Luís Miguel Torres Cadilhe
Protective activity of plant extracts
against genotoxicity of nitric oxide
Universidade do Minho
Escola de Ciências
Março de 2017 Luís Miguel T orr es Cadilhe P ro te c ti ve a c ti vi ty o f p la n t e x tr a c ts a g a in st g e n o to x ic it y o f n it ri c o x id e Minho | 20 1 7 ULuís Miguel Torres Cadilhe
Protective activity of plant extracts
against genotoxicity of nitric oxide
Universidade do Minho
Escola de Ciências
Março de 2017
Tese de Mestrado
Mestrado de Genética Molecular
Trabalho efetuado sob a orientação do
iii
Acknowledgements
To Prof. Rui Pedro Soares de Oliveira
, for the opportunity and pleasure to work under his supervision in something that incites my complete interest. Thank you for being a concerned supervisor, for your readiness to answer to all my doubts and curiosities, for all the knowledge I was able to gather throughout this work and most importantly for the thoughtfulness and the chances to expose my work.To Prof. Pier Parpot,
for his help and expertise regarding the chemical analysis of the extracts. The insights about the method itself were also an important input.To all my laboratory partners
, for creating such a pleasant environment to work in, for all the moments of friendship, for all your help and specially, for always sharing your material in the most desperate times. In other words, thank you for making this the most enjoyable experience so far.To my friends,
for sharing successes, failures and the same concerns as me, for constantly reminding me of what is at stake and to keep my focus and for all the relaxing moments in our short coffee breaks.To my family
, for making the physical distance that separates us feel irrelevant, for all your support, not only during this last year, but also in all the moments of my life, in the good and the less good. Thank you for your concern, for all the motivation, for all the help and for contributing so much to what I am today.v
Protective activity of plant extracts against genotoxicity of nitric oxide Abstract
A good reputation regarding the use of plant extracts has been growing consistently over the last years, the potential benefits of their use being heavily supported by the literature. However, as most studies aspire very little by investigating only their antioxidant activities, a lot of work remains to be done on other activities, such as antigenotoxicity. Excessive production of oxidative species, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), can reveal to be harmful to the cell. Amongst various targets, these molecules can affect DNA and may lead to the loss of its stability and integrity, imposing a very dangerous threat to survival. Base excision repair (BER) and homologous recombination (HR) are two major DNA repair mechanisms of the cell, and are responsible for the repair of several forms of DNA damage, including base modification and strand breakage.
SNP is a NO-releasing agent and can be used to simulate an excessive increase of NO production inside the cell. NO may oxidize in many different compounds, depending on the molecules it encounters within the cell. The most damaging oxidation products include peroxynitrite and dinitrogen trioxide, a very powerful oxidant and a very strong nitrosating agent, respectively, both being able to directly interact and modify DNA.
This project purposed to investigate the protective properties of extracts of Dittrichia viscosa (DVE) and
Ginkgo biloba (GBE) against NO-mediated genotoxicity and in what ways protection occurs by using the Schizosaccharomyces pombe model. In the end, the chemical profiles of DVE and GBE agreed with many published others, as most of the detected molecules were also detected in those studies. In vitro assays
revealed a relatively (to standard) low IC50 values of DVE in DPPH reduction and iron chelating activity and
GBE in DPPH reduction and NO scavenging. DVE did not provide viability protection against NO-mediated damage. GBE, however, was able to protect the studied BER and HR Sch. pombe mutants. Additionally, treatment with GBE alone significantly induced a quicker progression of cell cycle and slightly attenuated the delay produced by SNP treatment. Both extracts appear to activate oxidative-stress response through Pap1, although the prevention of oxidative stress by SNP was not achieved in either cases. In conclusion, GBE protection appears to be independent of BER and it may result from a combination of effects including the scavenging of NO, the generated ROS and RNS by flavonoids and the activation of
vii Actividade protectora de extractos vegetais contra a genotoxicidade do óxido nítrico
Resumo
Nos últimos anos, tem-se vindo a construir uma boa reputação no que toca ao uso de extractos vegetais, sendo os seus potenciais benefícios fortemente suportados pela literatura. No entanto, a maioria dos estudos aspira a muito pouco ao investigar apenas as suas actividades antioxidantes, pelo que há muito trabalho a ser feito em relação a outras actividades, como a antigenotoxicidade. A produção excessiva de espécies oxidativas, como as espécies reativas de oxigénio (ROS) e de nitrogénio (RNS), podem revelar-se prejudicial à célula. Entre os seus vários alvos, estas moléculas podem afectar o DNA, podendo levar à perda da sua estabilidade e integridade, impondo assim uma grave ameaça à sobrevivência. A reparação por excisão de bases (BER) e a recombinação homóloga (HR) são dois dos principais mecanismos de reparação de DNA da célula e são responáveis pela reparação de vários tipos de danos no DNA, incluindo modificações de bases e quebras de cadeia.
O nitroprussiato de sódio é um agente que liberta NO e pode ser utilizado para simular um aumento excessivo da produção de NO por parte da célula. O NO é capaz de oxidar em muitos compostos diferentes, dependendo das moléculas que encontra dentro da célula. Os produtos de oxidação mais prejudiciais são o peroxinitrito e o trióxido de dinitrogénio, um poderoso oxidante e um forte agente nitrosante, respectivamente, sendo ambos capazes de interagir e modificar directamente o DNA.
Este projecto pretendeu investigar as propriedades protectoras de extractos de Dittrichia viscosa (DVE)
e Ginkgo biloba (GBE) contra a genotoxicidade do NO e de que forma essa protecção ocorreria,
recorrendo ao modelo de Schizosaccharomyces pombe. No fim, os perfis químicos de DVE e GBE foram semelhantes a muitos outros publicados, pelo que a maioria das moléculas detectadas foram também
detectadas nesses estudos. Os ensaios in vitro revelaram valores de IC50 relativamente (ao padrão) baixos
para DVE na reducção do DPPH e na actividade quelante do ferro e para GBE na reducção do DPPH e na eliminação do NO. DVE não proporcionou protecção da viabilidade contra danos induzidos pelo NO.
GBE, no entanto, foi capaz de proteger estirpes de Sch. pombe mutantes na BER e HR. Ainda, o
tratamento apenas com GBE induziu uma progressão significativamente mais rápida do ciclo celular e atenuou ligeiramente o atraso produzido pelo tratamento com SNP. Ambos os extractos pareceram activar a resposta a stress oxidativo através da Pap1, embora a prevenção do stresse oxidativo induzido pelo SNP não tenha acontecido em nenhum dos casos. Concluindo, a protecção pelo GBE parece ser independente da BER e deve resultar de uma combinação de efeitos, incluindo a eliminação do NO, dos ROS e dos RNS gerados por parte dos flavonóides e a activação de proteínas de resposta a stress oxidativo.
ix
Table of Contents
Acknowledgements ... iii
Abstract ... v
Resumo ... vii
Abbreviations and Acronyms ... xi
List of figures ... xiii
List of tables ... xvii
1. Introduction ... 1
1.1. DNA damage and repair ... 1
1.2. Nitric oxide biochemistry ... 7
1.3. Dittrichia viscosa (L.) Greuter and Ginkgo biloba (L.) ...10
1.4. Schizosaccharomyces pombe ...12
1.5. Scientific problem and objectives ...14
2. Materials and Methods ...15
2.1. Yeast strains, media, and growth conditions ...15
2.2. Plant material and extracts preparation ...15
2.3. Electrospray Ionisation-Tandem Mass Spectrometry (ESI-MS/MS) ...16
2.4. In vitro assays ...16
2.5. Viability assays ...18
2.6. Cell cycle ...19
2.7. Fluorescence microscopy ...20
3. Results ...21
3.1. The chemical composition of DVE and GBE is similar to that of other described extracts ...21
3.2. DVE and GBE showed different in vitro activities ...24
3.3. GBE significantly protected viability loss caused by SNP ...24
3.4. GBE alone induces quicker progression in cell cycle and slightly attenuates SNP-induced delay ...29
3.5. Both extracts induce relocalization of oxidative-stress response proteins ...32
4. Discussion ...37
5. References ...43
xi
Abbreviations and Acronyms
3’-dRP 3’-deoxyribose phosphate 3’-P 3'-phosphate 5’-dRP 5’-deoxyribose phosphate 5’-P 5'-phosphate 6-4PP 6-4 photoproducts AP Apyrimidinic/apurinic
ATP Adenosine 5'-triphosphate
BER Base excision repair
CDK Cyclin-dependent kinases
CPD Cyclobutane pyrimidine dimer
DEA-NO Diethylamine NONOate
DNA Deoxyribonucleic acid
DPPH 2,2-diphenyl-1-picrylhydrazyl
DSB Double-strand break
DSBR Double-strand break repair
DVE Dittrichia viscosa extract
EDTA Ethylenediaminetetraacetic acid
EMM Edinburgh minimal medium
eNOS Endothelial nitric oxide synthase
GBE Ginkgo biloba leaf extract
GG-NER Global-genome NER
HJ Hollidayjunction
HR Homologous recombination
HU Hydroxyurea
IC50 Inhibition concentration 50
ICA Iron chelating activity
iNOS Inducible nitric oxide synthase
IUCN International Union for Conservation of Nature
MMR Mismatch repair
xii
NER Nucleotide excision repair
NHEJ Non-homologous end joining
NMMA NG-monomethyl-L-arginine
nNOS Neuronal nitric oxide synthase
NOS Nitric oxide synthase
OD Optical density
PCNA Proliferating cell nuclear antigen
PI Propidium iodide
RFC Replication factor C
RNA Ribonucleic acid
RNS Reactive nitrogen species
ROS Reactive oxygen species
SDSA Synthesis-dependent strand annealing
SLE Systemic lupus erythematosus
SNP Sodium nitroprusside
SPER-NO Spermine NONOate
SSB Single-strand break
TC-NER Transcription-coupled NER
UV Ultraviolet
UVB Ultraviolet-B
UVER Endonuclease-dependent excision repair
YD Yeast Extract Dextrose
xiii
List of figures
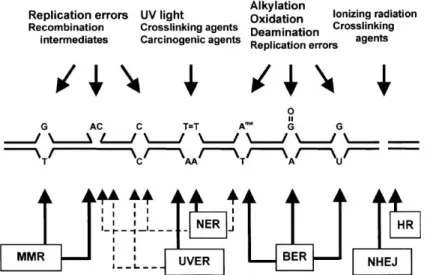
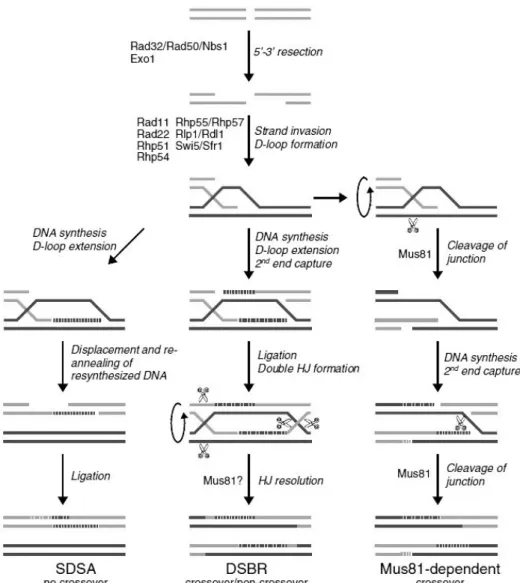
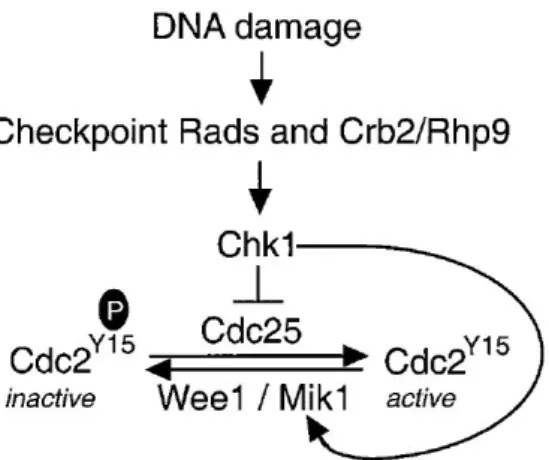
Figure 1 – DNA damage and respective repair pathways in Sch. pombe. Mismatched bases (except C/C) and loops that occur during replication and recombination are mainly repaired by mismatch repair pathway. Nucleotide excision repair and endonuclease-dependent excision repair pathways are assembled to remove bulky DNA adducts caused by exposure to UV radiation; these two pathways have also been associated with other types of DNA damage (dashed arrows). Base excision repair is the most competent pathway to repair non-bulky modifications that occur from base modifications or incorporation of modified bases. Finally, homologous recombination and non-homologous end joining repair double-strand breaks. From Egel, 2004. ... 1 Figure 2 – Base excision repair (BER) pathway in Sch. pombe. Apyrimidinic/apurinic sites are primarily repaired through short-patch BER (thicker arrows): Nth1 initiates repair by incising the AP site, which results in the formation of 3 -dRP and 5 -P ends; the blocked 3’-end is converted in a 3’-OH by Apn2; DNA polymerase and DNA ligase fill the gap and seal the nick, respectively. In long-patch BER, Apn2 is the initiator and incises the AP site, leaving a 5’-dRP end; DNA polymerase synthesizes 2-8 new nucleotides and displaces the damage-containing oligonucleotide, which is cleaved by Rad2; DNA ligase completes repair by sealing the nick. Besides synthesizing DNA, Pol4 is able to cleave 5’-dRP, only needing DNA polymerase and DNA ligase to complete the process. The AP endonuclease independent repair pathway is initiated by Nth1 and followed by Tdp1, which cleaves 3’-dRP, leaving a 3’-P to be processed by Pnk1 and completed by DNA polymerase and DNA ligase. From Nilsen et al., 2012. ... 3 Figure 3 – HR comprises SDSA, DSBR and Mus81-dependent pathways. In SDSA, the distension of the D-loop follows strand invasion, allowing DNA synthesis. The new DNA strand is displaced from the template and returned to the damaged molecule. DSBR pathway, proposed by Szostak et al. (1983), also involves D-loop distention, leading to the capture of a second 3’ overhang and formation of a double Holliday junction (HJ). The resolution of HJ results in gene conversion, with or without associated crossovers. In the last pathway, Mus81 cleaves the invaded DNA strand disrupting the D-loop. The second 3’ overhang is captured and DNA is synthesized. Lastly, Mus81 cleaves the strand of DNA keeping the homologs together, resulting in crossover of the strands. Edited from Raji and Hartsuiker, 2006. ... 5 Figure 4 – DNA damage checkpoint in Sch. pombe. Upon sensing of DNA damage and signal transduction, Chk1 indirectly induces the delay of cell cycle by phosphorylating Wee1 and Cdc25. Wee1 and Mik1 phosphorylate Y15 of Cdc2, resulting in its inactivation. Cdc25, on the other hand, dephosphorylates Cdc2 to activate it and subsequently, induce mitosis. From Baber-Furnari et al., 2000. ... 6
xiv Figure 5 – Chemical structure of quercetin. The three main features required for the antioxidant activity of flavonoids are the ortho-dihydroxy structure in the B ring which facilitates electron delocalization (A), 2,3-double bond in conjugation with the 4-oxo(=O) function in the C ring to allow electron delocalization from the B ring (B) and the hydroxyl groups at positions 3 and 5, which allow binding of hydrogen to the oxo group (C). From Procházková et al., 2011. ...11 Figure 6 – Viability of 972, Δchk1 and Δrhp51Sch. pombe cells when cultured on solid medium incorporated with (from left to right): water (negative control), 750 µg/mL DVE, 1 mM SNP and both 750 µg/mL DVE and 1 mM SNP. Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the top of the photos). The
plates were incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...25
Figure 7 – Viability of 972 and Δchk1 Sch. pombe cells when incubated for 0, 30, 60 and 90 min in liquid medium containing (from left to right): water (negative control), 750 µg/mL DVE, 300 mM SNP and both 750 µg/mL DVE and 300 mM SNP. Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the top
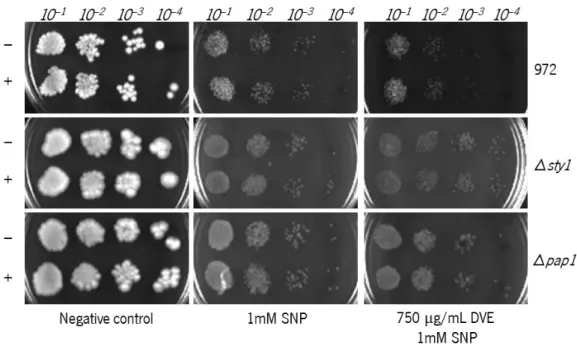
of the photos). The plates were incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...26 Figure 8 – Viability of 972, Δsty1 and Δpap1 Sch. pombe cells when pre-incubated in 750 µg/mL DVE-containing liquid medium and cultured in medium incorporated with (from left to right): water (negative control), 1 mM SNP and both 750 µg/mL DVE and 1 mM SNP. Cells subjected to pre-treatment without DVE are signalized by (-) and with DVE by (+). Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the top of the
photos). The plates were incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...27 Figure 9 – Viability of 972 Sch. pombe cells when incubated for 0, 30, 60, 90 and 120 min in liquid medium containing (from left to right): ethanol(negative control), 300 mM SNP and 1000 µg/mL GBE and 300 mM SNP. Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the top of the photos). The plates were
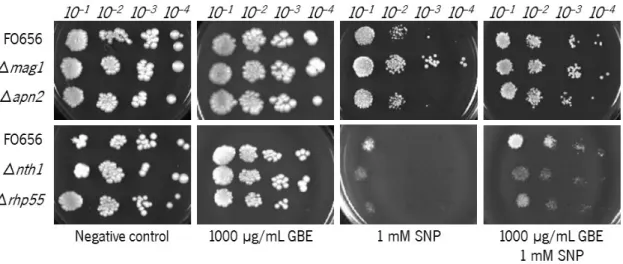
incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...27 Figure 10 – Viability of FO656, Δmag1, Δrhp51, Δnth1 and Δrhp55Sch. pombe cells when cultured on solid medium incorporated with (from left to right): ethanol(negative control), 1000 µg/mL GBE, 1 mM SNP and both 1000 µg/mL GBE and 1 mM SNP. Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the
top of the photos). The plates were incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...28
xv Figure 11 – Viability of 972, Δchk1, Δsty1 and Δpap1 Sch. pombe cells when cultured in solid medium incorporated with (from left to right): ethanol(negative control), 1000 µg/mL GBE, 1 mM SNP and both 1000 µg/mL GBE and 1 mM SNP. Cells were diluted up to 10–4 before pipetting onto the plates (indicated at the top of
the photos). The plates were incubated for 4 days at 30 °C. The photographs are representative of three independent experiments. ...29 Figure 12 – Cell cycle progression analysis of Sch. pombe cells under the different treatments. Before analysis, the cells were treated with hydroxyurea (HU) in order to synchronize their cell cycle and were compared with an asynchronous culture (top left). After washing HU, the cells were submitted to their respective treatments: water (negative control), 750 µg/mL DVE, 1 mM SNP and both 750 µg/mL DVE and 1 mM SNP. Every 30 min, a sample of each treatment was taken and processed for analysis. During the treatments, the cells were incubated at the standard conditions of 30 °C, 200 rpm. ...31 Figure 13 – Microphotographs of fluorescence microscopy of sty1::GFP transformed Sch. pombe after 0, 10 and 60 min of incubation in liquid minimal medium containing (from left to right): water (negative control), 750 µg/mL DVE, 4 mM SNP and both 750 µg/mL DVE and 4 mM SNP. The photographs are representative of three independent experiments. Magnification: 1000x. ...33 Figure 14 – Microphotographs of fluorescence microscopy of of pap1::GFP transformed Sch. pombe after 0, 10 and 60 min of incubation in liquid minimal medium containing (from left to right): water (negative control), 750 µg/mL DVE, 4 mM SNP and both 750 µg/mL DVE and 4 mM SNP. The photographs are representative of three independent experiments. Magnification: 1000x. ...34 Figure 15 – Microphotographs of fluorescence microscopy of of pap1::GFP transformed Sch. pombe after 0, 10 and 60 min of incubation in liquid medium containing (from left to right): water (negative control), 1000 µg/mL GBE, 4 mM SNP and both 1000 µg/mL GBE and 4 mM SNP. The photographs are representative of three independent experiments. Magnification: 1000x. ...35
xvii
List of tables
Table 1 – Genotypes of the strains used in this study. ...15 Table 2 – Complete list of the tentatively identified compounds for DVE. ...21 Table 3 – Complete list of the tentatively identified compounds for GBE. Unidentified compounds that were also detected in other studies have (?) as their identification. ...22 Table 4 – Values of IC50 (µg/mL) determined for DVE, GBE and standards in DPPH reduction, ICA, and NO
1
1. Introduction
1.1. DNA damage and repair
DNA damage is any chemical or physical alteration in DNA, such as base modification, helix breakage, adduct formation, among others. Any chemical agent able to cause any sort of DNA damage is termed as genotoxic. However, DNA damage does not arise exclusively from this kind of agents, since it may also be spontaneously generated within the cell during normal metabolism (Roger et al., 2006). The consequences that arise from DNA damage range between point mutations in the DNA sequence and chromosomal breakage, which might easily result in cell death. In case the existence of the several DNA repair pathways is not enough to prove the importance of this molecule, many researchers have extensively studied and documented the harmful effects of DNA repair impairments, proving that the maintenance of genome stability is crucial to survival. There are five major DNA repair pathways (Figure 1): base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), non-homologous end joining (NHEJ) and homologous recombination (HR); each one being more adequate than the others to repair a specific type of DNA damage. These repair pathways are highly conserved among eukaryotes,
as their central components have been identified in many organisms, including Schizosaccharomyces
pombe. In contrast, only a few organisms possess UV-damaged DNA endonuclease-dependent excision
repair (UVER), including Sch. pombe, which, on the other hand, lacks the ability to directly reverse UV
lesions by photolyase (Richard. Egel, 2004).
Figure 1 – DNA damage and respective repair pathways in Sch. pombe. Mismatched bases (except C/C) and loops that occur during replication and recombination are mainly repaired by mismatch repair pathway. Nucleotide excision repair and endonuclease-dependent excision repair pathways are assembled to remove bulky DNA adducts caused by exposure to UV radiation; these two pathways have also been associated with other types of DNA damage (dashed arrows). Base excision repair is the most competent pathway to repair non-bulky modifications that occur
2 from base modifications or incorporation of modified bases. Finally, homologous recombination and non-homologous end joining repair double-strand breaks. From Egel, 2004.
The BER pathway (Figure 2) is mainly directed at oxidized and alkylated bases, as well as some types of mismatched bases that result during replication or through deamination (Kanamitsu & Ikeda, 2010). BER is initiated by a DNA glycosylase, which recognizes and nicks the N-glycosylic bond between the damaged base and deoxyribose to release the damaged base, creating an apyrimidinic/apurinic (AP) site in the process (H E Krokan, Standal, & Slupphaug, 1997; Hans E Krokan & Bjørås, 2013). Apyrimidinic/apurinic sites can also occur by spontaneous hydrolysis, representing a DNA lesion as well (Richard. Egel, 2004). AP sites are resolved by cleaving the 5’ phosphodiester bond of the lesion, by activity of AP endonucleases or by bifunctional DNA glycosylases (both DNA glycosylase and AP lyase
activities). Then, a DNA polymerase β fills the gap and the process is concluded after ligation of the
remaining nick by a DNA ligase; this process comprises the short-patch BER. In long-patch BER, between 2 and 8 nucleotides 3’ of the lesion are removed instead and polymerase or , through DNA synthesis, create a flap structure, which is processed by flap endonuclease Rad2. Repair by long-patch BER is completed by DNA ligase (Richard. Egel, 2004). There is also a proposed repair pathway, independent of AP endonucleases, which is initiated by bifunctional DNA glycosylase nth1p, followed by tyrosyl phosphodiesterase Tdp1 and concluded by activity of DNA polymerase and DNA ligase (Nilsen, Forstrøm, Bjørås, & Alseth, 2012).
So far, there is knowledge of about six genes coding for BER glycosylases: ung1, thp1, mag1, mag2, myh1 and nth1, the latter being the only bifunctional DNA glycosylase in Sch. pombe; and two AP
endonucleases: apn1 and apn2 (Richard. Egel, 2004). Ung1 is suggested to remove misincorporated
uracil, while Thp1 is suggested to repair uracil residues resulting from deamination of cytosine (Richard. Egel, 2004). Mag1 is directed to repair alkylated bases (Memisoglu & Samson, 1996). Its paralogous, Mag2 presents high sequence and active site similarity to mag1, however, it was suggested that, alone, it might stall at the damaged site without tackling the lesion (Adhikary, Cato, McGary, Rokas, & Eichman, 2013). On the other hand, Mag2 appears to contribute in the resistance to MMS-induced damage as much as Mag1 (Kanamitsu, Tanihigashi, Tanita, Inatani, & Ikeda, 2007). Myh1 is suggested to prevent mutations by removal of misincorporated adenines from G/A, C/A and 8-oxoG/A mismatches (Lu & Fawcett, 1998). Lastly, Apn1, is the major AP endonuclease in Saccharomyces cerevisiae but not in Sch. pombe nor humans, where it serves as back-up for Apn2, the exact opposite of what happens in the budding yeast model (Ribar, Izumi, & Mitra, 2004).
3 Figure 2 – Base excision repair (BER) pathway in Sch. pombe. Apyrimidinic/apurinic sites are primarily repaired through short-patch BER (thicker arrows): Nth1 initiates repair by incising the AP site, which results in the formation of 3 -dRP and 5 -P ends; the blocked 3’-end is converted in a 3’-OH by Apn2; DNA polymerase and DNA ligase fill the gap and seal the nick, respectively. In long-patch BER, Apn2 is the initiator and incises the AP site, leaving a 5’-dRP end; DNA polymerase synthesizes 2-8 new nucleotides and displaces the damage-containing oligonucleotide, which is cleaved by Rad2; DNA ligase completes repair by sealing the nick. Besides synthesizing DNA, Pol4 is able to cleave 5’-dRP, only needing DNA polymerase and DNA ligase to complete the process. The AP endonuclease independent repair pathway is initiated by Nth1 and followed by Tdp1, which cleaves 3’-dRP, leaving a 3’-P to be processed by Pnk1 and completed by DNA polymerase and DNA ligase. From Nilsen et al., 2012.
MMR in not very well understood in Sch. pombe and consequently, its functioning will be resumed for eukaryotes in general. Repair is initiated by MutS (or MutS ) binding to base-base mismatches and small loops, following binding of MutL . This ternary complex suffers a conformational switch and unbounds the sliding clamp from the mismatch site. The clamp may slide upstream or downstream from the mismatch site and accordingly, specific factors direct repair. EXO1 is recruited to degrade the mismatch-containing DNA strand and MutL delimits the extension of EXO1 activity. Pol and DNA ligase I conclude the repair process (Jiricny, 2006).
The NER pathway is specialized on the removal of larger lesions capable of impairing DNA replication and transcription, and is particularly important in the prevention of damage by UV radiation. UV irradiation results in cyclobutane pyrimidine dimers (CPDs) and the 6-4 photoproducts (6-4 PPs) (Balajee, 1999; Cadet, Sage, & Douki, 2005; Roger et al., 2006), the latter being the most mutagenic (Roger et al., 2006).
4
Impairments in this repair pathway in humans is associated to some genetic disorders, including xeroderma pigmentosum. This disease is characterized by an extreme sensitivity to sunlight and by cancer predisposition (Roger et al., 2006).
The two sub-pathways that comprise NER are global-genome NER (GG-NER) and transcription-coupled NER (TC-NER). GG-NER is independent of transcription status and chromatin structure and it repairs the non-transcribed strand of genes and non-coding sequences in a very slow reaction. TC-NER, on the other hand, is responsible for the fast damage removal from active genes and is triggered when a lesion in the transcribed strand blocks RNA polymerase II (Boiteux & Jinks-Robertson, 2013; Richard. Egel, 2004). Several Sch. pombe NER factors required for damage repair have known homologs in other organisms, indicating this pathway is similar between eukaryotes (Richard. Egel, 2004).
UV-induced DNA damage may also be repaired through a NER-independent pathway, UVER, in which Uve1 is the key factor (Bowman et al., 1994; Yonemasu et al., 1997). Uve1 has endonuclease activity, being able to incise 5’ of CPDs, 6-4PP, AP sites, and less efficiently 1,2 GpG (cisplatin-induced diadduct between two adjacent guanines) intrastrand cross-links (Avery et al., 1999; Bowman et al., 1994; Yajima et al., 1995). Some reports state that some base modifications, base-base mismatches and loops are also substrates of Uve1 (Avery et al., 1999; Kaur & Doetsch, 2000; Kaur, Fraser, Freyer, Davey, & Doetsch, 1999). After Uve1 incision, Rad2 (partaker in BER) excises the damaged bases, leaving a gap to be filled in by DNA polymerase, with aid by accessory factors proliferating cell nuclear antigen (PCNA) and replication factor C (RFC), and sealed by DNA ligase (Richard. Egel, 2004).
DNA-damaging agents may also induce the indirect formation of double-strand breaks (DSB); as BER removes damaged bases, it creates single-strand breaks (SSB) that if close enough from each other may turn into a DSB. In addition, if SSB repair fails, a DSB will be produced when encountered by a replication fork. DSB may be harmful to the cell, as they can lead to the loss of large chromosome fragments and
destabilize genome integrity due to these large-scale deletions (Raji & Hartsuiker, 2006).
Repair of DSB is accomplished through one of two strategies. DSB ends can simply be joint together, which conduces to incorrect repair in case loss of base pairs have occurred. This strategy corresponds to NHEJ pathway, and due to this likelihood, it is considered error-prone. The second strategy corresponds to HR pathway, which really comprises several repair mechanisms, and it involves invasion of the homologous DNA strand, differing only in the processing of the intermediate structure that forms afterwards. Strand invasion occurs after nucleolytic resection of DNA, by action of Mre11-Rad50-Nbs1
(MRN) complex, Rad32-Rad50-Nbs1 in Sch. pombe, and Exo1, from which results a 3’ single-stranded
5
strand exchange with the homologous double-stranded DNA, serving as a primer for DNA synthesis (Raji & Hartsuiker, 2006). From that point on, HR divides in synthesis-dependent strand annealing (SDSA), double-strand break repair (DSBR) and mus81p-dependent pathways, each one with a specific mechanism of repair (see Figure 3 for more details).
Figure 3 – HR comprises SDSA, DSBR and Mus81-dependent pathways. In SDSA, the distension of the D-loop follows strand invasion, allowing DNA synthesis. The new DNA strand is displaced from the template and returned to the damaged molecule. DSBR pathway, proposed by Szostak et al. (1983), also involves D-loop distention, leading to the capture of a second 3’ overhang and formation of a double Holliday junction (HJ). The resolution of HJ results in gene conversion, with or without associated crossovers. In the last pathway, Mus81 cleaves the invaded DNA strand disrupting the D-loop. The second 3’ overhang is captured and DNA is synthesized. Lastly, Mus81 cleaves the strand of DNA keeping the homologs together, resulting in crossover of the strands. Edited from Raji and Hartsuiker, 2006.
It is noteworthy that Sch. pombe has a very long G2, during which, a sister chromatid is available to use as a repair template; that is, in an asynchronous culture, HR is the predominant repair pathway (Manolis et al., 2001; Raji & Hartsuiker, 2006). Being a haploid organism, Sch. pombe, during G1, lacks
6
a repair template, thus HR cannot be performed in this cycle phase (Prudden et al., 2003; Raji & Hartsuiker, 2006).
The coordination of cell cycle progression plays an important part on the response to DNA damage. Cell cycle checkpoint pathways monitor the integrity of DNA, preventing the cell from advancing before completing the previous stage of cell cycle or without repairing damaged DNA (Lehmann, 1996). In eukaryotes, cell cycle progression is induced by activity of cyclin-dependent kinases (CDKs) (Pines, 1995). Wee1 and Mik1 negatively regulate cdc2p, the CDK controlling cell progression into mitosis, by phosphorylating it at tyrosine 15 and, in turn, Cdc25 phosphatase dephosphorylates cdc2p, activating it (Figure 4) (Lundgren et al., 1991; MacNeill & Nurse, 1997; Millar, McGowan, Lenaers, Jones, & Russell, 1991; Tang, Coleman, & Dunphy, 1993). Upon DNA damage, Wee1 and Mik1 maintain Cdc2 phosphorylation, which led researchers to suggest them to be targets of the DNA damage checkpoint pathway (Capasso et al., 2002; Kharbanda et al., 1994; O’Connell, Raleigh, Verkade, & Nurse, 1997; Rhind, Furnari, & Russell, 1997). Chk1 is an essential kinase in the transduction of a delay signal to the cell cycle machinery upon DNA damage detection (Capasso et al., 2002). Chk1 phosphorylates Wee1and Cdc25, indirectly blocking entrance in mitosis until DNA repair is completed, which improves survival following DNA damage (al-Khodairy et al., 1994; O’Connell, Walworth, & Carr, 2000). It has been
demonstrated that cells lacking chk1, besides failing to delay cell cycle progression, have increased
sensitivity to DNA damaging agents (al-Khodairy et al., 1994; Walworth et al., 1993). Thereby, the intricate association between the mechanisms of DNA repair and cell cycle progression represent a well-organized system dedicated to preserve the DNA molecule against damaging effects.
Figure 4 – DNA damage checkpoint in Sch. pombe. Upon sensing of DNA damage and signal transduction, Chk1 indirectly induces the delay of cell cycle by phosphorylating Wee1 and Cdc25. Wee1 and Mik1 phosphorylate Y15 of Cdc2, resulting in its inactivation. Cdc25, on the other hand, dephosphorylates Cdc2 to activate it and subsequently, induce mitosis. From Baber-Furnari et al., 2000.
7
1.2. Nitric oxide biochemistry
NO is a free radical and a signalling messenger (Dawson, Dawson, & Snyder, 1992), having cytoprotective as well as tumour promoting activity (Habib & Ali, 2011). NO is produced within the cells through conversion of L-arginine to L-citrulline by nitric oxide synthase (NOS) enzymes. Sch. pombe was reported for possessing NOS-like activity, however the genes encoding for that function remain unclear (Astuti, Nasuno, & Takagi, 2016; Kig & Temizkan, 2009). There are three known isoforms of NOS in mammals: NOS1 or neuronal nitric oxide synthase (nNOS), NOS2 or inducible nitric oxide synthase (iNOS) and NOS3 or endothelial nitric oxide synthase (eNOS) (Habib & Ali; 2011).
Both nNOS and eNOS are Ca2+ dependent and constitutively expressed, whereas iNOS is Ca2+
independent and is activated by cytokine exposure (Moncada, Palmer, & Higgs, 1991). nNOS is expressed
in the neurons, more precisely, in its post synaptic terminals. It is activated by the influx of Ca2+ upon
binding of the neurotransmitter glutamate to receptors in the membrane and by membrane depolarization
through the opening of Ca2+ channels (Pfeiffer, Mayer, & Hemmens, 1999). eNOS is expressed in the
endothelium of blood vessels. After synthesis, NO diffuses into smooth muscle cells of blood vessels and induces smooth muscle relaxation resulting in increased blood flow (Habib & Ali, 2011). The last one, iNOS, is mainly expressed in macrophages and is induced by inflammatory stimuli, e.g. cytokines (Gordge, 1998), and has a very important role in organism defence.
Phagocytic cells, such as macrophages and neutrophils, are the leading peacekeepers regarding organism immunity. Upon recognition and internalization of a potential pathogen, these cells become activated and give rise to an antimicrobial response through the synthesis of both reactive oxygen species (ROS) and reactive nitrogen species (RNS), the latter being synthesized by the inducible nitric oxide synthase iNOS (Collette, Zhou, & Lorenz, 2014). NO is implicated in various health conditions, such as ulcerative colitis (Middleton, Shorthouse, & Hunter, 1993), psoriasis (Kolb-Bachofen, Fehsel, Michel, & Ruzicka, 1994), arthritis and systemic lupus erythematosus (SLE) (Belmont et al., 1997). The latter disorder is characterized by an immune activated state where iNOS level is increased in macrophages, splenic and renal tissues, and consequently, NO production is also increased (Weinberg et al., 1994). The immune activated state promotes induction of iNOS in response to cytokines, which are responsible for the regulation of immune and inflammatory responses, and the control of the amplitude and duration of the response (Van der Meide & Schellekens, 1996).
The reactivity of NO mostly relies on its physicochemical properties, such as its small size, lipophilicity and high diffusion rate (Habib & Ali, 2011). Now, the reactive behaviour of the NO molecule, due to its unpaired electron, allows it to react with other free radicals, transition metal ions and some
8
biological molecules, since most of the biological molecules have completely filled orbitals making NO unreactive towards them (Padmaja & Huie, 1993).
NO chemistry involves multiple pathways and processes, such as inhibition of DNA synthesis, damage to mitochondria, loss of cell membrane integrity, apoptosis, changes in cell cycle and occurrence of DNA strand breaks (Burney, Caulfield, Niles, Wishnok, & Tannenbaum, 1999). After trespassing the cell membrane by diffusion, NO can react with non-heme iron or quench tyrosyl radical of ribonucleotide reductase which may lead to inhibition of DNA synthesis (Kwon, Stuehr, & Nathan, 1991; Roy, Lepoivre, Henry, & Fontecave, 1995).
NO effects in the mitochondria involve irreversible inactivation of enzymes, reversible inhibition of respiration and induction of mitochondrial permeability (Murphy, 1999; Sarti et al., 2003). By reversibly
inhibiting the complex IV of the mitochondria, cytochrome c oxidase, leakage of superoxide (O2•) from the
electron transport chain may occur (Cleeter, Cooper, Darley-Usmar, Moncada, & Schapira, 1994). Other important proteins, such as p53, can be affected by this molecule and initiate programmed cell death. p53, also referred to as the guardian of the genome, also takes part in the maintenance of genome stability as exposure of DNA damaging agents cause the rapid increase and accumulation of p53 in the cell (Nicotera, Bonfoco, & Brüne, 1995). The accumulation of p53 precedes DNA fragmentation
and later apoptosis. NO inhibitors such as NMMA (NG-monomethyl-L-arginine) prevent both p53
accumulation and inducible NO generation thus avoiding to apoptosis (Nicotera et al., 1995).
As mentioned previously, the toxic effects of NO mostly involve its oxidation products. The reaction
of NO with superoxide results in peroxynitrite (ONOO-). This molecule may be easily formed in the
mitochondria where superoxide continuously result as a product of respiration. It is a potent oxidant and
can react with and oxidize almost all biological molecules (Pfeiffer et al., 1999). ONOO– inhibits
mitochondrial enzymes, such as complex I, complex II, cytochrome c oxidase, the ATP synthase, aconitase, Mn-SOD, creatine kinase, and probably many other proteins (Brown, 1999; Cassina & Radi, 1996; Murphy, 1999; Wolosker, Panizzutti, & Engelender, 1996).
The conjugate acid form (ONOOH) can diffuse through membranes and therefore cause damage at
a distance (Marla, Lee, & Groves, 1997). The formation of ONOO– is controlled by the relative amounts of
NO andsuperoxide, and in the excess of either one of these compounds ONOO– is converted to nitrogen
dioxide (NO2), a stable product (Habib & Ali, 2011).
NO also reacts with molecular oxygen (O2) either in gas or in aqueous phase, leading respectively, to
production of nitrogen dioxide or nitroxyl anion (NO-) and nitrate (NO
3-) (Martin N Hughes, 2008).
Ultimately, NO–may still react with O
9
More complex reactions involve the oxidation of catechol-estrogens adducts to quinones which are able to reduce oxygen to superoxide, or in the presence of NO-releasing compounds, such as sodium nitroprusside (SNP), diethylamine NONOate (DEA-NO) or spermine NONOate (SPER-NO), lead to
production of ONOO– (Yoshie & Ohshima, 1998). In a similar way, polyhydroxy aromatic compounds,
easily autooxidize into semiquinone radicals to react with dioxygen and generate superoxide, which results
in ONOO– if in combination with a NO-releasing compound (Yoshie & Ohshima, 1997).
Additionally, it has been reported that the quinone derivative of catechol-estrogen, produced by NO-mediated oxidation, may form adducts with nucleophilic groups of DNA (Dwivedy, Devanesan, Cremonesi, Rogan, & Cavalieri, 1992). The human uterus and breast are sites of production of catechol-estrogens (Yager & Liehr, 1996), and consequently, it was suggested a possible mechanism of hormonal carcinogenesis associated with these organs (Khan, Alam, & Moinuddin, 2007; Liehr et al., 1995; Liehr & Ricci, 1996). It has been shown that incubation with both NO-donor compound and polyhydroxy aromatic compounds resulted in strand breakage of DNA (Habib & Ali, 2011).
As it can be noticed already by the examples provided above, NO chemical network involves a surprisingly wide range of molecules and diverse direct and indirect reactions, evidencing its potential to disturb the functioning of the cell. The adverse effects of NO do not end here however, as it even affects DNA stability and its mechanisms of repair. NO exposure leads to deamination of DNA bases, causing conversion of cytosine to uracil, guanine to xanthine and oxanosine, adenine to hypoxanthine and 5-methylcytosine to thymine. The deamination process (T. Nguyen et al., 1992; D A Wink et al., 1991) is
caused by N2O3, a powerful nitrosating agent, formed by reaction of NO and molecular oxygen (Lee &
Pfeifer, 2007). Evidence indicates that xanthine is able to base-pair with all four natural DNA bases (Eritja et al., 1986; Kamiya, Shimizu, Suzuki, Inoue, & Ohtsuka, 1992). Hypoxanthine, on the other hand, is indicated to preferentially base-pair with cytosine (Vilaivan et al., 2013).
All the mispairing might lead to the transcription of mutant or aberrant proteins and affect the metabolism of the cell. Furthermore, the instability of both xanthine and hypoxhantine leads to their depurination and, consequently, single-strand breakage (Lindahl & Andersson, 1972). Amongst all of the
compounds oxidized by ONOO-, DNA bases may too suffer modification, preferentially guanine, producing
8-nitro-2’-deoxyguanosine (8-nitro-dG), which can depurinate and create an AP site; 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG), 5-guanidino-4-nitroimidazole, and 2,2-diamino-4-[(2-deoxy- -D-erythro-pentofuranosyl)amino]-5(2H)-oxazolone (oxazolone) (Dedon & Barth, 2007). 8-oxo-dG efficiently
undergoes further oxidation originating several other products with potential to cause G→T transversions,
10
RNS, in general, have high affinity to amino acids with thiol groups, making them capable of inhibiting
enzymes having these residues (David A. Wink et al., 1994). DNA-alkyltransferase, which repairs O6
-alkylguanine back to guanine, is inhibited by nitrosation of its thiol residue (Ling-Ling et al., 1992; Zak, Kleibl, & Laval, 1994), and so does, formamidopyrimidine glycosylase, which ends with the removal of Zn from its zinc finger (D A Wink & Laval, 1994). Another DNA repair enzyme inhibited by RNS is DNA ligase by nitrosation of lysine (D. Wink, 1998). DNA repair mechanisms may themselves create breaks in DNA after the removal of the damaged base, when AP endonucleases cleave DNA at the AP site (Eastman & Barry, 1992), creating single-strand breaks with 3’- or 5’- blocked ends that prevent DNA polymerase or DNA ligase to perform their activities (H E Krokan et al., 1997). With DNA repair mechanisms inhibited, single-strand breaks will eventually turn into double-strands breaks, further bringing the cell closer to death (Tamir, deRojas-Walker, Wishnok, & Tannenbaum, 1996).
It is important to recall the cytoprotective role of NO in the cell when present in physiological concentrations, as NO is capable of dealing with intracellular pathogens and block viral replication. NO is also reported to protect DNA against oxidative stress and can actually prevent ROS-mediated DNA damage by inhibiting Fenton reaction of hydrogen peroxide, which would lead to generation of the radical •OH. In turn, this extremely reactive molecule would be available to react with almost all cell components, including DNA (David A. Wink & Ford, 1995).
1.3. Dittrichia viscosa (L.) Greuter and Ginkgo biloba (L.)
D. viscosa and G. biloba, like many others, belong to a very interesting group of plants with fascinating properties. Altogether, they have grown a considerable reputation due to their potential benefits to human health. Plants with such properties are most of the times referred to as medicinal plants. The evidence gathered among all of them suggests various possible applications, regarding, for example, diabetes (Gushiken et al., 2016), gastro-duodenal disorders (Parolin, Ion Scotta, & Bresch, 2014) and age-related diseases (Apetz, Munch, Govindaraghavan, & Gyengesi, 2014; Bittencourt et al., 2013; Mahdy et al., 2012), making them interesting, but mostly useful, research targets.
D. viscosa is an herbaceous plant species, mostly common in Mediterranean areas. Due to its high resistance to adverse conditions and environmental stresses, it frequently occurs in poor or degraded soils, such as roadsides, abandoned fields and dry riverbeds (Parolin et al., 2014). D. viscosa has been used for many years in traditional medicine to treat wounds, bruises, and intestinal disorders (De Laurentis, Losacco, Milillo, & Lai, 2002). A wide range of activities has been reported for D. viscosa, which include inflammatory, antidiabetic, antipyretic, antiseptic, balsamic, viral, fungal and
anti-11
bacterial (Barrero, Herrador, Arteaga, & Catalán, 2008; Lauro & Rolih, 1990). This species is mostly rich in sesquiterpenes and flavonoids, the latter being associated with its antioxidant properties, through
which, D. viscosa is suggested to inhibit lipid peroxidation and also to scavenge superoxide radical
(Schinella, Tournier, Prieto, de Buschiazzo, & Rı́ os, 2002). The antioxidant activity of flavonoids (Figure
5) is dependent on the number as well as the arrangement of their hydroxyl groups (OH) (Heim, Tagliaferro, & Bobilya, 2002). Flavonoid polymers such as proanthocyanidins, were demonstrated to be powerful antioxidants in vitro, due to their high number of hydroxyl groups (Lotito et al., 2000). Some structural features such as ortho-dihydroxy structure in the B ring, 2,3-double bonds in conjugation with the 4-oxo group and hydroxyl groups at positions 3 and 5 were indicated to grant efficient radical scavenging activities (Procházková, Boušová, & Wilhelmová, 2011).
Figure 5 – Chemical structure of quercetin. The three main features required for the antioxidant activity of flavonoids are the ortho-dihydroxy structure in the B ring which facilitates electron delocalization (A), 2,3-double bond in conjugation with the 4-oxo(=O) function in the C ring to allow electron delocalization from the B ring (B) and the hydroxyl groups at positions 3 and 5, which allow binding of hydrogen to the oxo group (C). From Procházková et al., 2011.
Research involving D. viscosa has also revealed some less desirable effects. A study on root
meristem cells of Allium cepa showed chromosomal aberrations and micronuclei formation upon
treatment with D. viscosa extract. This extract also induced cell death, membrane damage, and the
formation of binucleated cells and ghost cells (Aşkin Celik & Aslantürk, 2010). It is known that D. viscosa
is capable of inhibiting seed germination and root growth of other plant species as a competitive advantage (Levizou, Karageorgou, Psaras, & Manetas, 2002; Parolin et al., 2014), which may be related to the cytotoxic and genotoxic activities referred before. By contrast, during the investigation of the epicuticular material of leaves of D. viscosa, it was determined that this material had strong absorbance in the ultraviolet and that its removal and subsequent leaf exposure to ultraviolet-B (UVB) radiation did not affect stomata function nor caused epidermal browning, suggesting resistance to UVB-induced damage (Stephanou, Manetas, Stephanou, & Manetas, 1995).
G. biloba, one of the few living fossils and the only survivor of the Ginkgophyta division, is popularly known for being among the few surviving species of the atomic bombings of Hiroshima and Nagasaki.
12
This species is currently classified as an endangered species by the IUCN Red List due to the uncertainty of its persistence in the wild and rarity. The plant itself is known to be highly resistant to environmental stresses and microbial diseases (Isah, 2015). Scientific research has produced an overwhelmingly large amount of reports describing its properties and medicinal uses. G. biloba extract (GBE), made from the leaves, is one of the most sold herbal remedies in the current market (Kressmann, Müller, & Blume, 2002), being usually prescribed to treat blood disorders, such as cerebral insufficiency and peripheral vascular disease, and to improve memory function (Abad, Bedoya, & Bermejo, 2010; Isah, 2015). The demand of G. biloba pharmaceutical formulations led to the large-scale cultivation of this tree in countries such as the United States of America, China and France (Isah, 2015).
Due to its many active compounds, G. biloba has attracted the interest of many researchers. G.
biloba is stated to protect against free radicals, delaying the progress of diabetes and dementia, improve the cognitive function of Alzheimer’s disease patients and to benefit the treatment of Parkinson’s disease (Cheng, Xu, & Wang, 2009; M.-S. Kim, Lee, Lee, & Kim, 2004; Weinmann, Roll, Schwarzbach, Vauth, & Willich, 2010; Yang, Wang, Sun, Zhang, & Liu, 2016; L. Zhou, Meng, Qian, & Yang, 2011). It is also suggested that it has some beneficial effects on patients suffering angina pectoris but there are also a number of reports defending that more clinical trials are needed to provide more conclusive evidence (Sun, Wang, & Xu, 2015). The antihypertensive effect of GBE has been verified in vitro and in vivo (Kudolo, 2001), which suggested it as a new alternative to the treatment of hypertension, although, its administration in humans did not reduce the incidence of hypertension (Brinkley et al., 2010).
Fairly recently, GBE was shown to protect S. cerevisiae against oxidant and genotoxic agent H2O2
(Marques, Azevedo, Johansson, & Oliveira, 2011), to decrease chromosomal damage induced by radiofrequency radiation (Esmekaya et al., 2011), and lastly, to be a potential preventive cancer agent in BRCA1-mutant ovarian epithelial cells by regulating cell proliferation, tumor suppression, and DNA damage repair pathways (Jiang et al., 2011).
1.4. Schizosaccharomyces pombe
Schizosaccharomyces pombe was originally isolated from millet beer in eastern Africa and first described by Paul Lindner in 1893 as a single-celled rod-shaped yeast. Lindner was able to point that Sch. pombe division precedes the appearance of a wall transversal to the cell, which leads to a constriction that completely separates the two parts. Additionally, because of this new yeast high variability
13
difference relative to Saccharomyces as well as its mean of division (fission) and their common features, such as spore formation and fermentation capacity (Lindner, 1893).
However, as one could possibly assume, the standard strains of today are not related to the strain isolated by Lindner. In 1924, A. Osterwalder isolated and described what he understood to be a new
species, which he named Schizosaccharomyces liquifaciens. Later on, it ended up being renamed
Schizosaccharomyces pombe, strain liquifaciens (Osterwalder). From the collection of this strain, Urs Leopold was able to isolate two homothallic clones, of which only one survived. This strain formed roughly
90% spores in pure culture and so, it was named h90. In the same year, 1950, Leopold also isolated two
heterothallic clones of opposite mating types, named h+ and h- (Munz, Wolf, Kohli, & Leupold, 1989).
In Sch. pombe, the mating-type of a cell is determined by the expression of the P (plus) or M (minus) allele (Richard Egel, 1989). In short, a cluster of genes comprising an expressing locus mat1, and two silent loci mat2-P and mat3-M control mating-type. The transposition of a copy of mat2-P or mat3-M, into the expressing mat1, results in the production of P or M pheromone, determining its mating-type (R Egel,
Beach, & Klar, 1984; Hayles & Nurse, 2016). Through this mechanism h90 cells are capable of switching
mating-types and so, a population of thisstrain comprises about an half of h+ and another half of h- cells,
meaning there is always an available partner to mate (Munz et al., 1989). On the other hand, cells of the
heterothallic strains h+ and h- require the complementary pheromone in order to conjugate, otherwise
vegetative growth is the only mean of propagation (Hayles & Nurse, 2016; Munz et al., 1989). The inability
of h+ and h- strains to switch mating-types is acknowledged to have resulted from single-step events from
a h90 strain, subsequently stabilizing mat1 due to aberrant recombination (Richard Egel, 1989; Munz et
al., 1989). Of a matter of curiosity, despite the cell considering DNA breaks as DNA damage, mating-type
switching involves a site-specific DNA break in the right border of mat1. (Nielsen & Egel, 1989).
The majority of the mutants used nowadays in genetics and molecular biology research are derived
from these strains of close origin, h90, h+ and h-, and are known as 968, 975 and 972, respectively (Munz
et al., 1989). Some of Sch. pombe most appealing characteristics include its ease of culture, short
generation time (2-4 hours), easy manipulation and safe work methods (Giga‐Hama & Kumagai, 1999).
The genome sequencing of Sch. pombe was a fundamental step to encourage research concerning this yeast due to the determination of highly conserved genes important for eukaryotic cell organization, regarding the cytoskeleton, compartmentation, cell-cycle control, protein phosphorylation and RNA splicing, etc. In fact, 67% of the annotated 5059 protein-coding genes are conserved in humans. Fifty of those genes are significantly similar to human-disease related genes, almost half being involved in DNA damage and repair, checkpoint controls and the cell cycle; all processes that highly contribute to maintain
14
genome stability (Wood et al., 2002). Sch. pombe chromosomal sequences did not reveal evidence of
large-scale genome duplications in any of its three chromosomes (Wood et al., 2002). Additionally, the telomeres, centromeres and origins of replication of Sch. pombe are more similar to higher eukaryotes than S. cerevisiae, the other most used yeast in research (Hayles & Nurse, 2016; Wood et al., 2002).
Throughout the years, Sch. pombe has proven itself as a remarkable model organism for the studies of the eukaryotic cell cycle and its regulation (Moser & Russell, 2000; Yamamoto, Imai, & Watanabe, 1997), and DNA repair (Elledge & Zhou, 2000) and recombination (Wixon, 2002; Wood et al., 2002). Sch. pombe has specially contributed to our understanding in microtubule formation (I. M. Hagan & Petersen, 2000), meiotic differentiation (Yamamoto et al., 1997), cellular morphogenesis (Brunner & Nurse, 2000) and polarity (Bähler & Peter, 2000) and stress response mechanisms (Toone & Jones, 1998).
1.5. Scientific problem and objectives
DNA is under constant attack from damaging agents, them being by-products of the cell metabolism or from exogenous origin (Helleday, Eshtad, & Nik-Zainal, 2014). If it were not for the development of DNA repair mechanisms, accumulation of DNA damage would quickly lead to serious cell dysfunction and death. DNA damage checkpoint proteins also play an important role in DNA repair by stalling cell cycle progression, which allows the operation of repair mechanisms. NO, when excessively produced, causes indirect DNA damage and inhibition of proteins involved in its repair. The fission yeast model have provided important insights in many cellular processes, regarding cell cycle and DNA repair, thus being, the righteous subject for this study.
Plant extracts have grown a significant reputation for their potential benefits in human health (Apetz et al., 2014; Bittencourt et al., 2013; Mahdy et al., 2012). Many plant extracts have been studied already and a good number of recent reports provide indications of their favourable effects on numerous conditions and diseases (Esfandiary et al., 2014; Hritcu et al., 2014; Ozarowski et al., 2013; Sutalangka, Wattanathorn, Muchimapura, & Thukham-mee, 2013). The accumulation of DNA damage throughout life may induce alterations in the biological functions of organisms. By changing the expression patterns of proteins, such as transcriptional factors, an abnormal regulation of signalling pathways may be induced resulting in different pathological processes (Sáez, 2016). Unfortunately, not much work have been addressed to the antigenotoxic potential of such extracts (Vilar, Leite, & Chen Chen, 2009). Therefore, the aim of this work, is to investigate if DVE and GBE confer protection against DNA damage upon exposure to NO, and, if possible, also clarify in which way DNA damage protection occurs.
15
2. Materials and Methods
2.1. Yeast strains, media, and growth conditions
In this study, Schizosaccharomyces pombe wild-type haploid strains 972 and FO656 were used;
including DNA repair, oxidative stress-response and DNA damage checkpoint mutants (Table 1). Strains FO656, FO763, RHP502 and FO767 were kindly provided by Ingrun Alseth (Department of Microbiology, Oslo University Hospital, Oslo, Norway); SF869 and SF315 by Stefania Francesconi (Genome Dynamics Unit, Pasteur Institute, Paris, France); and FO661 AV18, AV25, EHH5 and EHH14 provided by Elena Hidalgo (Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain). Strain stocks were kept in 50% glycerol at -80 °C and were cultured every week on solid YD medium (1% w/v yeast extract (Acros Organics), 3% w/v glucose, and 2% w/v agar) at 30 °C and stored at 4 °C after 3 days. All solid media contains 2% w/v agar. Media and growth conditions of each experiment is explained to the detail in the respective subsection.
Table 1 – Genotypes of the strains used in this study.
Strain Genotype Source
972 h–
FO656 h+ ura4-D18 leu1-32 his3-D1 arg3-D4 (Alseth et al., 2004)
FO763 h+ nth1::ura4+ ura4-D18 leu1-32 his3-D1 arg3-D1 (Alseth et al., 2004)
RHP502 apn2::KanMX ura4-D18 leu1-32 his3-D1 arg3-D4+ Magnar Bjørås
FO767 mag1::ura4+ ura4-D18 leu1-32 his3-D1 arg3-D4+ Magnar Bjørås
FO661 h– rhp55Δ::arg3+ 4ura4-D18 leu1-32 his3-D1 arg3-D4+ (Alseth et al., 2004)
SF869 h+ rhp51::ura4 ura4-D18 leu1-32 Stefania Francesconi
SF315 h+ chk1::ura4 leu1-32 ade ura4-D18 Stefania Francesconi
AV18 h– sty1::kanMX6 (Zuin et al., 2005)
AV25 h– pap1::kanMX6 (Zuin et al., 2005)
EHH5 h– leu1 sty1::GFP::kanMX6 (Zuin et al., 2005)
EHH14 h+ his2 ura4 pap1::ura4– leu1 nmt::GFP-pap1::leu1 (Zuin et al., 2005)
2.2. Plant material and extracts preparation
This study relied in the use of extracts prepared from the leaves of D. viscosa and G. biloba. D.
viscosa specimens were identified by Özkan Eren (Department of Biology; University of Adnan Menderes)
16
(November 2007). D. viscosa leaf extract was prepared as described by Çelik T. and Aslantürk O. (2010). The leaves were rinsed with water, dried in a ventilated oven at 55 °C for 24h and then reduced to a fine powder using a blender. The powder was stored at room temperature in the dark. The extract stock solution was prepared to a concentration of 50 mg/mL by boiling the powder with deionized water for 5 min and letting the solution cool down at room temperature for 10 min, followed by filtering with a 0.22 µm syringe filter unit. DVE stock solution was stored at -20 °C until use.
G. biloba leaves were collected at the University of Minho campus (Braga, Portugal) by the end of summer (September 2015) and left to dry out in the dark at room temperature. The leaves were considered dry when they became stiff and broke easily when bent. From that point on, approximately 5 g of leaves (with petiole) were triturated using a blender and put in a cellulose extraction thimble. The powder-containing thimble was immersed in approximately 100 mL of absolute ethanol inside a 250 mL laboratory bottle and left to incubate for 7 days at 30 °C, 200 rpm, in the dark. At the end of the incubation period, the thimble was removed and the ethanolic solution was filtered, followed by evaporation using a rotovapor (RE121+461, Buchi) at 40 °C, 40 rpm in the dark. Afterwards, a small amount of deionized water was added to the evaporated extract to collect any remaining residue and facilitate freezing at -80 °C. After freezing, the extract was put in a freeze dryer (Christ Alpha 2-4, B. Braun) for 4 days in the dark. The resulting powder was kept at -20 °C. GBE stock solution was prepared with absolute ethanol to a concentration of 50 mg/mL and stored at -20 °C until use.
2.3. Electrospray Ionisation-Tandem Mass Spectrometry (ESI-MS/MS)
Both GBE and DVE (100 ppm) were dissolved in 20% methanol and injected in a ion trap mass spectrometer (LXQ Linear, Thermo) equipped with a syringe pump at a flow rate of 5 µL/min and a
vaccum of 5x10–5 Torr. The analyses were performed in the negative selective ion (see Appendix 3 for
more details). The obtained [M-H]– masses were used in the tentative identification of the detected
compounds by literature comparison. The data from the chemical analyses were processed by Xcalibur software.
2.4. In vitro assays
Both DVE and GBE were tested for 2,2-diphenyl-1-picrylhydrazyl (DDPH) reducing, iron-chelating (ICA) and NO scavenging activities. Each extract was tested for five concentrations. The samples were read in a microplate reader (SpectraMaxPlus 284, Molecular Devices). All the assays were performed in triplicate.
17
2.4.1. DPPH assay
DVE and GBE were tested for anti-radical activity. DDPH solution was prepared just before the assay to a concentration of 0.004% (w/v) and kept protected from light at all times. Ten µL of DVE/GBE was added to DPPH-containing wells (reactional volume: 150 µL) and the plate was left in the dark for 60 min before reading at 515 nm. Gallic acid was used as standard. For the blank samples, water and ethanol were used in substitution to DVE and GBE, respectively. The percentage of DPPH reduction was calculated as follows:
% =� − � � ×
Ab – Blank sample absorbance (DPPH) As – Sample absorbance (DPPH and extract)
DPPH reduction percentages were represented in a linear progression with the corresponding extract
concentration and IC50 was calculated. IC50 corresponds to the concentration at which DPPH is reduced by
50%.
2.4.2. Iron-chelating activity
The ICA assay evaluates the capacity to chelate Fe2+. In reaction with
3-(2-pyridyl)-5,6.diphenyl-1,2,4-triazine-4´,4’-disulphonic acid sodium salt (ferrozine), Fe2+ forms a complex that can be quantified by
spectrophotometry. Ferrozine and FeSO4 solutions were prepared right before the experiment and kept
protected from light. Fifty µL of 0.3 mM ferrozine and 50 µL of extract were added to the plate. In order
to prevent bias due to reaction with the extract and ferrozine, Fe2+ is added in last place: 50 µL of 0.06
mM FeSO4 (reactional volume: 150 µL). Ethylenediaminetetraacetic acid (EDTA) was used as standard.
For the blank samples, water and ethanol were used in substitution to DVE and GBE, respectively. The samples were then incubated in the dark at room temperature for 10 min and read at 562 nm. Iron-chelating activity was calculated as follows:
� % =� − � − �� ×
Ac – Control absorbance (ferrozine, iron) Ab – Blank sample absorbance (extract)
18
ICA values were represented in a linear progression with the corresponding extract concentration
and IC50 was calculated. IC50 corresponds to the concentration at which formation of ferrozine-Fe2+ is
reduced by 50%.
2.4.3. NO scavenging
NO scavenging activity can be measured through Griess reaction: in acidic conditions, nitrite reacts
with sulfanilamide and N-1-napthylethylenediamine dihydrochloride (NED) to form an azo compound
detectable by spectrophotometry. All the solutions were prepared in the same day the assay was performed and were kept protected from light. Griess reagent was prepared by mixing 1% w/v
sulfanilamide (dissolved in 5% H3PO4 wt. %) and 0.1% w/v NED SNP in a 1:1 ratio. One hundred µL of
SNP (20 mM) and 100 µL of extract diluted in phosphate buffer KH2PO4 19 mM pH 7.4 were added to
the plate and left under the light for 60 min. One hundred µL of Griess reagent was then added to the samples (reactional volume: 300 µL) and incubated at room temperature in the dark for 10 min and
absorbance read at 562 nm. For the blank samples, 2% H3PO4 was used in substitution to DVE and GBE,
respectively. NO scavenging activity was calculated as follows:
% = � − � − �� ×
Ac – Control absorbance (buffer, SNP and Griess reagent)
Ab – Blank sample absorbance (extract, SNP and H3PO4)
As – Sample absorbance (extract, SNP and Griess reagent)
NO scavenging values were represented in a linear progression according to the corresponding
extract concentration and IC50 was calculated. IC50 corresponds to the concentration at which Griess
reaction is reduced by 50%. 2.5. Viability assays
Cells were grown on YES liquid medium (0.5% yeast extract, 3% glucose and 225 mg/L supplements – adenine, histidine, leucine, lysine hydrochloride and uracil) and incubated at 30 °C, 200 rpm to an
OD600 of 0.4. Cells were submitted to four treatments, consisting in a negative control, extract control,
positive control and co-incubation. Treatments involved incorporation of the compounds in the cooling solid medium, addition to the cellular suspensions or cells pre-treatment with extract and pipetting onto treated medium. The extracts concentration was the same 750 µg/mL for D. viscosa and 1000 µg/mL for G. biloba in all viability assays. For the medium incorporation and pre-treatment assays, 1 mM of SNP
19
was incorporated in the medium and 300 mM was used in the suspension assays. Cell cultures were
serially diluted up to 10–4 before pipetting on the plate. Deionized water was used to replace D. viscosa
extract and SNP, and absolute ethanol was used to replace G. biloba extract, in the corresponding
controls. 2.6. Cell cycle
Cells were grown overnight in YES liquid medium to a concentration of 3–4x106 cells/mL. A sample
of 500 µL was taken from the inoculum, for control purposes, and centrifuged (14,100 rcf), following the discard of the supernatant and fixation of the cells in 500 µL of cold (4 °C) ethanol 70 %. The sample was then stored at 4 °C in the dark until further processing. Every sample in this experiment was processed has previously described.
Afterwards, the cell culture was synchronized in S phase by addition of hydroxyurea (HU) (Sigma) to a final concentration of 12 mM and let to incubate at 30°C, 200 rpm for 4 hours. At the end of incubation time, another control sample was taken. The culture was then centrifuged and washed twice with sterile deionized water to remove HU and another sample was taken for control. After HU removal, the culture was separated in four aliquots and prepared for the four described treatments (GBE at 1000 µg/mL and SNP at 1 mM). Samples of 500 µL were taken from all the treatments every 30 min, including time 0, up to 120 min. After collecting, the samples were washed twice with 50 mM sodium citrate pH 7.5 (4°C). In this step, centrifuging was performed at 4 °C, 2600 rpm for 4 min. The samples remained in 50 mM sodium citrate pH 7.5 and RNAse A (Macherey Nagel Bioanalysis) was added to a final concentration of 0.1 mg/mL, following overnight incubation at 37 °C. Before storage at 4 °C, the samples were sonicated twice for 10 seconds at medium intensity.
Just before analysis, propidium iodide (PI) (Acros Organic, Fisher) was added to a final concentration of 4 µg/mL. In an orderly manner, the three samples taken before the beginning of the treatments served as references for an asynchronous culture, synchronized culture and hydroxyurea-free culture. In addition, a nitrogen starved cell culture was also prepared as a control. Cell cycle analysis was performed in a cytometer (Epics XL, Beckman Coulter) (300 events/s). The results of the experiment were processed using Flowing Software 2.5.1.
20
2.7. Fluorescence microscopy
Cells were grown in EMM (3 g/L potassium hydrogen phthalate, 2.2 g/L Na2HPO4, 5 g/L NH4Cl,
glucose 2% w/v, 52.5 g/L MgCl2.6H2O, 0.735 g/L CaCl2.2H2O, 50 g/L KCl, 2 g/L Na2SO4, 1 g/L
pantothenic acid, 10 g/L nicotinic acid, 10 g/L inositol, 10 mg/L biotin, 5 g/L boric acid, 4 g/L MnSO4,
4 g/L ZnSO4.7H2O, 2 g/L FeCl2.6H2O, 0.4 g/L molybdic acid, 1 g/L KI, 0.4 g/L CuSO4.5H2O and 10 g/L
citric acid) medium to OD600 of 0.4. Before applying treatments, a sample was taken from the inoculum
and prepared for observation in the fluorescence microscope (DM5000B+CTR5000+ebq100, Leica) for control purposes. The cell suspension was divided into 4 aliquots and each was given a different treatment. During the experiment, the four aliquots were incubated at 30 °C, 200 rpm. Samples were taken at 10 and 60 min and immediately observed in the fluorescence microscope.