U
NIVERSIDADE DE
L
ISBOA
F
ACULDADE DE
C
IÊNCIAS
D
EPARTAMENTO DE
Q
UÍMICA E
B
IOQUÍMICA
Establishment of a suppression therapy for beta-
thalassemia due to a nonsense mutation
Patrícia Sofia Martins Dias
Mestrado em Bioquímica
Especialização em Bioquímica Médica
Dissertação orientada por:
Doutora Luísa Maria Ferreira Romão Loison
“If you want to be successful. It is just this simple. Know what you are doing. Love what you
are doing. And believe in what you are doing.” – Will Rogers
Acknowledgments
Após um ano de grandes sentimentos, aquele que por fim sobressai é a gratidão. Desejo a todos os que de uma forma direta ou indireta fizeram parte deste caminho que, apesar de difícil, acabou por ser o melhor que já percorri, um futuro com muito sucesso e alegria.
Em primeiro lugar queria agradecer à Faculdade de Ciências da Universidade de Lisboa por me ter acolhido em sua casa. A todos os professores que durante estes 5 anos estiveram sempre presentes e que da melhor forma conseguiram transmitir o seu conhecimento. Aos meus amigos de faculdade com quem desfrutei as melhores e as piores experiências, amizades que espero manter o resto da vida. Alguém um dia me disse “O tempo de Faculdade é o melhor, aproveita!”. A todos um muito obrigada! De todas as pessoas que conheci na Faculdade quero principalmente agradecer ao meu amigo e companheiro de aventuras Tiago. Quando te conheci soube que iríamos construir uma grande amizade. Agora que estes 5 anos acabaram e cada um vai seguir o seu caminho, espero que me guardes no teu coração como eu guardar-te-ei no meu. Para onde fores, eu irei visitar-te, lembra-te disso.
Obrigada ao Instituto Nacional de Saúde Doutor Ricardo Jorge por ter aceitado que durante este ano pertencesse à sua casa e à sua família. Quero agradecer ao Senhor Presidente Fernando de Almeida e a todas as identidades desta organização pelo apoio prestado. Ao Doutor João Lavinha e à Doutora Glória Isidro por me permitirem fazer parte do Departamento de Genética Humana.
Um especial agradecimento à Doutora Luísa Romão por me ter aceitado, acolhido e integrado no seu grupo e por todos os conhecimentos que me transmitiu. Sempre acreditou no meu trabalho e sempre me orientou da melhor forma. Muito obrigada. Estarei sempre grata.
Quero agradecer também aos meus colegas de laboratório, pois não são apenas colegas, mas grandes amigos. Andreia, Paulo, Joana, Rafael, Rafaela, Ana e Zé, vocês ensinaram-me tanto, que este parágrafo não é suficiente para agradecer tudo. Crescer cientificamente foi muito importante e tentaram sempre dar conselhos, sugestões e ver as bandas invisíveis que eu teimava em querer ver. Mas sobretudo ensinaram-me a ser melhor pessoa. Todos os Team Buildings que passaram e todos os convívios que fizemos irão deixar saudades. Ao Paulo, por estar presente e ajudar, foste essencial, todas as “paulisses” acabaram por tornar os dias cinzentos muito melhores. À Joana, por ser a minha companhia fit e me dar bons conselhos e mimos quando precisava. Ao Rafael, por ser o fofo que é, e alinhar nas cantorias e danças. À Rafaela, por conseguir ser tão chorona como eu, fazemos uma boa dupla. À Ana, por me deixar usar a sua bancada quando ocupo as duas, pelos duetos desafinados (da minha parte) e por após dias de trabalho até às tantas ainda me convencer a ir passear ou comer sushi. Ao Zé, por ser vizinho de bancada, por toda a sua boa disposição e alegria, e por ser o salvador dos problemas no Departamento. A todos, um até já!
Andreia, como sabes não poderia deixar de te agradecer. Ensinaste-me tudo e por isso sinto-me preparada para enfrentar as dificuldades que virão. Foste o meu grande apoio este ano, nas coisas boas e menos boas estavas lá, para aplaudir ou repreender (num bom sentido). Tudo aquilo que fizeste por mim não será esquecido. Desejo-te o melhor e tenho a certeza que te tornarás uma grande cientista. Como eu costumava dizer: “Tive muita sorte, não poderia ter tido melhor”.
Quero agradecer aos meus pais e à minha irmãzinha que sempre estiveram presentes. Ensinaram-me tudo o que sei hoje e permitiram que seguisse aquilo que sonhava sem nunca duvidar que era capaz. Espero que estejam orgulhosos de mim como eu estou orgulhosa de vocês. Mãe és a mulher mais forte que conheço, sempre lutaste para me dares tudo e muitas vezes não te disse o quanto grata estou da vida que me deste. Obrigada mamã. Pai, obrigada por te preocupares com o meu futuro e desejares o melhor para mim, és um exemplo a seguir. Fazes-me todos os dias acreditar que consigo mais e melhor. Obrigada papá. Maninha, és o meu orgulho, foste o melhor presente que recebi na minha
vida e espero nunca te desiludir. Muito obrigada pela ajuda a corrigir o Português, és a melhor. Amo-vos.
Por fim, queria agradecer a uma pessoa especial. És o meu companheiro, melhor amigo e confidente. Não mudava nada em ti ou em nós. Por tudo o que fizeste por mim, pelas palavras de apoio, abraços de conforto e sentimentos que nunca serão esquecidos, um muito obrigada a ti Rui.
Abstract and Key-words
β-thalassemia is a genetic disorder caused by the absence or reduction of human β-globin protein levels, which leads to a reduction on hemoglobin synthesis. This reduction can be caused by a premature termination codon (PTC) in the human β-globin gene. The messenger ribonucleic acid (mRNA) containing a PTC can be recognized and degraded by the nonsense-mediated decay mechanism (NMD), if the PTC is located 50-54 nucleotides upstream of the last exon-exon junction. If the PTC is located near to the initiation codon or downstream of the last exon-exon junction, then this mRNA is not degraded by NMD. In the first case a small peptide is degraded by proteolysis and in the second case a long truncated protein is synthesised. These proteins aggregate and precipitate, leading to a severe damage of erythroid precursors.
The conventional therapies for β-thalassemia temporarily restore the levels of human β-globin protein, however, these therapies have secondary effects that lead to the deterioration of the patient health. Given this, a therapy that could restore the levels of human β-globin protein by inducing the readthrough of the PTC in human β-globin mRNA, would be an advantage to these patients. Suppression therapy has been studied in detail for genetic diseases caused by PTCs. Salvatori et al. demonstrated the ability of G418 to induce the readthrough of a PTC at codon 39 of human β-globin mRNA, however, the ability of suppression by this aminoglycoside at other codons of human β-globin mRNA, and the use of other suppression compounds, have not yet been investigated. Suppression therapy consists in the readthrough of a nonsense codon into a sense codon, which allows the mRNA to be totally translated, resulting in a complete protein with partial or complete function. There are some compounds, including aminoglycosides and non-aminoglycosides, that bind to the decoding center of the ribosome and allow the near-cognate aminoacyl-tRNA to bind, resuming translation.
The main aims of this project were to understand if G418, an aminoglycoside, or PTC124, a non-aminoglycoside, produce efficient levels of PTC readthrough in human β-globin mRNA, and to understand how different PTCs on β-globin mRNA respond to the suppression therapy. Therefore, HeLa and HEK293 cells were transiently transfected with constructs containing the first 55 codons of human β-globin gene fused to firefly luciferase gene without any PTC (βWT-FLUC) or carrying a PTC at codon 15 (TGA) or 39 (TAG) (β15-FLUC or β39-FLUC). The results were obtained by Western blot and bioluminescence assays.
The results obtained by Western blot seem to indicate that G418 induce the readthrough of PTCs at codon 15 or 39 of the human β-globin mRNA in HEK293 cells treated with concentrations higher than 400 µg/mL. Additionally, a PTC at codon 15 of human β-globin mRNA seems to respond more efficiently to the readthrough than a PTC at codon 39. The results obtained after PTC124 treatment are not conclusive, however, PTC124 might be inducing the readthrough of a PTC at codon 15 of human β-globin mRNA in HEK293 cells treated with 5 µM, and at codon 39 of human β-globin mRNA in HEK293 cells treated with 5, 10 and 15 µM.
Since G418 revealed to be able to suppress a PTC at codon 15 or 39 of the human β-globin mRNA and restore the full-length human β-globin protein in constructs containing a PTC at codon 15 and 39 of human globin gene, it arises as a promising compound to be used in a future therapy for β-thalassemia. Before that, it is important to study the effect of NMD in suppression therapy and how its inhibition can enhance the suppressive effect.
Resumo e palavras-chave
A célula necessita de manter os níveis de proteína regulados, uma vez que estas possuem funções que determinam a sobrevivência e adaptação da célula em resposta a diferentes estímulos. De forma a regular estes níveis a célula altera os padrões de expressão génica, controlando vários processos celulares, nomeadamente, a transcrição e a tradução. Uma desregulação nestes processos pode levar a inúmeras doenças, como as doenças genéticas.
A β-talassemia é um exemplo de uma doença genética, caraterizada pela redução ou ausência de β-globina, a qual leva consequentemente a anemia. A redução dos níveis de proteína pode ser causada pela presença de codões prematuros da tradução (PTCs) no seu mRNA. O mRNA que contenha estes PTCs pode ser envidado para o mecanismo de degradação mediado por mutações nonsense (NMD) caso cumpra os requisitos necessários. O NMD é um mecanismo de controlo de qualidade da célula, uma vez que tem um papel protetor contra erros genéticos. Durante o splicing são adicionados complexos exão-exão (EJCs) a 20-24 nucleótidos a montante das junções exão-exão-exão-exão. Durante a ronda pioneira da tradução todos os EJCs adicionados são removidos. No entanto, na presença de um PTC localizado a 50-54 nucleótidos a montante da junção exão-exão, o EJC não é removido e interage com outros complexos proteicos de forma a recrutar endonucleases e exonucleases, os quais levam à degradação do mRNA. Os mRNAs que contenham PTCs próximos do codão de iniciação são uma exceção a esta regra, assim como mRNAs que contenham PTCs a jusante da ultima junção exão-exão. Quando o mRNA que contenha o PTC não é enviado para NMD pode ocorrer a produção de pequenos péptidos, que são degradados por proteólise, ou de longas proteínas truncadas, que agregam e precipitam, levando à destruição dos percursores eritroides e ao agravamento do fenótipo do doente.Sendo assim, tanto o tipo de mutação com a sua posição no gene influenciam o fenótipo e a severidade da β-talassemia. Esta doença apresenta três fenótipos, a β-talassemia suave, a intermédia e a grave. Os pacientes que apresentam β-talassemia suave são assintomáticos, no entanto aqueles cujo fenótipo é mais grave, apresentam anemias severas com necessidade de transfusões sanguíneas recorrentes. A β-talassemia intermédia apresenta sintomas com grau de severidade entre a β-talassemia suave e β-talassemia grave. Apesar de não existir uma cura para a β-talassemia existem algumas terapias convencionais, como por exemplo, as transfusões sanguíneas que permitem restaurar temporariamente os níveis de β-globina no sangue. No entanto, estas terapias possuem efeitos secundários, nomeadamente a deposição de elevados níveis de ferro em diferentes órgãos e tecidos que prejudicam a saúde do doente. Desta forma, é importante estudar uma terapia que permita restaurar os níveis da proteína β-globina, assim como a sua função. Existem diversos artigos que relatam uma terapia de supressão onde são restaurados os níveis e a função de proteína em doenças causadas pela presença de um PTC, como na fibrose cística ou na distrofia muscular Duchenne. Esta terapia consiste na releitura de um codão que levaria à terminação prematura da tradução. Este processo permite que o mRNA seja totalmente traduzido, resultando na síntese de uma proteína completa e com pelo menos alguma da sua função restaurada. De forma a ser possível esta releitura, alguns compostos aminoglicósidos e não-aminoglicósidos permitem a ligação de um aminoacil-tRNA com duas bases compatíveis com o codão stop prematuro, anulando a ligação dos fatores de libertação da tradução ao PTC. Assim sendo, ocorre a incorporação de um novo aminoácido e a tradução continua.
A terapia de supressão tem sido estudada em diversas doenças genéticas causadas pela presença de PTCs em genes, que levam à terminação prematura da tradução. Salvatori et al. demonstrou a capacidade do composto aminoglicósido G418 de induzir a releitura de um PTC no codão 39 no mRNA da β-globina humana. No entanto, a capacidade de supressão deste composto, assim como de outros
agentes supressores, ainda não foi estudada na releitura de PTCs em outros locais do mRNA da β-globina humana.
Assim, o objetivo principal deste projeto foi estudar que compostos aminoglicósidos e não-aminoglicósidos permitiam obter níveis eficientes de supressão de codões prematuros da tradução no mRNA da β-globina humana. Um segundo objetivo foi compreender como diferentes PTCs em diferentes posições do mRNA respondem a esta terapia de supressão. De forma a ser possível observar a proteína β-globina humana por Western blot, delineou-se uma estratégia que consistiu em clonar a open reading frame (ORF) do enhanced green fluorescent protein (EGFP), sem os codões de iniciação e terminação, no exão 3 do gene da β-globina. Apesar de inúmeras tentativas, não foi possível obter nenhum clone positivo. Dado que a estratégia anterior se mostrou difícil de alcançar, foi delineada uma segunda estratégia. Utilizaram-se construtos que continham 55 codões da β-globina humana em fusão com o gene da firefly luciferase. Estes construtos apesar de não sensíveis ao NMD por não conterem junções exão-exão, permitiram no entanto, compreender as características da terapia de supressão na releitura dos PTCs no mRNA da β-globina humana. De forma a alcançar os objetivos propostos, as células HeLa ou HEK293 foram transfectadas com os constructos referidos, sendo que o gene da β-globina não apresentava mutações (βWT-FLUC) ou possuía uma mutação nonsense no codão 15 (TGA) (β15-FLUC) ou 39 (TAG) (β39-FLUC). As células foram tratadas com G418 (aminoglicósido) ou PTC124 (não-aminoglicósido) e analisou-se a ocorrência de supressão por ensaios de bioluminescência e por Western blot.
Após a análise dos resultados obtidos para as células HeLa transfectadas com constructos contendo um PTC no codão 39 no gene da β-globina humana, observou-se que o G418 levou ao aumento da atividade relativa da firefly luciferase na maioria das concentrações testadas. Os resultados obtidos para as células HEK293 aparentam indicar a presença de supressão de um PTC localizado no codão 15 ou 39 no mRNA da β-globina humana, quando estas células são tratadas com G418. No Western blot observou-se a presença de uma banda com tamanho similar à βWT-FLUC quando as células HEK293 transfetadas com construtos contendo um PTC localizado no codão 39 são tratadas com 500, 750, 1000 e 1250 µg/mL. Relativamente às células HEK293 transfetadas com constructos contendo uma mutação no codão 15 pudemos observar a presença de uma banda com tamanho similar ao βWT-FLUC para as células tratadas com G418 a 400, 500, 750, 1000 e 1250 µg/mL. Adicionalmente, foi observado um aumento da atividade relativa da firefly luciferase nestas condições. Este composto não parece influenciar a expressão do constructo βWT-FLUC em nenhuma concentração testada, em ambas as linhas celulares. Relativamente à eficiência de supressão, o composto G418 parece ser mais eficiente na releitura do PTC no codão 15 no mRNA da β-globina humana, relativamente à releitura do PTC no codão 39. Os resultados apresentados para o não-aminoglicósido PTC124 não são claros devido ao facto de as bandas presentes nos Western blots serem difusas. No entanto, é possível que o PTC124 induza a releitura do PTC no codão 15 do mRNA da β-globina humana, quando as células HEK293 são tratadas com PTC124 a 5 µM. Pareceu ser possível a releitura do PTC no codão 39 do mRNA da β-globina humana, quando as células HEK293 são tratadas com PTC124 a 5, 10 ou 15 µM.
A eficiência da terapia de supressão pode ser influenciada pelo tripleto que compõe o codão prematuro da tradução ou pela sequência flanqueadora deste codão. Os resultados obtidos permitiram compreender que ambas as sequências flanqueadoras da mutação no codão 15 e no codão 39 permitem que ocorra a releitura do PTC, embora haja uma maior eficiência na releitura do PTC no codão 15 do mRNA da β-globina humana utilizando o composto G418. Apesar de estes resultados positivos indicarem um avanço para estabelecer uma terapia de supressão para a β-talassemia, existem várias vertentes desta terapia que ainda necessitam de ser exploradas. Dado que o NMD pode limitar os níveis de mRNA disponíveis para a releitura do codão, é importante testar a sua inibição (parcial) neste tipo
de terapia, utilizando compostos que alteram a eficiência do NMD. Adicionalmente, é importante estudar a função da proteína sintetizada, dado que, após a reinserção de um novo aminácido, é possível que a sua função não seja completa.
Palavras-chave:
β-globina; G418; mecanismo de degradação mediado por mutações nonsense; PTC124; codão prematuros da tradução
Index
Acknowledgments III
Abstract and Key-words V
Resumo e palavras-chave VI
Figure Index XI
Table Index XIII
Symbol and Abbreviation Index XIV
Chapter 1 – Introduction 1
1.1. mRNA synthesis and associated components 1
1.2. Translation 1
1.2.1. The pioneer round 1
1.2.2. Translation stages 2
1.3. Nonsense codons 5
1.4. The nonsense-mediated mRNA decay 5
1.4.1. NMD functions and mechanism 5
1.4.2. Exceptions to the rule 7
1.5. β-Thalassemia 7
1.5.1. The human β-globin gene 7
1.5.2. The genetic disease 8
1.5.3. Clinical genotype/phenotype 8
1.5.4. NMD and β-thalassemia 9
1.6. The conventional β-thalassemia therapy 10
1.7. Suppression therapy 11
1.7.1. Mechanism of PTC suppression 11
1.7.2. Compounds with suppressive properties 12
Aminoglycosides 12
Non-aminoglycosides 14
1.7.3. Dificulties in establishing a suppression therapy 16
Chapter 2 - Aims 17
Chapter 3 - Materials and methods 18
3.1. Cloning of EGFP gene into exon 3 of human β-globin gene using the pTRE-2pur plasmid 18
3.1.1. pTRE-2pur constructs 18
3.1.2. Molecular strategy 18
3.1.3. Screening by digestion and colony PCR 20
3.2. Cloning of firefly luciferase ORF into exon 3 of β-globin gene, using the pGL2TRE plasmid 20 3.2.1. pGL2TRE plasmid containg firefly luciferase gene and pGL4TRE plasmid containing
Renilla luciferase gene 20
3.2.2. Cell culture and transfection 22
3.2.3. Drug treatment and cell lysis 22
3.2.4. SDS-PAGE and Western blot 22
3.2.6. Statistical analysis 23
Chapter 4 - Results 24
4.1. Cloning EGFP ORF into exon 3 of the human β-globin gene 24
4.2. pGL2TRE constructs containing the firefly luciferase gene fused to a part of the human
β-globin gene 25
4.2.1. Confirmation of pGL2TRE constructs containing the firefly luciferase gene fused to the
human β-globin gene by DNA sequencing 25
4.2.2. Firefly luciferase gene fused to the human β-globin gene accumulates to significant levels at 12, 24, 48 and 72 hours post-transfection, in HeLa cells. 27 4.2.3. G418 at 300µg/mL and PTC124 at 150µM do not induce readthrough of a PTC in the β39-FLUC mRNA and do not increase relative firefly luciferase activity, in HeLa cells 29 4.2.4. Testing the effect of different concentrations of G418 on the efficiency of readthrough
of PTCs in human β-globin mRNA expressed in HeLa cells 30
4.2.5. Testing the effect of increasing concentrations of G418 on the efficiency of readthrough
of PTCs in the human β-globin mRNA expressed in HEK293 cells 33
4.2.6. Testing the effect of increasing concentrations of PTC124 on the efficiency of readthrough of PTCs in the human β-globin mRNA expressed in HeLa cells 37 4.2.7. Testing the effect of increasing concentrations of PTC124 on the efficiency of readthrough of PTCs in the human β-globin mRNA expressed in HEK293 cells 39
Chapter 5 - Discussion and future directions 43
Chapter 6 - References 47
Chapter 7 - Annexes 51
7.1. Primers 51
7.2. Sequence of the human β-globin gene with EGFP tag inserted into exon 3, cloned in pTRE-2pur
plasmid 51
7.3. EGFP ORF amplification 52
7.4. DNA digestion 53
7.5. DNA ligation reaction 54
7.6. Colony PCR 54
7.7. Sequence of the human β-globin gene fused with firefly luciferase gene cloned in pGL2TRE 56
7.8. DNA sequencing 57
Figure Index
FIGURE 1.1 – PIONEER ROUND OF TRANSLATION (A) AND THE STEADY STATE TRANSLATION (B)
COMPLEXES ... 2
FIGURE 1.2–REPRESENTATION OF THE TRANSLATION INITIATION PROCESS. ... 3
FIGURE 1.3-REPRESENTATION OF TRANSLATION ELONGATION. ... 4
FIGURE 1.4-REPRESENTATION OF THE NMD MECHANISM ... 6
FIGURE 1.5- REPRESENTATION OF A TRANSCRIPT WITH A PTC CLOSE TO THE AUG. ... 7
FIGURE 1.6- REPRESENTATION OF THE β-GLOBIN GENES CLUSTER. ... 8
FIGURE 1.7-THE β-THALASSEMIA PHENOTYPE IS INFLUENCED BY THE NMD MECHANISM. ... 10
FIGURE 1.8 - TRANSLATION TERMINATION AT NORMAL STOP CODONS AND AT PREMATURE TERMINATION CODONS. ... 12
FIGURE 1.9-GENTAMICIN AND ITS CONGENERS PRODUCED DURING ITS SYNTHESIS. ... 13
FIGURE 1.10 - CONVENTIONAL AMINOGLYCOSIDES AND NOVEL SYNTHETIC AMINOGLYCOSIDES DEVELOPED FOR PTC SUPPRESSION IN EUKARYOTIC CELLS. ... 14
FIGURE 1.11-NON-AMINOGLYCOSIDES COMPOUNDS THAT HAVE PTC SUPPRESSION PROPERTIES. .... 15
FIGURE 3.1-CLONING STRATEGY OF EGFP INTO THE HUMAN Β-GLOBIN EXON 3 PREVIOUSLY CLONED INTO THE PTRE-2PUR PLASMID. ... 18
FIGURE 3.2-FUSION GENE BETWEEN THE β-GLOBIN AND EGFP GENES CLONED INTO PTRE-2PUR. .... 19
FIGURE 3.3–SCHEMATIC REPRESENTATION OF THE PGL2TRE CONTAINING THE Β-GLOBIN GENE. ... 21
FIGURE 3.4- SCHEMATIC REPRESENTATION OF THE FUSION GENES BETWEEN Β-GLOBIN GENE (ΒWT, Β15 OR Β39) AND FIREFLY LUCIFERASE ORF(FLUC). ... 21
FIGURE 4.1-PLATES OBTAINED DURING THE PROCESS OF CLONING EGFPORF INTO EXON 3 OF HUMAN β-GLOBIN GENE, IN PTRE-2PUR PLASMID. ... 24
FIGURE 4.2– SCREENING OF TRANSFORMED COLONIES WITH THE PLASMID CONTAINING THE FUSION GENE BETWEEN OF EGFP AND THE HUMAN β-GLOBIN GENE. ... 25
FIGURE 4.3-DNA SEQUENCING OF β15-FLUC(A) AND β39-FLUC(B). ... 26
FIGURE 4.4-DNA SEQUENCING OF βWT-FLUC IN CODON 15(A) AND CODON 39(B). ... 27
FIGURE 4.5–FLUC,βWT-FLUC, AND β39-FLUC PROTEIN ACCUMMULATES TO SIGNIFICANT LEVELS AT 12 HOURS POST-TRANSFECTION AND ACHIEVES THE HIGHER CONCENTRATION AT AROUND 48 HOURS POST-TRANSFECTION, IN HELA CELLS. ... 29
FIGURE 4.6-G418 AT 300µG/ML AND PTC124 AT 150µM DO NOT INDUCE READTHROUGH OF A PTC AT CODON 39 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HELA CELLS. ... 30
FIGURE 4.7- G418 DOES NOT SEEM TO INDUCE THE READTHROUGH OF A PTC AT CODON 39 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HELA CELLS, AT ANY TESTED CONCENTRATION. ... 31
FIGURE 4.8-G418 DOES NOT SEEM TO INDUCE THE READTHROUGH OF A PTC AT CODON 15 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HELA CELLS, AT ANY TESTED CONCENTRATION. ... 32
FIGURE 4.9-G418 DOES NOT SEEM TO SIGNIFICANTLY CHANGE THE LEVELS OF FIREFLY LUCIFERASE ACTIVITY OF βWT-FLUC PROTEIN EXPRESSED IN HELA CELLS, AT ANY OF THE CONCENTRATIONS TESTED. ... 33
FIGURE 4.10-G418 SEEMS TO INDUCE THE READTHROUGH OF A PTC AT CODON 39 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HEK293 CELLS, AT 500,750,1000 AND 1250µG/ML. ... 34
FIGURE 4.11-G418 SEEMS TO INDUCE THE READTHROUGH OF A PTC AT CODON 15 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HEK293 CELLS, AT 750,1000 AND 1250µG/ML. ... 35
FIGURE 4.12 - G418 DOES NOT SEEM TO INDUCE CHANGES IN βWT-FLUC PROTEIN EXPRESSED IN HEK293 CELLS TREATED WITH G418 AT THE TESTED CONCENTRATIONS. ... 36
FIGURE 4.13–PTC124 DOES NOT SEEM TO INDUCE THE READTHROUGH OF A PTC AT CODON 39 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HELA CELLS, AT ANY TESTED CONCENTRATION. ... 38 FIGURE 4.14- PTC124 DOES NOT SEEM TO INDUCE THE READTHROUGH OF A PTC AT CODON 15 OF HUMAN β-GLOBIN MRNA EXPRESSED IN HELA CELLS, AT ANY TESTED CONCENTRATION. ... 39 FIGURE 4.15-PTC124 SEEMS TO INDUCE THE READTHROUGH OF A PTC AT CODON 39 OF HUMAN
β-GLOBIN MRNA EXPRESSED IN HEK293 CELLS, AT 5,10 AND 15µM. ... 40 FIGURE 4.16-PTC124 SEEMS TO INDUCE THE READTHROUGH OF A PTC AT CODON 15 OF HUMAN
β-GLOBIN MRNA EXPRESSED IN HEK293 CELLS, AT 10µM. ... 42 FIGURE 7.1-SEQUENCE OF HUMAN Β-GLOBIN GENE WITH EGFP TAG INSERTED INTO EXON 3, CLONED IN PTRE-2PUR. ... 51
FIGURE 7.2-AMPLIFICATION OF EGFP FRAGMENT CONTAINING BSTXI RESTRICTION SITES. ... 53
FIGURE 7.3-DIGESTION OF PTRE-2PUR. ... 54
FIGURE 7.4-SEQUENCE OF HUMAN Β-GLOBIN GENE (1 TO 55 CODONS) FUSED TO FIREFLY LUCIFERASE
Table Index
TABLE 1.1- AMINO ACIDS INCORPORATED IN THE NASCENTE PEPTID DEPENDING ON THE TRIPLET OF
THE PTC49. ... 11
TABLE 7.1-PRIMERS USED IN THIS PROJECT. ... 51
TABLE 7.2 - MELTING AND ANNEALING TEMPERATURE OF THE PRIMERS USED IN EGFP FRAGMENT AMPLIFICATION. ... 52
TABLE 7.3-PCR REACTION CONDITIONS USED FOR EGFP FRAGMENT AMPLIFICATION. ... 52
TABLE 7.4-PCR REACTION CONDITIONS USED FOR EGFP FRAGMENT AMPLIFICATION. ... 52
TABLE 7.5-PCR CYCLING PARAMETERS USED FOR EGFP FRAGMENT AMPLIFICATION. ... 52
TABLE 7.6 - OPTIMIZED CONDITIONS USED IN THE DIGESTION OF PTRE-2PUR βWT AND EGFP FRAGMENT. ... 53
TABLE 7.7- PTRE-2PUR VECTOR AND EGFP INSERT AMOUNT TO OBTAIN THE MOLAR RATIOS OF 1:3,1:5, 1:6, 1:8 AND 1:10, USING THE LIGATION CALCULATOR FROM IN SILICO (HTTP://WWW.INSILICO.UNI-DUESSELDORF.DE/). ... 54
TABLE 7.8-PRIMERS SEQUENCE, MELTING AND ANNEALING TEMPERATURE. ... 55
TABLE 7.9-COLONY PCR CONDITIONS ... 55
TABLE 7.10–COLONY PCR REACTION CONDITIONS ... 55
TABLE 7.11-PCR CYCLING PARAMETERS USED FOR COLONY PCR. ... 55
TABLE 7.12–DNA SEQUENCING REACTION. ... 57
TABLE 7.13–PCR CYCLING PARAMETERS USED FOR DNA SEQUENCING REACTION. ... 57
TABLE 7.14-ACRILAMIDE GEL COMPOSITION. ... 58
Symbol and Abbreviation Index
α alpha
β beta
β0 Absent production of β-globin protein
β+ Reduced production of β-globin protein
β5 Nonsense mutation in codon 5 of β-globin
β15 Nonsense mutation in codon 15 of β-globin
β17 Nonsense mutation in codon 17 of β-globin
β39 Nonsense mutation in codon 39 of β-globin
β127 Nonsense mutation in codon 127 of β-globin
βWT Wild type β-globin
40S Ribosomal subunit 40S 60 S Ribosomal subunit 60S 80S complex Ribosome A Adenine AG Aminoglycoside AP Alkaline phosphatase
APC Adenomatous polyposis coli
A-site Aminoacyl site
ATPase Adenylpyrophosphatase
AUG Initiation codon
BSA Bovine serum albumin
C Cytosine
CBC Cap-binding complex
CBP20 Cap-binding protein with 20 kilo Daltons
CBP80 Cap-binding protein with 80 kilo Daltons
cDNA Complementar DNA
CF Cistic fibrosis
CFTR Cistic fibrosis transmembrane conductance regulater
CO Carbon moxide
CO2 Carbon dioxide
DCP1 Decapping protein 1
DCP2 Decapping protein 2
DECID Decay-inducing complex
DMD Duchenne muscular dystrophy
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
dNTP Deoxynucleotide
ECL Enhanced chemiluminescence
eEF1 Eukaryotic elongation factor 1
eEF1A Eukaryotic elongation factor 1A
eEF2 Eukaryotic elongation factor 2
eIF1 Eukaryotic initiator factor 1
eIF1A Eukaryotic initiator factor 1A
eIF3 Eukaryotic initiator factor 3
eIF4A Eukaryotic initiator factor 4A
eIF4E Eukaryotic initiator factor 4E
eIF4F Eukaryotic initiator factors 4F
eIF4G Eukaryotic initiator factor 4G
eIF5B Eukaryotic initiator factor 5B
eRF1 Eukaryotic release factor 1
EJC Exon junction complex
ER Endoplasmic reticulum
E-site Exit site
FBS Fetal bovine serum
FLUC Firefly Luciferase
G Guanine
G418 Aminoglycoside
EGFP Enhance green fluorescent protein
GTP Guanine triphosphate
HBA Hemoglobin A
hCMV Human cytomegalovirus
HEK293 Human embryonic kidney cells 293
HeLa Henrietta Lacks
kDa Kilo Daltons
L Lysine
LB Luria-Bertani
m7G 7-methylguanosine
miRNAs Micro RNAs
MPS I-H Mucopolysaccharidosis type I
mRNA Messenger ribonucleic acid
mRNP Messenger ribonucleoprotein particle
NB30 Synthetic aminoglycoside
NB54 Synthetic aminoglycoside
NB58 Synthetic aminoglycoside
NO2 Nitrogen dioxide
NMD Nonsense mediated decay
NMDI-1 NMD inhibitor 1
nt Nucleotide
O2 Oxygen
ORF Open reading frame
P-site Peptidyl site
PABPC Cytoplasmic poly-A binding protein
PABPC1 Cytoplasmic poly-A binding protein 1
PABPN1 Nuclear poly-A binding protein 1
PAP Poly(A) polymerase
Pi Inorganic phosphate
PIC Pre initiation complex
PCR Polymerase chain reaction
PLB Passive Lysis Buffer
PTC Premature termination codon
PTC124 Ataluren - Non-aminoglycoside
RNA Ribonucleic acid
ROS Reactive oxygen species
RT13 Non-aminoglycoside
RT14 Non-aminoglycoside
SDS Sodium dodecyl sulfate
SMG Serine/Threonine protein kinase
SURF SMG1/UPF1/Release factors complex
T Tymine
t1/2 half-life time
TBS-T Tris-buffered saline Triton
TC Ternary complex
TC Termination codon
tRNA Transfer ribonucleic acid
XRN1 5’ to 3’ exoribonuclease 1
U Uracil
UAG, UAA or UGA Termination codon
UPF1 Up-frameshift factor 1
UPF2 Up-frameshift factor 2
UPF3 Up-frameshift factor 3
Chapter 1 – I
ntroduction
1.1. mRNA synthesis and associated components
Deoxyribonucleic acid (DNA) encodes the genetic information that leads to protein production. The type and level of specific proteins are extremely important since they define cell function. The cell changes the type and the abundance of proteins by altering, for example, the genetic expression patterns, depending on which stimulus it receives. These changes allow the cell to adapt and survive to several environmental conditions1,2.
DNA is transcribed into pre-mRNA (pre-messenger ribonucleic acid) by the enzyme RNA polymerase II. At the 5’ end, a 7-methylguanosine cap structure (m7GpppN) is integrated3. The cap is
bound to CBC (cap-binding complex) that has two polypeptides, one with 20 kilodaltons (kDa) (CBP20) and one with 80 kDa (CBP80)4,5. In 3’ end of the pre-mRNA, the poly(A) polymerase (PAP) adds a stretch of adenosines, also known as the poly-A tail6. Both the cap structure and poly-A tail protect the mRNA from degradation and are important regulators of translation efficiency7. This pre-mRNA is
single-stranded with introns and exons. By definition, introns are non-coding regions and exons are coding regions. Introns of pre-mRNA are removed by splicing and flanked exons are bound. Exon junction complexes (EJC) are placed 20 nucleotides (nt) upstream of the exon-exon junction and its location is independent of the sequence context1,8,9. These complexes have different roles in the nonsense-mediated mRNA decay (NMD), as it will be discussed, but also in mRNA splicing, transport, and translation10. The messenger ribonucleic acid (mRNA) associates with complementary non-coding
mRNAs, such as micro-RNAs (miRNAs), and proteins. This complex is called messenger ribonucleoprotein particle (mRNP) and its contents determine the localization and fate of the mRNA in the cytoplasm1,11,12.
1.2. Translation
1.2.1. The pioneer roundIn mammalian cells, the newly synthesized transcripts are processed, exported from the nucleus to the cytoplasm, and translated. During the pioneer round of translation (first round) it is ensured the quality control of the transcript 13. mRNAs bound to CBC are associated with at least one EJC, if they
experienced splicing, and with poly-A-binding proteins (PABP), nuclear PABPN1 and cytoplasmic PABPC1. In the pioneer round of translation, CBC can interact directly with the eukaryotic initiator factor 4G (eIF4G)14. It has been proposed that eIF4G can interact with PABPC1, allowing the 5’ end of the transcript to interact with its 3’ end, and circularizing the mRNA (closed-loop complex)15. During the pioneer round, translational ribosomes remove the EJCs and associated factors, and the nuclear PABPN1 bound to the poly-A is replaced by the cytoplasmic PABPC114. After this round, CBC can be
replaced by the eukaryotic initiator factor 4E (eIF4E), which directs steady-state rounds of mRNA translation (Figure 1.1). eIF4E is a subunit of a complex named eIF4F. This complex is formed by the eukaryotic initiator factor 4A (eIF4A), that is an ATP-dependent RNA helicase, and the eIF4G that is a scaffold protein1,16,17.
The circularization of the mRNA is not essential for the translation process, however, since it brings the ends together, it enhances the recycling of translation components back to the 5’ end16,18.
Figure 1.1 – Pioneer round of translation (A) and the steady state translation (B) complexes
In the pioneer round of translation, CBC attached to eIF4G or CTIF (that is a component of the CBP8025) binds to the 5’ end
of the transcript. The 3’ end has the poly-A tail bound to nuclear proteins, PABPN1 (N1). The ribosome translate the open reading frame (ORF) and displaces the EJC (bound to up-frameshift factor 3 (UPF3) and UPF2) from the mRNA until it finds a termination codon (TC). The nuclear proteins PABPN1 are replaced by the cytoplasmic proteins, PABPC1 (C1). At the end of the pioneer round of translation, CBC is replaced by eIF4E. The 5’ end interacts with the 3’ end because eIF4G is a scaffold protein that brings together the CBC or the eIF4E and the PABPC1, allowing the mRNA to circularize.1,15–17,19
Image adapted fromRufener 2013 19
1.2.2. Translation stages
Translation has four stages: initiation, elongation, termination and ribosome recycling16.
Translation initiation
Translation initiation starts with the formation of the ternary complex (TC). The TC is a trimeric complex comprised by eukaryotic initiator factor 2 (eIF2), the initiator methionyl transfer RNA (tRNAiMet) and a guanine triphosphate (GTP)16,20. Then, occurs the assembly of the 40S, with its
associated initiator factors (i.e. eIF3, eIF1, eIF1A), with the ternary complex (TC). With this association, the 43S pre-initiation complex (43S PIC) is formed21. The 43S PIC recruitment to mRNA is achieved
by the interaction with translation initiation factors, as the eIF3 and eIF4F 16,22,23. These interactions
result in the formation of the 48S complex which scans the 5’ UTR of the mRNA (5’-to-3’). After the recognition of the AUG by the tRNAiMet, the 60S ribosomal subunit is recruited, GTP is hydrolysed and
the initiator factors are dissociated. Finally, this leads to a complete 80S ribosome where the tRNAiMet
accommodates in the P (peptidyl) site of the ribosome17,21,24. (Figure 1.2)
Figure 1.2 – Representation of the translation initiation process.
The steps of translation initiation include: 1) formation of the trimeric complex (TC) (Met-tRNAi, GTP and eIF2); 2) association
of the TC with the ribosomal subunit 40S and initiator factors, forming the 43S pre-initiation complex (43S PIC); 3a) mRNA bound to eIF4G and PABP form an activated messenger ribonucleoprotein (mRNP); 3b) activated mRNP attachment to the 43S PIC; 4) PIC scans along the 5’ UTR to find the initiation codon (AUG). 5) AUG is recognized, eIF1 is released, and GTP-eIF2 is converted into GDP-GTP-eIF2. 6) eIF5B-GTP and the subunit 60S of the ribossome joins to the complex, eIF5B-GTP binds to the Met-tRNAi and eIF2 and eIF5 are removed; 7) GTP is hydrolysed by eIF5B, eIF1A and eIF5B dissociate, the 80S
initiation complex is formed, and Met-tRNAi accommodates in the P site of the ribosome21,25.
Translation elongation
Translation elongation comprises three steps: decoding of an mRNA by the cognate aminoacyl-tRNA, formation of the peptide bond and translocation of the peptidyl-tRNA from the A (aminoacyl) site to P site of the ribosome. When this last step occurs, a new codon becomes accessible to the A site21. The elongation phase begins with the formation of the 80S on the mRNA. The anticodon of the Met-tRNAi is in the P site and the second codon of the open reading frame (ORF) is in the A site expecting
the cognate aminoacyl-tRNA to bind21,26. The eukaryotic elongation factor 1 (eEF1) binds
aminoacyl-tRNA and guides it to the A site of the ribosome, in a GTP-dependent manner. When the codon is recognized by its tRNA, the eukaryotic elongation factor 1A (eEF1A) is responsible for GTP hydrolysis. Then, the aminoacyl-tRNA can be accommodated at the A site26. Finally, the peptide bond is formed between the aminoacyl-tRNA and the peptidyl-tRNA21,27. The ribosomal subunits rotate to each other until the eukaryotic elongation factor 2 (eEF2) bound to GTP stabilizes them. The hydrolyses of the GTP allows the translocation of the 3’ end of the tRNA from P and A sites to E (exit) and P sites, respectively21,28 (Figure 1.3). When a termination codon enters to the decoding site, the elongation stops and the translation termination begins21.
Figure 1.3 - Representation of translation elongation.
In the first step, the eEF1-GTP binds aminoacyl-tRNA and guides it to the A site of the ribosome.When the anticodon of this tRNA base-pairs to its specific mRNA codon, it is possible to form a new peptide bond between the aminoacyl-tRNA and the peptidyl-tRNA. Then, the subunits rotate (represented by the blue colors), the eEF2-GTP binds and stabilizes them. Then, the hydrolyses of the GTP allows the translocation of the 3’ end of the tRNAs from P and A sites to E and P sites, respectively. Finally, the release of the tRNA in the E site initiates a new cycle28.
Image adapted fromLareau 2014 28
Translation termination and the ribosome recycling
The translation termination begins when the termination codon in the mRNA (UAG, UAA or UGA) enters to the A site of the ribosome and is recognized. When this occurs, a near-cognate aminoacyl-tRNA (two of three anticodons complementary to a stop codon) competes with eukaryotic release factors 1 and 3 complex (eRF1/3) for A-site binding. Normally, the eRF1/3 binding overcomes the aminoacyl-tRNA and an appropriate termination translation is achieved29. Thus, the eRF1 and eRF3 bind to the ribosome as a complex, associated with GTP, and induces conformational changes21. The eRF1 has a three-dimensional structure that mimics the size and shape of an aminoacyl-tRNA molecule and it recognizes all three termination codons in the A site of the ribosome. It also allows the release of
the nascent peptide2,21,29. For the efficient hydrolysis of the peptidyl-tRNA and the release of the peptide,
GTP bound to the release factors is hydrolysed by eRF3. This hydrolysis allows the accommodation of the eRF1 in the peptidyl transferase center21.
After the translation termination, the ribosome is recycled. However, when the aminoacyl-tRNA binding overcomes the eRF1/3 binding on the A site, in the presence of a termination codon, it results in a stop codon suppression (or readthrough of this codon)18.
1.3. Nonsense codons
Nonsense mutations are nucleotide changes within the coding sequence of a gene that form a PTC or a nonsense codon. They are a result of RNA editing or errors of the editing processes, inefficient RNA splicing, non-productive DNA rearrangements or a mutation of the DNA30. There are 23 different
nucleotide substitutions that lead to nonsense codons, however, the most frequent are CGA to TGA (21%) and CAG to TAG (19%)31.
The mRNA containing a PTC can be targeted to decay by the NMD, or escape from it. In the second case, when the ribosome finds a PTC in the mRNA, the translation termination occurs and the mRNA is translated into a peptide, that can be or not degraded by proteases. When these truncated proteins are not degraded by proteases, they might not be functional, and so, they aggregate and cause endoplasmic reticulum (ER) stress. The ER is responsible for the synthesis, folding, and assembly of proteins that are designed for secretion or for the membrane of the cell34. The absence of functional
proteins lead to numerous diseases including genetic disorders (i.e. cancer32, cystic fibrosis (CF)18, Duchenne muscular dystrophy (DMD)18, and thalassemia33).
1.4. The nonsense-mediated mRNA decay
1.4.1. NMD functions and mechanismNMD is a conserved mechanism found in all eukaryotes, and it can detect and degrade mRNAs carrying PTCs. The main function of this mechanism is to ensure that these mRNAs are guided to rapid decay, avoiding the translation of truncated and potentially harmful proteins2,11,35. Moreover, NMD also regulates the steady-state levels of wild-type transcripts arising as a mechanism of gene expression regulation11.
In mammalian cells, the NMD pathway is dependent on the deposition of a protein complex during the splicing process. This complex, the EJC, is placed 20-24 nucleotides upstream of the exon-exon junction. During the pioneer round of translation, the translating ribosomes displace the EJC from the ORF, as it was previously mentioned. However, if the mRNA contains a PTC more than 50-54 nucleotides upstream of the last exon-exon junction, at least one EJC will remain in the mRNA after its translation, targeting the mRNA for degradation by NMD11. EJC anchors the NMD-specific factors
UPF2, loaded into the mRNA during the cytoplasmic transportation, and UPF3, loaded during the splicing, in the nucleus2,11,34.
During premature translation termination, UPF1, an RNA-dependent adenylpyrophosphatase (ATPase) and a 5’ to 3’ helicase, interacts with the serine/threonine protein kinase 1 (SMG-1), the eukaryotic release factor 1 (eRF1), and the eRF3 to form a complex named SURF (SMG-1/UPF1/Release factors)35,11. Then the UPF1 (in the SURF complex) interacts with the EJC-bounded UPF2 forming a complex named decay-inducing complex (DECID) 2,11. SMG1 phosphorylates UPF1 leading to the recruitment of SMG5, SMG6 and SMG7. SMG6 is responsible for the endonucleolytic RNA cleavage whereas SMG5 and SMG7 are responsible for the recruitment of general decay factors10.
Once the mRNA is targeted for decay, it can be degraded through: 1) decapping by DCP1/DCP2 decapping complex followed by 5’ to 3’ exonucleolytic activity of exoribonuclease 1 (XRN1), or 2) deadenylation followed by 3’ to 5’ exonucleolytic degradation through an exosome complex2 (Figure
1.4).
Figure 1.4 - Representation of the NMD mechanism
During splicing EJC complexes associate with the mRNA 20-24 nucleotides upstream of the exon-exon junctions11,34. During
the pioneer round of tranlation the ribosome displaces all the EJC complexes. However, if a PTC is located 50-54 nt upstream of the last EJC complex, this complex is not removed. UPF1 associates with SMG1 and with eIF1/eIF3 to form the SURF complex. The interaction of the UPF1 (in the SURF complex) with UPF2 (bound with EJC) leads to the formation of the DECID complex36. This interaction leads to the phosphorylation of UPF1 mediated by SMG1 and the dissociation of
eRF1-eRF3. Addicionaly, the phosphorylation of UPF1 leads to the recruitment of SMG6, SMG5 and SMG7. SMG6 has endonuclease activity, cleaving the mRNA, while SMG7 and SMG5 proteins recruit the decay factors. The degradation of the mRNA can be achieved either by decapping followed by 5’ to 3’ exonucleolytic activity by XRN1 or deadenylation followed by 3’ to 5’ exonucleolytic activity by exosome complex2.
Image from Paulo J. da Costa 201737
1.4.2. Exceptions to the rule
Some transcripts, despite having a PTC located 50-54 nucleotides upstream of the last EJC, can be resistant to NMD38. This theory was proposed by Inácio at al. that have shown that transcripts with nonsense mutations in close proximity to the AUG fail to trigger NMD34.This could be explained by the closed proximity of PABPC1 to the eRF3 during the premature translation termination process. In this situation,there is a competition mechanism in which eRF3 can interact with UPF1 or with PABPC1. When a PTC is closed to the AUG, PABPC1 is favourably located to interact with eRF3 bound to the PTC, avoiding eRF3-UPF1 interaction, and NMD is inhibited11,39. When a PTC is not located in proximity to AUG, PABPC1 can not interact with eRF3, so UPF1 interacts with this release factor, leading to NMD activation. Therefore, UPF1 and PABPC1 have opposite effects. This phenomenon occurs due to the fact that the mRNA can circularize, providing the necessary conformation for the interaction of eRF3 and PABPC111,35 (Figure 1.5).
Figure 1.5 - Representation of a transcript with a PTC close to the AUG.
When a PTC is close to the AUG, PABPC1 can interact with eRF3, inhibiting the interaction of UPF1 with this realease factor and NMD activation39.
Image fromPeixeiro 2012 39
1.5. β-Thalassemia
1.5.1. The human β-globin gene
The human β-globin gene is one of the models used to study NMD due to its small size and its nonsense mutations associated with disease. It is also considered to be an attractive model to study suppression therapy 40. The human β-globin gene is located in the human chromosome 11, in a cluster
containing five genes2. The cluster of human β-globin genes (HBB) consists of the embryonic epsilon
gene (ε), fetal G-gamma(Gγ) gene, the A-gamma (Aγ) gene, the adult delta (δ)and β-globin genes. (Figure 1.6)41.
Figure 1.6 - Representation of the β-globin genes cluster.
The human chromosome 11 has the cluster of human β-globin genes (HBB) that consists of the embryonic epsilon gene (ε), fetal G-gamma(Gγ) gene, the A-gamma (Aγ) gene, the adult delta (δ) and β-globin genes. There is also a regulatory element, the locus control region (LCR), that contains four erythroid-specific DNAse hypersensitive sites (HS). ERV-9 is a retrotransposon and is located upstream of the LCR41,42.
Image adapted from Cao 2002 42
Since the human β-globin pre-mRNA has two introns and three exons, after transcription and splicing, two EJCs are assembled on the mRNA2. The human β-globin mRNAs are stable since globin
proteins are extremely needed and abundant, having half-lives that vary from 10 to 24 hours2. Human
α-globin and β-globin proteins have similar levels in the cell, as two α-globin and two β-globin chains associate in a tetrameric and functional unit, named hemoglobin43. The major function of the human
hemoglobin is to transport oxygen (O2) from the lungs to the tissues. However, it has other functions,
such as the transport of carbon dioxide (CO2), carbon monoxide (CO), and nitric oxide (NO2)43. When
there is some deregulation in the levels of one of the units, it may result in a disease. For example, when β-globin is not produced at the same level of α-globin, for instance, due to a genetic mutation, this results in β-thalassemia, as it will be explained in the following subchapter.
1.5.2. The genetic disease
“Thalassemia” name was implemented in Greek and it is the conjugation of the name thalassa (sea) and haima (blood)44. β-thalassemia is a genetic disorder with an annual incidence of 1 in 100000 individuals in the world and 1 in 10000 in Europe43. It is predominant in regions where there is a selective pressure from Plasmodium falciparum as Cyprus, Sardinia, and Southeast Asia. The population migration and the fact that people from different ethnic groups have descendants introduce β-thalassemia in most countries44. This genetic transmission is usually autosomal recessive. When parents are heterozygous for a β-globin mutation, their child has 25% of chance of having the disease, 50% of chance of being a carrier and 25% of being not affected43. The types of β-thalassemia are very
different at a molecular level. There are more than 200 mutations identified and the majority of β-thalassemia is caused by a point mutation by substitution. Insertion or deletion of a single nucleotide is also common. However, deletion of a part of the β-globin gene on chromosome 11 is rare38,43. This
syndrome is characterized by a reduction (β+) or an absence (β0) of β-globin protein which causes a reduction on hemoglobin synthesis44. Even that β-globin protein is not produced, α-globin chains continue to be synthesized, which leads to its accumulation and precipitation within the erythroid precursors, causing intramedullary destruction of the latter38.
1.5.3. Clinical genotype/phenotype
There are three clinical conditions recognized for thalassemia, the thalassemia minor or β-thalassemia carrier state, the β-β-thalassemia intermedia, and the β-β-thalassemia major41. β-thalassemia
minor is a condition of heterozygous patients, with one normal allele (β/β+ or β/β0), and in most cases,
differences in severity, ranging from β-thalassemia minor to β-thalassemia major. The phenotypic diversity of β-thalassemia intermedia can be explained by the differences on the amount of β-globin production (β+/β+ or β+/β0). These patients have a moderate anemia but they do not require regular
transfusions, however, they can have for example, osteoporosis, masses of erythropoietic tissue that primarily affect the spleen, liver, and lymph nodes, painful leg ulcers and the increase of the possibility of a thrombotic event43,44. In β-thalassemia major, patients do not produce β-globin protein (β0/β0),
which means they do not produce hemoglobin (α2β2)38. This causes a severe anemia that appears in the
first 6 months of the child life and when it is not treated, the child die in the first two years of their life38.
These patients depend on regular blood cell transfusions, and present pale, feeding problems, diarrhea, irritability, fever, and a progressive enlargement of the abdomen41,43,44. The β-thalassemia major, when
untreated, leads to growth retardation, pallor, jaundice, poor musculature, hepatosplenomegaly, leg ulcers and skeletal changes due to an expansion of the bone marrow. Even when the patient with β-thalassemia has a therapeutic approach being applied, this therapy leads to numerous difficulties, for example endocrine complications (i.e. growth retardation, failure of sexual maturation, diabetes mellitus) due to iron overload in different organs and tissues, caused by the regular transfusions43,44.
1.5.4. NMD and β-thalassemia
Some of the mutations associated with β-thalassemia are nonsense mutations. In these cases, the reduction of the β-globin protein may be due to the activation of NMD2,35,41. One of the most known
examples is a nonsense mutation at codon 39 (β39) of the β-globin gene. In this case, the CAG codon is mutated to a TAG stop codon. The corresponding mRNA is targeted to NMD and will be degraded2.
Other mutations at codons 15, 37, 59, or 127 can also be found in association with β-thalassemia36. As
it was described, there are some exceptions to the NMD activation, for instance, nonsense codons in proximity to the AUG45. Several studies proposed that in the human β-globin mRNA, a nonsense mutation in the 5’ half of the first exon [i.e. nonsense mutation at codon 5 (β5), β15, or β17] may result in high levels of mRNA34,40. It was also shown that the half-life time (t1/2) of these transcripts are lower
than the normal control but higher than t1/2 of β39 mRNA, which is committed to NMD40. Given this,
NMD has a crucial role in the the definition of the phenotype of β-thalassemia, as well as in other genetic diseases2. If a PTC is located in a position that leads to the NMD activation, the individuals have β-thalassemia minor and are heterozygous and asymptomatic. In this case, the mRNA is degraded and there is no protein from the corresponding allele, causing a decrease in the amount of human β-globin in about 50%, but near-normal hemoglobin levels are maintained by the normal allele. If a PTC is located at a position that does not activate the NMD, there are two different situations. If a PTC is located at codons 1-23 (exon 1) or at the 5’ end of the exon 3, a small truncated protein is produced and degraded by effective proteolysis. So, the phenotype is similar to when the NMD is activated. But if the PTC is located at the 3’ end of the exon 3, a the long truncated protein is synthesized and thus, proteolysis is ineffective. Therefore, these proteins accumulate, as well as the human α-globin protein in excess, which leads to a dominant and severe phenotype, like one of the phenotypes of β-thalassemia intermedia2
Figure 1.7 - The β-thalassemia phenotype is influenced by the NMD mechanism.
The PTCs located at codons 1-23 (AUG-proximal) do not activate the NMD. In this case, there is an efficient degradation of the short proteins produced. PTCs at codons 24-87 (50-54nt upstream of EJC) activate NMD, and there is no protein produced. When the PTC is located at the 5’ end of the exon 3, the NMD is not activated but the proteins produced are short and can be effectively degraded. In these three cases the heterozygous are asymptomatic since the levels of globin are mantained by the normal allele. When the PTC is located at the 3’ end of the exon 3, the NMD is not activated and long truncated nonfunctional β-globin proteins are produced. These proteins are not degraded by proteolysis, instead, there is precipitation of insoluble globin chains, and the heterozygotes are severely affected2.
Image from Peixeiro 2011 2
1.6. The conventional β-thalassemia therapy
β-thalassemia can not be cured, however, there are some therapies which allow the regularization of hemoglobin levels. One of the conventional options for thalassemia, normally in β-thalassemia intermedia, is splenectomy, in order to avoid the consequences of erythroid precursor accumulation46,44. This therapy has been proposed in most cases of β-thalassemia intermedia whose
individuals contain β-globin truncated proteins, that lead to a high damage of erythroblasts and erythrocytes. However, patients that do this therapy have a high risk of venous thromboembolism, pulmonary hypertension, leg ulcers, silent cerebral infarcts, and risk of infections46,47.
Other therapy used in β-thalassemia, normally in β-thalassemia major, is regular transfusions to maintain the oxygen delivery. This therapy leads to fewer complications as thrombosis or extramedullary hematopoiesis, however, it leads to a higher iron overload46. Other concerns about this therapy are the risk of transfusion-related acute lung injury, the hemolytic transfusion reactions, and the incompatible blood transfusions44,48. The transfusion therapy and the increase in intestinal iron
absorption lead to high levels of iron in the blood, since the human body can not excrete it. The iron in excess accumulates in the tissues and causes, for example, heart failure and liver cancer44. For this
reason, these transfusions are often combined with iron chelation therapy46.
Since the majority of the conventional therapies used for the treatment of β-thalassemia have a lot of secondary effects, and some of them can be dangerous to the patient, a deeper study of this disease and treatments should be an advantage. So, a new therapy with less secondary effects, that allows the globin protein to be completely synthesized and functional, would be of extremely importance for β-thalassemia patients carrying nonsense mutations. Within this concept, suppression therapy is being investigated as an alternative to conventional therapies, specifically when transcripts carry nonsense codons.
1.7. Suppression therapy
Suppression therapy was first described in 1996 when it was reported that the use of low doses of geneticin (G418) and gentamicin (aminoglycosides compounds) could suppress PTCs in the mRNA of cystic fibrosis transmembrane conductance regulator (CFTR) and restore the levels of the full-length protein, in cultured Henrietta Lacks (HeLa) cells18,49. After that discovery, a lot of studies with other
compounds have been described in 40 different diseases, not only in culture cells but also in animal models, and some of these compounds were already used in clinical trials50,55.
1.7.1. Mechanism of PTC suppression
The suppression of a termination codon can occur naturally, when a near-cognate aminoacyl-tRNA binds to the stop codon instead of the eRF1/eRF3. This is mediated by the mispairing of a near-cognate aminoacyl-tRNA, that contains two complementary nucleotides with the stop codon. This occurs under basal conditions in 1% of the translation events in case of a PTC and in 0,1% in case of a natural termination codon. In the case of PTCs, their recoding allows translation to resume and the production of a complete protein, opposing the effect of a nonsense mutation29. In case of a normal
termination codon suppression, other in-frame stop codons may occur in the 3’ UTR, that reduce the possibility of translation to continue. If it continues, the mRNA is targeted to degradation29. The frequency of PTC suppression under basal conditions is not enough in case of a disease and so, the goal of the suppression therapy is to mimic this recoding of the PTC into a sense codon, but in a higher frequency29,18. Suppression therapy efficiency depends on the sequence context of the PTC, on the nature
of the PTC codon and on the amino-acids at the C-terminus of the nascent polypeptide. Suppression therapy has better efficiency at UGA stop codons, followed by UAG and UAA, due to the hydrogen bonds between eRF1 and these codons, and is enhanced when a PTC is followed by a cytosine51. There are some amino acids that are more likely to be incorporated in the nascent peptide, depending on the triplet of the PTC 52,29. This evidence is showed in table 1.1.
Table 1.1 - Amino acids incorporated in the nascente peptid depending on the triplet of the PTC49.
The fact that a normal stop codon is less influenced by the suppression therapy than a PTC, suggests that there are differences in the termination events after their recognition29. Several experiments were performed to explain this, and it was realized that there is a much longer ribosomal pause at a PTC than at a normal stop codon29. When in the presence of a normal stop codon, the interaction of PABP with eRF3, stimulates the release of the peptide chain and an efficient translation termination18 (Figure
1.9A). However, when the translation termination occurs at a PTC, eRF3 is unable to interact with PABP, causing a ribosomal pause during termination30 (Figure 1.9B). This ribosomal pause makes the
PTC more susceptible to be recognized by a near-cognate aminoacyl-tRNA18.
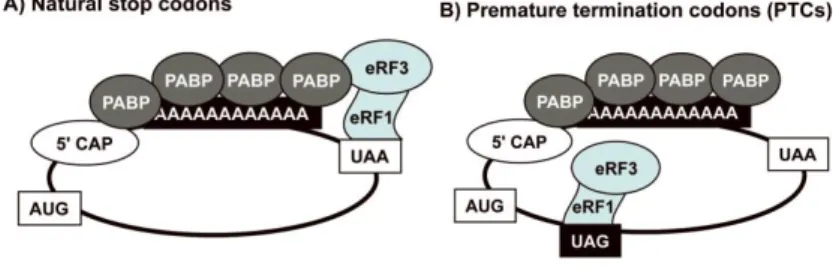
Figure 1.8 - Translation termination at normal stop codons and at premature termination codons.
A) In normal situations, the eRF1/eRF3 complex is bound to the natural stop codon (i.e. UAA). As this codon is near to the 3’ end, there is an interaction between the complex and PABP that enhances the termination. B) In case of a PTC, the eRF1/eRF3 complex is not near to the 3’ end, avoiding its interaction with PABP. Therefore, the termination is not efficient, which leads to a pause in the ribosome that makes PTC more susceptible to suppression29.
Image from Keeling 201129
1.7.2. Compounds with suppressive properties
There are several low molecular weight compounds that have been shown to increase the readthrough of a PTC into a sense codon. One of the most studied classes of compounds is aminoglycosides. Other compounds non-related to aminoglycosides are the non-aminoglycosides29.
Aminoglycosides
Aminoglycosides are a class of antibiotics used to treat Gram-negative bacterial infections. They are also known to induce the suppression of premature termination codons18,53,54. They are generally
composed of two or three amino sugars associated with a glycosidic linkage to a 2-deoxystreptamine ring. These compounds bind to the decoding center of the ribosome, which has a proofreading activity that allows the right codon-anticodon base pairing. It ensures that the right aminoacyl-tRNA is accommodated in the A site of the ribosome18,53. However, when aminoglycosides bind to the decoding center, they reduce the fidelity of the ribosomal proofreading activity and lead to the misincorporation of a near-cognate aminoacyl-tRNA. This misincorporation allows the translation to continue to the 3’ end of the mRNA54. It has been reported that aminoglycosides do not bind to the eukaryotic decoding
center so strongly as they bind to the prokaryotic, since the eukaryotic ribosomal RNA sequence differs from the prokaryotic at two key nucleotides18,53. This allows ribosomal pausing without stopping the translation29.
There are numerous obstacles when using these aminoglycosides in the suppression therapy, since they are influenced by the sequence nearby the PTC. So, a therapy involving an individualized genome sequencing and personalized medicine would be beneficial. Besides that, the use of aminoglycosides for a long term will induce nephrotoxicity and ototoxicity29.
The aminoglycoside gentamicin is the most well-studied compound and it is known to suppress the PTC in sequence context-dependent manner (Figure 1.9)29. Numerous positive results have been
reported when using this compound as a treatment of several diseases (i.e. DMD and CF). However, it leads to secondary effects, such as hearing loss and kidney damage18.
Figure 1.9 - Gentamicin and its congeners produced during its synthesis.
Gentamicin is an antibiotic composed of two amino sugars associated by a glycosidic linkage to a 2-deoxystreptamine ring. It
is the most studied aminoglycoside used in the suppression of PTCs. During its synthesis, five isoforms are generated that have different toxicity and PTC suppression capability29.
Image from Keeling 2011 29
The cellular aminoglycosides toxicity can be explained by their off-target effects in the cell. One of the biggest problems of these compounds is its capability to become positively charged when they are endocytosed, since the lysosome has a low pH. This means that these compounds can bind to the negative-charged phospholipids in the membrane of the lysosome and interfere with a lot of cell signalling pathways, enzymatic reactions or lead to phospholipidosis29. Another problem related to their
charge is the formation of reactive oxygen species (ROS) that can damage the DNA, proteins, and lipids. ROS are also involved in several processes including proliferation, growth, and apoptosis29,55. To overcome these secondary effects, gentamicin can be co-administrated with poly-L-aspartame or daptomycin to prevent their binding to the lysosome membranes, preventing nephrotoxicity, and enhancing the nonsense suppression. The administration of antioxidants, such as D-methionine and melatonin, prevents the damage promoted by ROS18,29.
Because of the reduced suppression efficiency at low doses and the toxicity due to its long-term administration, other synthetic compounds have been developed from conventional aminoglycosides29.
For example, the NB30 in the study of Rett syndrome56 and Usher syndrome57, the NB54 in CF, DMD
and Mucopolysaccharidosis type I (MPS I-H)18, or the G418 in β-thalassemia36 (Figure 1.10). In a first development, NB54 showed an improvement in PTC readthrough efficiency with less toxicity when compared to gentamicin56. After these positive and promising results, new generations of these synthetic
aminoglycosides were developed and they showed even better results in the PTC suppression in comparison with the previous ones. For example, NB58 is more effective than NB54 in the treatment of MPS I-H.
Figure 1.10 - Conventional aminoglycosides and novel synthetic aminoglycosides developed for PTC suppression in eukaryotic cells.
The conventional aminoglycosides as the paromomycin, amikacin, and G418 have been used in the PTC suppression in several genetic disorders, however, they have toxic properties. To overcome this issue, novel synthetic aminoglycosides have been developed from the conventional ones using their structural bones. NB30 was derivated from paromomycin using the structural represented as A. NB54 was derivated from both paromomycin and amikacin with the structures A and B. NB54 is composed by the three conventional aminoglycosides represented, paromomycin, amikacin, and G418, and has the three structures, A, B, and C, as represented29.
Image from Keeling 2011 29
Among all the conventional aminoglycosides, G418 (also known as geneticin or G418 sulfate) is the one showing the highest PTC readthrough activity. However, G418 is also very cytotoxic, a fact that can be related to its specificity to mammalian ribosome58. G418 seems to induce the readthrough of the PTC in recessive proximal renal tubular acidosis at 75 µg/mL59, in spinal muscular atrophy (SMA)
at 150, 300 and 600 µg/mL60 and in CF at 100 and 200µg/mL61.
Non-aminoglycosides
There are also some non-aminoglycosides that have PTC suppression properties (Figure 1.11). One example of the non-aminoglycosides is negamycin. This compound is a dipeptide antibiotic and has shown to suppress PTCs. It allows the restoring of protein function of adenomatous polyposis coli (APC) gene associated with colon cancer, and the restoring of dystrophin protein, with less toxicity than gentamicin18,29. Even that the binding mechanism of this compound has not been elucidated yet, it is
known that it binds to the decoding site of the eukaryotic ribosome29. Other antibiotics, as spiramycin,
josamycin, and tylosin, have been shown to suppress APC nonsense mutations and restore the APC protein levels in culture cells18. There are other non-aminoglycosides that are not antibiotics but that
suppress PTCs. These compounds were obtained from high-throughput screenings: RT13 and RT14 in the dystrophin gene in myoblasts derived from a mouse model of DMD50 and PTC124 (or Ataluren) for DMD and CF29,62.