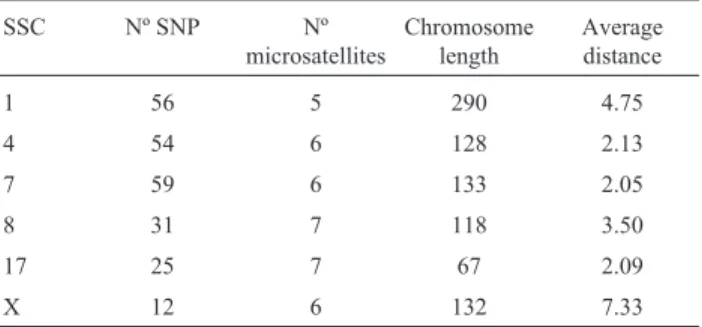
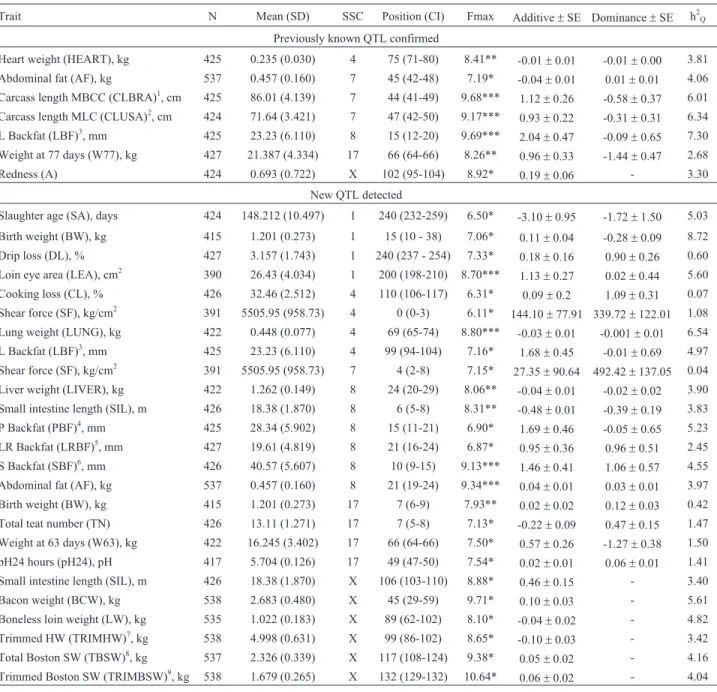
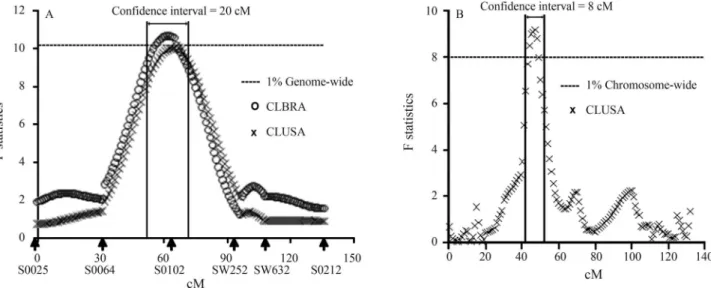
Fine mapping and single nucleotide polymorphism effects estimation on pig chromosomes 1, 4, 7, 8, 17 and X
Texto
Imagem
Documentos relacionados
É nesta mudança, abruptamente solicitada e muitas das vezes legislada, que nos vão impondo, neste contexto de sociedades sem emprego; a ordem para a flexibilização como
De acordo com a ANQIP, Associação Nacional para a Qualidade das Instalações Prediais, a certificação e rotulagem da eficiência hídrica dos produtos vai permitir reduzir o
Therefore, the objective of this study was to verify the effects of different cryoprotectants with and without 1% phloroglucinol and pre-cooling periods on
This study aimed to evaluate the effect of different managements with pig slurry (PS) and cattle slurry (CS) on wheat dry matter and grain yields, and the potential to estimate this
The objective of this study was to examine the effects of suckling schemes (continued, controlled, and total separation) and the type of pregnancy (single and twin) on
The purpose of this study is, therefore, to evaluate the influence of pig slurry applied to agricultural areas on soil chemical and physical properties and on the mobility of
The purpose of this study was to evaluate the effects of a dietary added formaldehyde-propionic acid blend on feed enterobacteria counts and on growing pig performance and
The aim of the present study was to evaluate the effects of different aeration frequencies on biomass temperature and gas emissions during composting of pig and poultry carcasses
