fisiopatologia da AF, estando envolvida na hipertensão pulmonar, no priapismo e nas crises vaso-oclusivas (CVO) (KATO; GLADWIN; STEINBERG, 2007; THAKUR et al., 2014; VIGNON-ZELLWEGER, 2009; WOOD; HSU; GLADWIN, 2008).
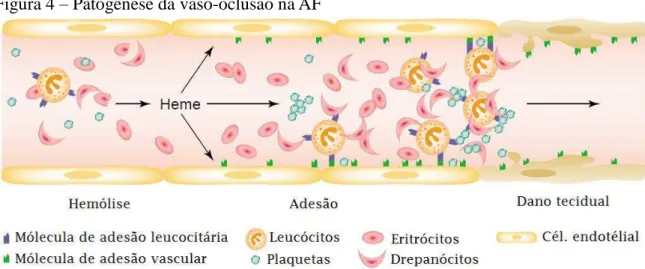
Figura 4 – Patogênese da vaso-oclusão na AF
Fonte: Adaptado de Wagener et al. (2001). Os eritrócitos falcizados quando sofrem hemólise liberam hemoglobina e heme livre no meio extracelular. Este fenômeno induz a expressão de moléculas de adesão na superfície das células endoteliais, leucócitos e reticulócitos. As células aderidas ao endotélio causam ativação dos fatores de coagulação e bloqueio do fluxo.
1.4.Inflamação na AF
A AF é uma doença caracterizada por um estado inflamatório crônico com episódios agudos evidenciados por um aumento da contagem de leucócitos e meia vida leucocitária diminuída. A adesão de células ao endotélio causa redução do fluxo sanguíneo, falcização das hemácias, hipóxia local, e aumento de moléculas pró-inflamatórias. Ao mesmo tempo ocorre a ativação da cascata de coagulação e ativação de leucócitos que são atraídos para o foco inflamatório causando obstrução e dano ao endotélio vascular (HEBBEL, ROBERT P; OSAROGIAGBON; KAUL, 2009; INWALD et al., 2000; WUN et al., 2002).
O perfil inflamatório presente em pacientes com AF resulta da adesão do eritrócito falcizado juntamente com outras células sanguíneas ao endotélio, de lesões de isquemia-reperfusão, das hemólises e das infecções (MADIGAN; MALIK, 2006; ZAGO, MARCO ANTONIO; PINTO, 2007). O endotélio ativado é responsável pela produção de uma série de citocinas e quimiocinas que contribuem para ativação das células e das plaquetas contribuindo para a produção aumentada de moléculas inflamatórias (LANARO et al., 2009; PROENÇA-FERREIRA et al., 2014).
(LANARO et al., 2009). A migração das células inflamatórias através da barreira endotelial contribui para manifestações da doença como as úlceras de perna (SMITH, PHILIP COLERIDGE, 2006). Além disto, as plaquetas, estimuladas pelo fator de ativação plaquetária (PAF) produzido pelas células endoteliais ativadas, liberam trombospondina e fibronectina (FBS) que participam das interações celulares com o endotélio e contribuem para a oclusão dos vasos (GALLEY; WEBSTER, 2004).
A interação dos leucócitos, hemácias, plaquetas, o aumento dos fatores de crescimento e citocinas, o aumento da viscosidade do leito vascular e a adesão ao endotélio resultam na oclusão do fluxo nas vênulas pós-capilares e isquemia, vaso-oclusão, que pode ser seguida do restabelecimento do fluxo (Figura 5) (FRENETTE, 2002; TURHAN et al., 2002). Este evento cíclico chamado de isquemia-reperfusão aumenta o estresse oxidativo no local e causa dano ao tecido endotelial também contribuindo para perpetuação do perfil inflamatório crônico (HABARA; STEINBERG, 2016; HEBBEL, 2014).
Figura 5 – Isquemia-reperfusão na AF
1.5.Dano endotelial
A disfunção endotelial manifestada pela ativação das células endoteliais é comumente caracterizada pelo aumento de moléculas de adesão na superfície das células endoteliais e está envolvida no desenvolvimento de patologias vasculares. A lesão de reperfusão, a adesão dos leucócitos e o insulto reológico causado pela falcização dos eritrócitos podem conduzir a danos endoteliais e descolamento de células do endotélio, como relatado em outras doenças isquêmicas (KAUL; HEBBEL, 2000; RUDNICKI et al., 2016; STRIJBOS et al., 2009).
Os radicais livres de oxigênio (ROS) são moléculas ativas contendo átomos de oxigênio e persistem fisiologicamente como um produto do metabolismo e como um mecanismo de defesa utilizado pelo sistema imunitário para neutralizar ameaças bacterianas. No entanto, quando ROS são produzidos em quantidades que sobrecarregam as moléculas antioxidantes endógenas, podem resultar em dano celular significativo (LAPLANTE et al., 2005). Estas moléculas estão implicadas em doenças como hipertensão, doença renal crônica e na AF (KUPESIZ et al., 2012).
Após os eventos de isquemia-reperfusão acontece a re-oxigenação da área que sofreu hipóxia e aumento da geração de ROS. A ocorrência repetida e aleatória destes eventos afeta negativamente a função das células do endotélio vascular e contribui para o dano de múltiplos órgãos (HABARA; STEINBERG, 2016; HEBBEL, 2014). Foi demonstrado em modelos animais que a ET-1, potente vasoconstritor, tem efeitos adversos potentes na AF por potencializar a lesão causada pela isquemia-reperfusão estimulando a produção de ROS (HEIMLICH et al., 2015; SABAA et al., 2008).
com quantidades aumentadas de H2O2 e moléculas de adesão em sua superfície, liberando
ROS que provocam danos oxidativos (OKPALA, 2004; WUN et al., 2002). Além disso, pacientes com AF têm diminuição da capacidade antioxidante, interferindo nos mecanismos endógenos de proteção para evitar lesões induzidas pelo estresse oxidativo (ALMEIDA et al., 2010).
1.6.Eventos clínicos na AF
Foram descritos dois modelos de subfenótipos associados ao quadro inflamatório dos pacientes com AF: O fenótipo vaso-oclusivo e o fenótipo da disfunção endotelial causada por hemólise. No primeiro modelo os pacientes com maior concentração de HbS teriam maior capacidade de desenvolver vaso-oclusão devido a da maior adesão endotelial causada pela polimerização da HbS. Deste fenótipo resultariam manifestações clínicas como osteonecrose, síndrome torácica aguda e maior quantidade de episódios dolorosos. O modelo causado pela hemólise, pacientes com menor concentração de HbS e níveis aumentados de marcadores de hemólise como contagem aumentada de reticulócitos, desidrogenase lática sérica, hemoglobina livre, heme, arginase e diminuição de NO teriam uma anemia mais acentuada, complicações clínicas provenientes da hemólise e também disfunção endotelial. Nesses pacientes as manifestações clínicas mais comuns são hipertensão pulmonar, acidente vascular cerebral, úlceras de perna e priapismo (KATO; GLADWIN; STEINBERG, 2007).
Dor é a complicação clínica mais frequente nestes pacientes e a elevada frequência de crises é uma característica marcante da doença sendo associada a morte precoce e maior causa de hospitalizações em pacientes maiores de 20 anos. A dor intensa é uma consequência das CVO e costuma ceder ao longo de dias ou semanas. Apesar das CVO serem auto limitadas podem resultar em dano permanente aos órgãos. Fatores como hematócrito elevado e baixa concentração de HbF estão associados com um aumento na frequência de CVO. Outra intercorrência de relevância para as CVO é a elevada suscetibilidade à infecções bacterianas, principalmente pneumococos e Haemophilus influenzae (DARBARI et al., 2012; PLATT et al., 1991; REES; WILLIAMS; GLADWIN, 2010; SMITH, TERIKA P et al., 2015; SMITH, WALLY R, 2014).
peniana e impotência. A presença dos episódios pode estar relacionada ao baixo fluxo venoso e isquemia tecidual (BURNETT; BIVALACQUA, 2007; ROGERS, 2005)
A síndrome torácica aguda (STA) é a segunda maior causa de hospitalizações entre os pacientes com AF. É uma forma de lesão pulmonar aguda e é definida como o desenvolvimento de infiltrado pulmonar alveolar envolvendo pelo menos um segmento do pulmão. Esta síndrome é causada por uma combinação de infecções, embolia e vaso-oclusão da vasculatura pulmonar. A gravidade é variável, cerca de 13% dos pacientes necessitam de ventilação mecânica e a taxa de mortalidade chega a 3%. O tratamento envolve amplo espectro de antibióticos, broncodilatadores e suporte de oxigênio (MILLER; GLADWIN, 2012; NOVELLI; GLADWIN, 2016).
A hipertensão pulmonar e a disfunção diastólica do ventrículo esquerdo são encontradas em até 18 a 32% dos adultos com AF e estão associadas com tolerância reduzida a exercícios físicos e a mortalidade. A pressão pulmonar aumenta durante as CVO e ainda mais durante a síndrome torácica aguda. Pacientes com ambas as doenças, hipertensão pulmonar e disfunção diastólica, estão em risco particularmente elevado de óbito (DAMY et al., 2015; NIU et al., 2009; REES; WILLIAMS; GLADWIN, 2010).
Osteonecrose da cabeça do fêmur é uma complicação grave da AF. Dependendo do genótipo e da gravidade a prevalência da necrose varia de 3 a 50% entre os pacientes. A necrose femoral é causada pelo congestionamento de células na medula óssea. A combinação de patologias vasculares e ósseas contribui para o desenvolvimento de osteonecrose e leva a reparação óssea inadequada que pode evoluir com fraturas ósseas. Outra complicação óssea é a dactilite (síndrome de mão-pé), que se apresenta como um processo de necrose e inflamação da medula óssea das extremidades dos membros, frequente nos dois primeiros anos de vida, desaparecendo com o aumento da idade (DALTRO et al., 2015; ZAGO, MARCO ANTONIO; PINTO, 2007).
O acidente vascular cerebral (AVC) agudo e a isquemia cerebral crônica estão entre as principais causas de incapacitação dos pacientes falciformes. Apesar dos mecanismos ainda não serem totalmente conhecidos alguns fatores são associados a este evento como anemia, leucocitose, dano endotelial e deficiência de NO. Cerca de 17% dos pacientes com AF são afetados por um AVC, podendo até mesmo chegar a 80% no período de 3 anos após o primeiro evento. O risco de AVC é maior durante a primeira década de vida, sendo mais significativo entre as idades de 2 a 5 anos (MENAA, 2013; NOVELLI; GLADWIN, 2016; REES; WILLIAMS; GLADWIN, 2010).
1.7.Angiogênese
A angiogênese é um processo dinâmico de diferenciação e proliferação endotelial, caracteriza-se pela formação de novos capilares a partir de vasos pré-existentes e é um evento essencial para os processos de desenvolvimento, reprodução e reparação. Os estímulos fisiológicos importantes para a angiogênese são principalmente a isquemia, hipóxia tecidual e inflamação. Alguns fatores específicos são conhecidos por estimularem ou inibirem a angiogênese, entre estes estão os fatores de crescimento vasculares, citocinas inflamatórias, moléculas de adesão e o óxido nítrico (DUITS; RODRIGUEZ; SCHNOG, 2006a; NIU et al., 2009).
O processo de angiogênese envolve interações celulares de diversos tipos e mecanismos, onde a organização das células nos vasos sanguíneos e a germinação do lúmen são impulsionadas pela proliferação e migração das células endoteliais em resposta aos mediadores angiogênicos. Alterações na angiogênese têm sido associadas com algumas condições patológicas, particularmente doenças inflamatórias crônicas e respostas celulares dependentes de oxigênio (COSTA; INCIO; SOARES, 2007; FAM et al., 2003; LOPES et al., 2015; NILAND; EBLE, 2012).
processos angiogênicos na patofisiologia da doença (LOPES et al., 2015; NIU et al., 2009; SUNDARAM et al., 2010).
1.8.Papel do VEGF na AF
Uma baixa tensão de oxigênio afeta a fisiologia celular endotelial estimulando a transcrição de fatores que promovem a vasoconstrição como um mecanismo de melhor distribuição de oxigênio. Paralelamente, os fibroblastos e a liberação de fatores de crescimento para células endoteliais são estimulados com a função de remodelar os vasos e promover a angiogênese. Um dos principais fatores de crescimento envolvido na angiogênese é o VEGF (FALLER, 1999; LOPES et al., 2015).
O VEGF é um mitógeno de células endoteliais derivado das artérias, veias e vasos linfáticos e liberado por células endoteliais, fibroblastos, células do musculo liso, plaquetas, neutrófilos e macrófagos (BAO et al., 2009; FERRARA, 1999). Tem como função a quimiotaxia de leucócitos, aumento da permeabilidade vascular e da adesividade de células ao endotélio por aumentar a expressão de ICAM-1 e VCAM-1. Este mitógeno também induz a produção, pelas células endoteliais, de fatores pró-coagulantes e de NF-κB, conhecido como um mediador da inflamação vascular. O VEGF é responsável por regular a angiogênese, a embriogênese e a angiogênese patológica associada a tumores e neovascularização intraocular (BROCK; DVORAK; SENGER, 1991; KIM, I et al., 2001; NOURSE et al., 2009; STANNARD et al., 2007; SULLIVAN; BREKKEN, 2010).
al., 2011; FERRARA; GERBER; LECOUTER, 2003; RUGGIERO et al., 2011; SOBOLEWSKI; GACKO, 2011; WATSON et al., 2000). A inativação de apenas um alelo do VEGF-A ou a hiperexpressão desta molécula provoca a formação de vasos sanguíneos anormais e leva a letalidade embrionária. As concentrações de VEGF-A são rigidamente controladas para que ocorra a angiogênese fisiológica (AL-HABBOUBI, HEBA H. et al., 2011; FERRARA; GERBER; LECOUTER, 2003; MIQUEROL; LANGILLE; NAGY, 2000; OLSSON et al., 2006).
Um dos principais fatores que estimulam a expressão do VEGF é o fator induzido por hipóxia (HIF-1α), um fator de transcrição nuclear importante encontrado em células de mamíferos. Condições de hipóxia favorecem a ativação do HIF-1α e a consequente transcrição de vários genes cujas proteínas estão envolvidas em mecanismos homeostáticos, fisiológicos e patológicos, incluindo genes que codificam a eritropoietina, transferrina, endotelina e VEGF (WENGER et al., 1997; ZIELLO; JOVIN; HUANG, 2007).
Para que ocorra a manutenção da homeostase é necessário que exista um equilíbrio entre os sinais pró-angiogêncos e anti-angiogênicos. Este equilíbrio não é mantido na AF (DUITS; RODRIGUEZ; SCHNOG, 2006a). Um dos antagonistas primários e reguladores da função do VEGF in vivo é o sFlt-1, o receptor de VEGF solúvel. Esta molécula se liga a VEGF e impede a interação com os receptores, assim, controlando o potencial angiogênico do VEGF. Normalmente as concentrações de sFlt-1 acompanham o aumento das concentrações de VEGF, mas este aumento de sFlt-1 não é visto na AF, o que pode alterar o equilíbrio da angiogênese (MOHAN et al., 2005).
Níveis anormalmente aumentados de VEGF foram encontrados em várias doenças como enfisema pulmonar (KURTAGIC; JEDRYCHOWSKI; NUGENT, 2009), câncer (JAIN; MUNN; FUKUMURA, 2002), artrite reumatoide (GUO et al., 2016), retinopatia diabética (ABHARY et al., 2009; UCHIDA; HAAS, 2009) e AF (AL-HABBOUBI, HEBAH H et al.,
β01β; GÜRKAN; TANRIVERDI; BAŞδAεIŞδI, 2005).
Distúrbios patológicos característicos de uma angiogênese desequilibrada são encontrados na AF como a retinopatia falciforme e a hipertensão pulmonar. Assim, além de promover a ativação das células endoteliais e adesão de leucócitos e drepanócitos ao endotélio o VEGF também pode estar envolvido nas malformações vasculares observadas na AF (BRITTAIN; PARISE, 2007).
principais complicações da retinopatia falciforme são hemorragia vítrea, descolamento da retina e perda visual. O uso de fármacos anti-VEGF, como o bevacizumab e ranibizumab, tem mostrado resultados positivo na regressão de neovascularização e resolução de hemorragia vítrea em pacientes com AF (ELAGOUZ et al., 2010; MITROPOULOS et al., 2014; OLULEYE; BABALOLA, 2014; SHAIKH, 2008; SIQUEIRA et al., 2006).
O VEGF também desempenha um papel vital para a sobrevivência de células endoteliais através da indução de vários fatores anti-apoptóticos como Bcl-2 e a proteína A1 (FERRARA, 2001). Concentrações alteradas de VEGF foram associadas a um estado anti-apoptótico do endotélio em pacientes com AF, elevando o tempo de sobrevivência destas células. Um estudo conduzido por Bishop e colaboradores, em 1995, mostrou que apenas 30% das células endoteliais circulantes (CEC) encontravam-se em apoptose nos pacientes com AF contra 60% das CEC em indivíduos saudáveis. A apoptose exerce um papel regulador no crescimento de tecidos, alterações nesta regulação podem interferir na angiogênese e na remodelação vascular o que pode acarretar em retinopatia (BISHOP; BRIGGS; KELLEHER, 1995; SOLOVEY et al., 1999). Kim e colaboradores, 2003, observaram a expressão elevada de VEGF e análises imuno-histoquímicas de tecido de retina em pacientes com retinopatia falciforme (KIM et al., 2003)
O aumento do VEGF pode resultar em fibrose e hiperplasia da camada íntima dos vasos, interferindo nas complicações da AF à medida que se desenvolve trombose destes sítios hiperplásicos. O AVC é um exemplo clínico desta interferência (BAO et al., 2009; BLANN et al., 2008). Concentrações elevadas de VEGF também foram associadas a disfunção diastólica ventricular em crianças e adolescentes com AF, um grupo de risco para desenvolvimento de hipertensão pulmonar (NIU et al., 2009).
1.9.Moduladores genéticos da AF
Apesar de todos os pacientes possuírem o mesmo defeito genético, a incidência e a gravidade das CVO variam entre os pacientes e durante o período de vida destes. Além da complicada fisiopatologia que é influenciada geneticamente, os pacientes com AF têm uma grande variedade fenotípica. A concentração de HbF, a presença simultânea com talassemias e os haplótipos do cluster da globina tem papel relevante na gravidade da evolução clínica (ASHLEY-KOCH; YANG; OLNEY, 2000; STEINBERG, MARTIN H.; SEBASTIANI, 2012).
A HbF compõe cerca de 80 a 90% da hemoglobina total ao nascimento, esta porcentagem decresce na infância chegando a 0-2% na vida adulta. Esta hemoglobina tem maior avidez pelo oxigênio que a HbA. A manutenção de altos níveis de HbF tem benéficos clínicos comprovados e ajuda a reduzir as concentrações de HbS nos eritrócitos falcizados inibindo a polimerização da HbS devido à formação de um híbrido com as duas cadeias de globina que não se incorpora ao polímero. A característica não é encontrada nas outras hemoglobinas variantes (HbC, HbD e HbE) que apenas diluem as concentrações de HbS (NGO et al., 2013; STEINBERG, MARTIN H.; SEBASTIANI, 2012). Muitas drogas atuam como agentes indutores da expressão de HbF como, hidroxiuréia (HU), agentes demetiladores de DNA (5-azacitidina), fatores de crescimento hematopoético como a EPO, ácidos graxos de cadeia curta (butiratos e derivados) e os inibidores da histona deacetilase (STEINBERG, MARTIN H et al., 2003).
Outros genótipos da HbS são apresentados por concomitância da doença com
-talassemias. Com exceção do HbS/ 0
talassemia, as associações com esta doença apresentam manifestações clínicas mais brandas da doença falciforme (STEINBERG, M H et al., 2001). As manifestações clínicas mais brandas estão associadas a inibição da formação do polímero de HbS e redução de hemólise intravascular resultando em um curso mais brando da doença (ASHLEY-KOCH; YANG; OLNEY, 2000; ZAGO, MARCO ANTONIO; PINTO, 2007).
Os haplótipos do grupamento gênico da globina localizado no cromossomo 11
determinados através da analise dos sítios polimórficos por meio das endonucleases de restrição ( /HindII, G /HindIII, A /HindIII, 3'ψ /HindII e /AvaII) (OGEDEGBE, 2007).
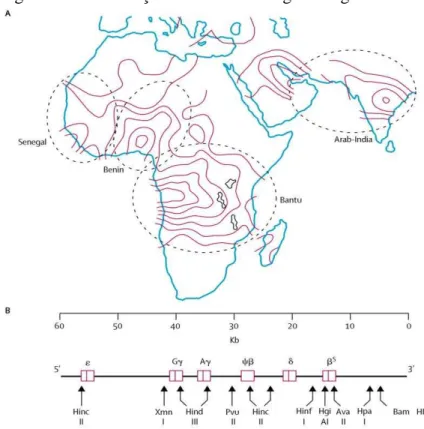
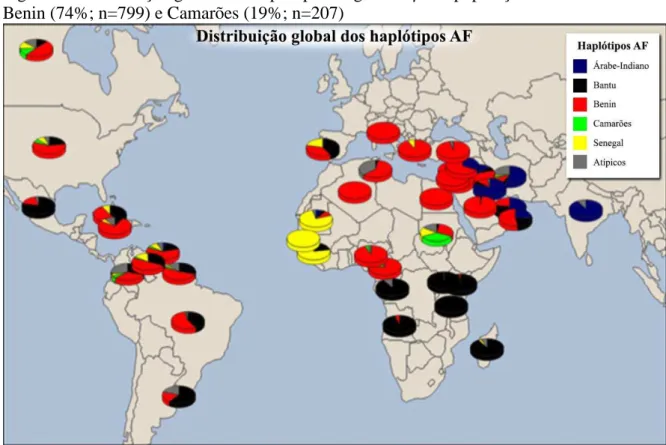
Foram relatados os 5 haplótipos mais comuns do gene da HbS, os quais refletem as regiões de origem em diferentes partes da África, Arábia-Saudita e Índia (Figura 6) (ZAGO, MARCO ANTONIO; PINTO, 2007). O Haplótipo Bantu ou República Centro Africana é originário da África Central, está associado a baixos níveis de HbF e desenvolvimento da forma mais grave da doença. O Senegal, de Senegal, esta associado a níveis elevados de HbF e curso mais brando da doença juntamente com o haplótipo asiático Árabe-indiano, da Arábia-Saúdita e norte da África. O Benin, originário do centro-oeste da África, e o Camarões, originário de Camarões, estão associados a níveis intermediários de HbF (BHAGAT, 2013; LABIE et al., 1985; MONTH et al., 1990; STUART; NAGEL, 2004).
Aproximadamente 5% dos cromossomos estão relacionados com haplótipos de menor prevalência, que são denominados atípicos. Estes podem ser produzidos por substituição de um nucleotídeo em um dos sítios polimórficos de restrição; recombinação entre dois haplótipos S típicos ou conversão não recíproca entre cromossomos de uma sequência de DNA, dentre estes, o mecanismo de recombinação parece ser o mais comum (ZAGO et al., 2001).
Figura 6 – Distribuição das áreas de origem do gene da HbS
contínuas) (B) Haplótipo do gene da globina identificado por sítios polimórficos no DNA de acordo com os sítios de clivagens pelas endonucleases de restrição
Em um estudo de 2015, Bitoungui e colaboradores revisaram a frequência dos
haplótipos da globina S
na população mundial afetada pela AF. Seus estudos concluíram que os haplótipos Benin e Camarões são os mais prevalentes. Os resultados encontrados foram compatíveis com os conhecimentos históricos da migração da população africana pelo mundo. No Ceará, em estudos conduzidos em Fortaleza, foi relatado o haplótipo de maior prevalência sendo o Bantu (63%), seguido por Benin (25%) e atípico (12%) (SILVA; GONÇALVES, 2009). Estes resultados estão em conformidade com a distribuição dos haplótipos observada para a população brasileira onde os haplótipos Benin e Bantu são os mais prevalente (Figura 7) (BITOUNGUI et al., 2015; SILVA, 2009).
O estudo dos haplótipos de gene S fornecem importantes indicadores para a condução da terapêutica. Diversos estudos foram realizados utilizando a associação dos
haplótipos da globina com o perfil oxidativo (SANTOS et al., 2012), com o fator de necrose tumoral (TNF-α) (ROCHA et al., 2014), com fator de Von Willebrand (VAN DER LAND et al., 2013), com os níveis de HbF (BHAGAT, 2013), com o perfil inflamatório (BANDEIRA et al., 2014) e com a STA (BEAN et al., 2014).
Figura 7 – Distribuição global dos haplótipos da globina S na população com AF. Benin (74%; n=799) e Camarões (19%; n=207)
1.10. Tratamento da AF
A maioria das terapias oferecidas para pacientes com AF são de suporte e incluem o uso de antibióticos, vitaminas, antioxidantes e analgésicos. Além destas medidas, em casos de anemia grave, é utilizada transfusão sanguínea como forma de aumentar a concentração de HbA nestes pacientes. O uso de HU é indicado para redução da frequência de crises dolorosas em casos de pacientes que tenham manifestações clínicas moderadas ou graves. O único tratamento que cura os pacientes com AF é o transplante de células tronco hematopoiética (IANNONE et al., 2005; QUINN; ROGERS; BUCHANAN, 2004).
A HU é um antimetabolito citotóxico e citoredutor, que atua na via da síntese de DNA inibindo a ribonucleotídeo redutase, empregado para o tratamento de doenças mieloproliferativa como Policitemia vera, Trombocitemia Essencial e Mielofibrose e doenças neoplásicas. (FRENETTE; ATWEH, 2007; KOVACIC, 2011; SARACENO; TEOLI; CHIMENTI, 2008). Este medicamento tem sido usado com sucesso na terapia da AF e é atualmente o único fármaco aprovado pela Food and Drug Administration (FDA) para uso nesta doença. No Brasil o uso de HU para pacientes com AF só foi aprovado em novembro de 2002, através da portaria 872 do Ministério da Saúde (CANÇADO et al., 2009; WONG et al., 2014).
As indicações de uso da HU na AF são: pacientes com três ou mais admissões hospitalares por crises vaso-oclusivas nos últimos 12 meses; adultos ou crianças com um ou mais episódios de STA nos últimos 24 meses e pacientes com disfunções orgânicas graves (CANÇADO et al., 2009; DAVIES; GILMORE, 2003; FIGUEREDO, 2007). A administração de HU aumenta significativamente a produção de HbF, inibindo a polimerização da HbS, e melhora os sintomas clínicos da doença. Esta droga atua na redução da expressão de moléculas de adesão, com propriedades anti-inflamatórias e anti-agregantes, reduzindo a frequência de STA, das CVO e das hospitalizações. Os dados também sugerem que o tratamento HU pode gerar NO e reduzir a contagem de leucócitos em pacientes com AF (LANARO et al., 2009; WONG et al., 2014). Além do dito acima, em estudos “in vitro”e “in vivo” foram encontradas evidências que sugerem que a HU tem propriedades anti-angiogênicas (DA GUARDA et al., 2016; LOPES et al., 2014, 2015).
relatadas (SIMÕES et al., 2010). Além dos efeitos adversos os pacientes em tratamento apresentam uma variabilidade na resposta ao tratamento com HU que provavelmente esta ligada aos moduladores genéticos da AF ou variações na metabolização do fármaco (IOLASCON; ANDOLFO; RUSSO, 2015).
1.11. Justificativa
Estudos recentes buscam associar a participação de mediadores inflamatórios e marcadores de dano endotelial aos moduladores genéticos da AF. A hipóxia tecidual causada pela vaso-oclusão, principal evento clínico na doença, pode levar a uma resposta angiogênica. Na patofisiologia da AF o processo de angiogênese é pouco estudado. O VEGF é considerado um promotor de angiogênese e neovascularização em uma variedade de processos fisiológicos e patológicos. Concentrações alteradas de moléculas pró-angiogênicas como o VEGF foram relatadas em pacientes com AF, mas a associação desta com moduladores genéticos e com o uso de HU, principal medicamento utilizado na AF e com possível ação anti-angiogênica, ainda precisa de mais esclarecimentos.
O Laboratório de Pesquisa em Hemoglobinopatias e Genética das Doenças Hematológicas (LHGDH) vem promovendo estudos visando melhorar a qualidade de vida e a identificação precoce dos eventos clínicos dos pacientes com AF. Estudos vêm sendo desenvolvidos na avaliação do papel do estresse oxidativo, dos biomarcadores inflamatórios, moduladores genéticos tais como os haplótipos do gene da beta globina S e BCL11A e HBSIL-MYB na modulação da HbF.
2. OBJETIVOS
2.1.Objetivo geral
Determinar a associação dos haplótipos do gene da globina s com as concentrações plasmáticas do fator de crescimento endotelial vascular-A e com os eventos clínicos em pacientes com AF.
2.2.Objetivos específicos
Identificar as características demográficas (Idade e sexo) e laboratoriais (Hb, Ht, VCM, CHCM, leucócitos, plaquetas e reticulócitos) dos pacientes com AF incluídos no estudo em uso ou não de HU;
Avaliar as concentrações plasmáticas do VEGF-A em pacientes com AF, em uso ou não de HU, e comparar com um grupo de indivíduos saudáveis;
Identificar os haplótipos do cluster da globina S em pacientes com AF incluídos no estudo;
Avaliar a associação das concentrações plasmáticas de VEGF-A com as manifestações clínicas da AF em pacientes em uso ou não de HU;
Avaliar a associação das concentrações plasmáticas de VEGF-A com os haplótipos do cluster da globina S nos pacientes com AF;
Avaliar a correlação entre as concentrações de HbF, HbS, reticulócitos e VEGF-A nos pacientes com AF.
3. CASUÍSTICA E MÉTODOS 3.1.Desenho do estudo
O estudo foi do tipo transversal analítico observacional.
3.2.Casuística
Foram convidados a participar da pesquisa, de forma voluntária, pacientes adultos de ambos os sexo, com diagnóstico clínico e laboratorial de AF (HbSS), atendidos no ambulatório de hematologia do HUWC e indivíduos saudáveis (HbAA), de ambos os sexos, doadores de sangue do hemocentro do estado do Ceará (HEMOCE).
Foram coletadas 112 amostras de sangue periférico, sendo destas 51 de pacientes com AF e 61 de indivíduos sem hemoglobinopatias após a obtenção do termo de consentimento livre e esclarecido (TCLE) (APÊNDICE A). Ficando assim estratificada a amostra.
Grupo I composto por pacientes com AF em tratamento com HU com dose média de 617 mg/dia (8,8 mg/kg/dia admitindo um peso médio de 70kg por pessoa) (Grupo HU), grupo II por pacientes com AF sem tratamento com HU (Grupo s/HU) e grupo III com indivíduos saudáveis (Grupo Controle).
As amostras foram estratificadas em relação a concentração de VEGF-A: pacientes com concentrações de VEGF ≤ 120,0 pg/mL e pacientes com concentrações de VEGF > 120,0 pg/mL de acordo com valores médios encontrados na população saudável que constituiu o grupo controle, que corrobora com os valores definidos para indivíduos saudáveis (128,9 pg/mL) encontrados no prospecto do kit de VEGF-A utilizado (Human VEGF-A platinum ELISA) (ANEXO B) fornecido pela empresa eBioscience®.
Quanto à quantidade de CVO por ano relatadas na avaliação ambulatorial os pacientes foram estratificados em pacientes com CVO < 3 e pacientes com CVO ≥ 3 (DARBARI et al., 2012; KINNEY et al., 1999; PERELMAN et al., 2003).
3.3.Local do Estudo
3.4.Seleção das amostras
3.4.1. Critérios de inclusão
Foram convidados a participar do estudo pacientes adultos em acompanhamento no ambulatório de Hematologia do HUWC da UFC com diagnóstico clínico e molecular de AF (HbSS). Os participantes do estudo se encontravam no estado estacionário da doença, segundo os critérios estabelecidos por Ballas (2012): ausência de episódios dolorosos e/ou doenças intercorrentes, tais como infecções e inflamações nas quatro semanas anteriores ao estudo; não admissões hospitalares nos últimos 2-3 dias antes do estudo e ausência de transfusão de sangue nos quatro meses anteriores ao estudo.
Como grupo controle foram selecionados de forma aleatória doadores regulares de sangue do HEMOCE, maiores de 18 anos, com parâmetros hematológicos (Hb, Ht, VCM, CHCM, Leucócitos, plaquetas e reticulócitos) normais.
3.4.2. Critérios de exclusão
Pacientes em uso de vitaminas antioxidantes; tabagistas e etilistas; gestantes; com sorologia positiva para HIV; com diabetes mellitus e/ou com algum quadro de insuficiência renal ou hepática.
No grupo controle foram excluídos os indivíduos em uso de vitaminas antioxidantes; tabagistas e elitistas e/ou em uso de medicamentos.
3.4.3. Coleta das amostras biológicas
Foram coletados 4mL sangue venoso por indivíduo, em tubos contendo ácido etilenodiamino tetra-acético (EDTA) como agente anticoagulante, para extração do DNA genômico, determinação dos haplótipos por Restriction Fragment Length Polymorphism
(PCRRLFP) e determinação dos níveis de VEGFA. O DNA genômico foi armazenado a -20ºC e o plasma a -80ºC para posterior análise.
3.4.4. Coleta dos dados
3.4.5. Definição das variáveis avaliadas nos grupos
3.4.5.1. Variáveis demográficas
Idade: intervalo entre a data de nascimento e dezembro de 2015. Sexo: duas categorias: masculino/feminino.
3.4.5.2. Variáveis laboratoriais
As variáveis laboratoriais Hb (g/dL), Ht (%), VCM (fL), CHCM (g/dL), HbF (%), HbS (%), contagem de leucócitos (x103/L), contagem de plaquetas (x106/L) e reticulócitos (%) foram variáveis numéricas contínuas obtidas paralelamente a coleta do material biológico. As variáveis laboratoriais Hb (g/dL), Ht (%), VCM (fL), CHCM (g/dL), contagem de leucócitos (x103/L), contagem de plaquetas (x106/L) e reticulócitos (%) do grupo controle foram realizadas no LHGDH.
3.4.5.3. Dados clínicos
Os dados clínicos (litíase biliar, priapismo, STA, úlceras de perna, cardiopatia, necrose femural, AVC e CVO) foram coletados retrospectivamente no período de 2000 a 2015 por consulta aos prontuários médicos, excetuando a frequência de CVO que foram avaliadas nos últimos 12 meses (DARBARI et al., 2012; KINNEY et al., 1999; PERELMAN et al., 2003).
3.5.Análises laboratoriais
3.5.1. Análises moleculares
O estudo dos haplótipos da globina S
foi realizado primeiramente através da extração do DNA, seguida da técnica de reação em cadeia mediada pela polimerase e PCR-RFLP.
3.5.1.1. Extração do DNA genômico
3.5.1.2. PCR-RFLP
A análise dos haplótipos da globina ßS foi realizada por meio da técnica da reação em cadeia mediada pela polimerase e utilização de enzimas de restrição para identificação do polimorfismo dos comprimentos nos fragmentos de restrição. A técnica utiliza enzimas de restrição para detecção de mutações e polimorfismos genéticos. As enzimas de restrição reconhecem sítios específicos na sequência do DNA amplificada, que é clivada somente quando o sítio está presente, gerando fragmentos de vários tamanhos que são separados e analisados por eletroforese, sendo posteriormente detectados pela coloração com brometo de etídio ou outro corante fluorescente (CLARK; THEIN, 2004).
3.5.1.3. Análise dos haplótipos da mutação da globina ßS
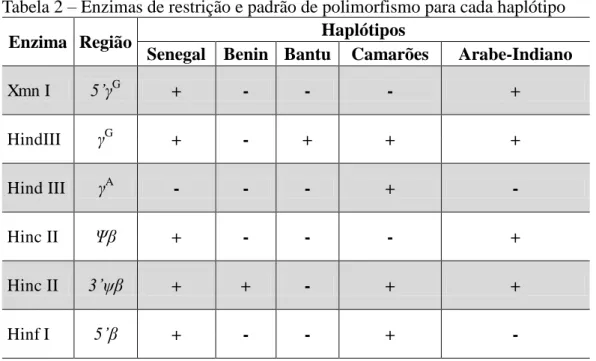
Foram analisados seis sítios polimórficos de restrição, seguindo a metodologia de Sutton, Bouhassi, Nagel (1989): 1. XmnI-5’ G, 2. Hind III- G, 3. Hind III- A, 4. Hinc II- ψ , 5. Hinc II-γ’ψ γ’, 6. Hinf I-5’ß. As informações sobre as enzimas de restrição utilizadas, as regiões de clivagem no DNA, o tamanho dos fragmentos após amplificação e após a clivagem com seus respectivos padrões de haplótipos estão representadas na tabela 1.
Tabela 1 – Enzimas de restrição utilizadas para a detecção de haplótipos do cluster do
gene S
, regiões de sítios polimórficos, tamanho dos fragmentos de DNA antes e após a clivagem
Oligonucleotídeos Enzima Região
Tamanho do fragmento Fragmento após clivagem H0: 5’AACTGTTGCTTTATAGGATTTTγ’
Xmn I 5’ G 650pb 450pb+200pb
H1: 5’AGGAGCTTATTGATAACCTCAG3'
Hβ: 5’AAGTGTGGAGTGTGCACATGAγ’
HindIII G 780pb 430pb+340pb+10pb
Hγ: 5’TGCTGCTAATGCTTCATTACAAγ’ Hγ: 5’TGCTGCTAATGCTTCATTACAAγ’
Hind III A 760pb 400pb+360pb
H4: 5’TAAATGAGGAGCATGCACACACγ’ H5: 5’GAACAGAAGTTGAGATAGAGAγ’
Hinc II ψ 701pb 360pb+340pb+1pb
H6: 5’ACTCAGTGGTCTTGTGGGCTγ’ H7: 5’TCTGCATTTGACTCTGTTAGCγ’
Hinc II γ’ψ 590pb 470pb+120pb
H8: 5’GGACCCTAACTGATATAACTAγ’ H9: 5’CTACGCTGACCTCATAAATGγ’
Hinf I 5’ 380pb 240pb+140pb
H10: 5’CTAATCTGCAAGAGTGTCTγ’
O PCR thermo-shaker utilizado nos protocolos foi da empresa VHD® (0,025 de Taq DNA Polimerase; 2 mM de MgCl2; 0,2 mM de cada dNTP) e as reações de amplificação
foram desenvolvidas no termociclador master cycler da Eppendorf®.
Os resultados da amplificação foram verificados pela corrida eletroforética em gel de agarose a 1,5%, sob corrente constante de 80 V por 15 minutos, e visualizados em câmara de ultravioleta (UV) após coloração com brometo de etídio.
Os resultados da digestão enzimática foram verificados pela corrida eletroforética em gel de agarose a 1,5%, sob corrente constante de 80 V por 30 minutos, e visualizados em câmara de UV após coloração com brometo de etídio.
Os resultados da digestão enzimática das amostras foram analisados de acordo com o padrão de polimorfismos para cada haplótipo que está representado na Tabela 2.
Tabela 2 – Enzimas de restrição e padrão de polimorfismo para cada haplótipo
Enzima Região Haplótipos
Senegal Benin Bantu Camarões Arabe-Indiano
Xmn I 5’ G + - - - +
HindIII G + - + + +
Hind III A - - - + -
Hinc II Ψ + - - - +
Hinc II γ’ψ + + - + +
Hinf I 5’ + - - + -
Fonte: Adaptado de Stuart e Nagel (2004).
3.5.2. Dosagem de VEGF-A
A dosagem de VEGF-A foi realizada em plasma de pacientes e indivíduos saudáveis pela técnica de Enzyme-Linked Immunosorbent Assay (ELISA) especifica para moléculas humanas de acordo com o protocolo do kit (Human VEGF -A platinum ELISA) (ANEXO C) fornecido pela empresa eBioscience®, seguido por leitura em densidade óptica utilizando filtro de 450nm.
A média das concentrações de VEGF-A nos indivíduos saudáveis participantes do estudo foi de (120,0 ± 79,11 pg/mL) (média ± desvio padrão).
3.6.Descarte de material biológico
O descarte do material biológico foi realizado segundo a resolução da diretoria colegiada 306, de 7 de dezembro de 2004 da Agência Nacional de Vigilância Sanitária.
3.7.Critérios éticos
Os protocolos desenvolvidos neste trabalho foram submetidos a apreciação ética e aprovados pelo Comitê de Ética em Pesquisa da Universidade Federal do Ceará sob parecer 706.154 na data de 02/06/2014 (ANEXO A). Neste termos a equipe executora comprometeu-se a cumprir todas as diretrizes e normas reguladoras descritas na Resolução 466 de 12/12/2012 do Conselho Nacional de Saúde.
3.8.Análise estatística
A análise estatística dos dados foi realizada mediante a utilização do programa estatístico GraphPad Prism versão 5.0. Utilizou-se o teste D’Agostino e Pearson para verificar a normalidade dos dados. Em seguida utilizou-se análise de variância ANOVA com pós-teste de Turkey para análises com 3 ou mais grupos. Para as análises com até dois grupos foi utilizado os testes estatísticos de Mann Whitney (quando variáveis não paramétricas) e teste T não pareado (para variáveis paramétricas).
4. RESULTADOS
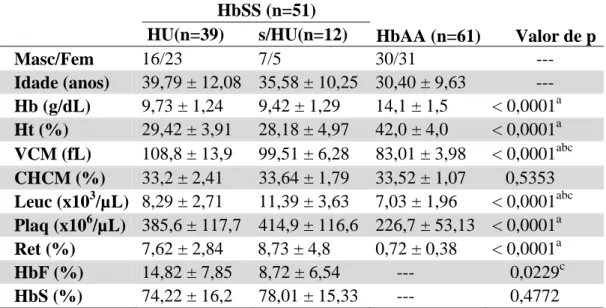
A população em estudo foi constituída por 51 pacientes com AF com idade variando de 23 a 69 anos, com média de 39,45 anos, sendo destes 39 em uso de HU e 12 sem tratamento com HU. Em relação ao sexo, no grupo em uso de HU 23 (58,97%) foram do sexo feminino e 16 (41,03%) do sexo masculino, no grupo que não fez uso de HU 5 (41,67%) foram do sexo feminino e 7 (58,33%) do sexo masculino. O grupo controle foi constituído por 61 indivíduos sem hemoglobinopatias com idade variando de 18 a 60 anos, com média de 30,4 anos. Em relação ao sexo 31 (50,81%) foram do sexo feminino e 30 (49,18%) do sexo masculino. As características demográficas e hematológicas estão apresentadas na tabela abaixo:
Tabela 3 – Características demográficas e dados hematológicos dos pacientes com AF, em uso ou não de HU, e grupo controle
HbSS (n=51)
HbAA (n=61) Valor de p HU(n=39) s/HU(n=12)
Masc/Fem 16/23 7/5 30/31 ---
Idade (anos) 39,79 ± 12,08 35,58 ± 10,25 30,40 ± 9,63 --- Hb (g/dL) 9,73 ± 1,24 9,42 ± 1,29 14,1 ± 1,5 < 0,0001a Ht (%) 29,42 ± 3,91 28,18 ± 4,97 42,0 ± 4,0 < 0,0001a VCM (fL) 108,8 ± 13,9 99,51 ± 6,28 83,01 ± 3,98 < 0,0001abc CHCM (%) 33,2 ± 2,41 33,64 ± 1,79 33,52 ± 1,07 0,5353 Leuc (x103/µL) 8,29 ± 2,71 11,39 ± 3,63 7,03 ± 1,96 < 0,0001abc Plaq (x106/µL) 385,6 ± 117,7 414,9 ± 116,6 226,7 ± 53,13 < 0,0001a Ret (%) 7,62 ± 2,84 8,73 ± 4,8 0,72 ± 0,38 < 0,0001a
HbF (%) 14,82 ± 7,85 8,72 ± 6,54 --- 0,0229c
HbS (%) 74,22 ± 16,2 78,01 ± 15,33 --- 0,4772
Nota: Os valores acima foram apresentados como média ± desvio padrão. Valores de p obtidos através do One way análise de variância (ANOVA) com pós-teste de Turkey e teste t não pareado com significância ao nível de p ≤ 0,05. (a): p ≤ 0,05 entre os grupos HbSS (HU) e HbAA; (b): p ≤ 0,05 entre os grupos HbSS (s/HU) e HbAA; (c): p ≤ 0,05 entre os grupos HbSS (HU) e (s/HU) . Fonte: Elaborado pelo autor.
grupo com AF em uso de HU e o grupo controle (HU: 7,62 ± 2,84% e HbAA: 0,72 ± 0,38%). Verificou-se diferença nas concentrações de HbF quando comparada entre os pacientes com AF em uso de HU (14,82 ± 7,85%) e sem uso de HU (8,72 ± 6,54%). Não foram encontradas diferenças nas concentrações de HbS (HU: 74,22 ± 16,2% e s/HU: 78,01 ± 15,33%) nos pacientes estudados.
Gráfico 1 – Concentrações de VEGF-A entre os pacientes com AF e grupo controle
HbAA HbSS
0 200 400
600
p= 0,0002*
a
V E GF -A p g /m L
Nota: Valores de p obtidos através do Teste T não pareado com significância ao nível de p ≤ 0,05. (a): p
≤ 0,05 em relação ao grupo HbAA, n=61; grupo HbSS, n=51. Diferença significativa entre os grupos AF HU e Controle. Fonte: Elaborado pelo autor.
Gráfico 2 – Concentrações de VEGF-A entre os pacientes com AF em uso de HU, sem uso de HU e grupo controle
HbAA HbSS (HU) HbSS (s/HU) 0
500 1000 1500
2000
p= <0,0001*
ab
a
V E GF -A ( p g /m L )Tabela 4 – Distribuição dos genótipos e haplótipos da globina da população estudada
Genótipos n (%)
Haplótipos (n)
Bantu Benin Atípico
Bantu/Bantu 20 (51,28%) 40 --- ---
Bantu/Benin 10 (25,64%) 10 10 ---
Benin/Benin 2 (5,13%) --- 4 ---
Bantu/Atípicos 4 (10,26%) 4 --- 4
Benin/Atípicos 1 (2,56%) --- 1 1
Atípicos/Atípicos 2 (5,13%) --- --- 4
Total 39 (100%) 54 (69,2%) 15 (19,3%) 9 (11,5%) Nota: Analisados pacientes em uso de HU. Fonte: Elaborada pelo autor.
Com relação aos genótipos da globina S
, foram encontrados 20 casos (51,28%) de Bantu/Bantu, 10 (25,64%) de Bantu/Benin, 2 (5,13%) de Benin/Benin, 4 (10,26%) de Bantu/Atípicos, 1 (2,56%) de Benin/Atípicos e 2 (5,13%) de Atípicos/Atípicos. Dos haplótipos analisados 69,2% foram do tipo Bantu, 19,3% do tipo Benin e 11,5% do tipo atípico (Tabela 4).
Não houve diferença estatisticamente significativa na comparação entre as médias das concentrações de VEGF-A nos grupos de haplótipos estudados (p > 0,05) (Tabela 5) (Gráfico 3).
Não foi encontrada associação entre as concentrações de VEGF-A e as manifestações clínicas da AF na população estudada (Tabela 6). Excetuando-se quando avaliadas as concentrações de VEGF-A em relação à quantidade de CVO por ano, onde foi observado que o grupo de pacientes que apresentaram mais de 3 crises por ano (416,4 ± 477,0 pg/mL, n=12) apresentaram concentrações de VEGF-A maiores que os pacientes com menos de 3 crises por ano (204,6 ± 138,3 pg/mL, n=29) . Observou-se que o uso de HU não teve influência nas concentrações de VEGF-A no grupo com CVO ≤ γ (HU: 198,78 ± 148,67pg/mL, n=22; s/HU: 221,84 ± 108,98 pg/mL, n=7), mas no grupo com maior quantidade de CVO (HU: 243,94 ± 172,99 pg/mL, n=17; s/HU: 693,788 ± 711,89 pg/mL, n=5) o uso de HU é responsável pelas concentrações elevadas de VEGF-A deste grupo (Tabela 7 e Gráfico 4).
Tabela 5 –Concentrações de VEGF-A dos pacientes com AF em uso de HU de acordo com os haplótipos do cluster da globina S
Bantu/Bantu (n=20)
Bantu/Benin (n=10)
Benin/Benin
(n=2) Valor de p VEGF-A
(pg/mL) 313,7 ± 251,3 164,4 ± 134,6
a
218,1 ± 34,93ab 0,2150
Nota: Analisados pacientes em uso de HU, n=32. Resultados expressos em valores de média ± desvio padrão. Valor de p obtidos através do One way análise de variância (ANOVA) com pós-teste de Turkey