Efeito de dois Fármacos Antineoplásicos (Cisplatina e
Dimetilaminopartenolídeo) a nível dos Parâmetros de Stresse
Oxidativo no Fígado de Murganhos com Cancro da Bexiga
Induzido com N-Butil-N-(4-Hidroxibutil)-Nitrosamina
Dissertação de Mestrado em Bioquímica
TÂNIA ALEXANDRA FERREIRA DINIS
Orientador: Professor Doutor Carlos Alberto e Silva Venâncio Coorientadora: Professora Doutora Maria Manuel Oliveira
Efeito de dois Fármacos Antineoplásicos (Cisplatina e
Dimetilaminopartenolido) a nível dos Parâmetros de Stresse
Oxidativo no Fígado de Murganhos com Cancro da Bexiga
Induzido com N-Butil-N-(4-Hidroxibutil)-Nitrosamina
Dissertação de Mestrado em Bioquímica
TÂNIA ALEXANDRA FERREIRA DINIS
Orientador: Professor Doutor Carlos Alberto e Silva Venâncio Coorientadora: Professora Doutora Maria Manuel Oliveira
Este trabalho foi expressamente elaborado como dissertação original para o
efeito da obtenção do grau de Mestre em Bioquímica na Universidade de
AGRADECIMENTOS
Ao longo deste percurso académico, principalmente nesta última etapa, foram várias as pessoas que de uma maneira ou de outra me ajudaram a concretizar os meus objetivos.
Agradeço em especial aos dois professores que me acompanharam ao longo deste projeto. Ao Professor Doutor Carlos Venâncio como orientador desta dissertação, pela disponibilidade prestada sempre que necessária. Agradeço também as críticas, comentários e sugestões, assim como todos os conhecimentos transmitidos. À Professora Doutora Maria Manuel Oliveira, como co-orientadora, todo o trabalho e tempo dedicado ao longo destes meses. Agradeço ainda, aos dois, todo o apoio, paciência, compreensão e boa disposição, assim como as críticas e conselhos que garantiram as condições necessárias para a melhoria e concretização deste trabalho. Um muito obrigado!!
À Professora Doutora Carla Amaral, diretora de Licenciatura e de Mestrado em Bioquímica, pela oportunidade de ingressar neste Mestrado que termino com a realização desta dissertação. Agradeço-lhe ainda toda a disponibilidade dedicada ao longo do meu estágio de Licenciatura, no papel de minha orientadora. Foi essencial!
Ao Professor Doutor Francisco Peixoto, à Professora Doutora Amélia Silva e ao Professor Doutor Luís Félix por toda a disponibilidade e ajuda e por todos os conhecimentos que me transmitiram ao longo destes anos. Ao Professor Doutor Rui Costa por toda a ajuda e disponibilização das amostras.
Aos meus amigos e colegas de laboratório, à Andreia e à Daniela em especial por todas as horas que passamos juntas! Ao Tiago, à Mónica e ao Cláudio. Com vocês tudo se tornou mais fácil!
A todos os meus amigos por todo o apoio que me deram nos momentos mais difíceis, brindando-me diariamente com a amizade que nos une, agradeço em particular à Andreia Veloso pelas horas passadas ao meu lado no laboratório e por toda a ajuda.
À minha família, aos meus pais e irmão pelo apoio, compreensão, confiança e esforço que fizeram para eu concluir esta etapa. São tudo para mim!
A todos aqueles que me acompanharam nesta etapa da minha vida, e me fizeram sorrir e aprender, proporcionando-me momentos únicos e que nunca esquecerei.
bexiga representa 7% de todos os cancros, e aproximadamente 2 a 5% de todas as neoplasias malignas. A cisplatina é um dos compostos quimioterapeuticos mais antigos a ser utilizado, nomeadamente no cancro da bexiga. Contudo, o seu uso apresenta limitações, como são exemplo a indução de hepatotoxicidade e o desenvolvimento de resistência das células cancerígenas à sua ação. Estes efeitos secundários estão associados a um aumento das espécies reativas de oxigénio (ROS) nos tecidos. Recentemente foi descoberto que o dimetilaminopartenolídeo (DMAPT), um composto extraído da planta medicinal Tanacetum
parthenium, possui propriedades promissoras antitumorais. A co-administração destes dois
fármacos (cisplatina e DMAPT) suscitam o interesse no tratamento do cancro da bexiga. Neste contexto, o principal objetivo deste trabalho foi verificar se a co-administração da cisplatina e DMAPT não provoca um nível de hepatotoxicidade superior ao observado com a administração da cisplatina, no tratamento do cancro da bexiga de murganhos. Para obter este modelo oncológico, foi utilizado a N-butil-N-(4-hidroxibutil)nitrosamina (BBN), um dos compostos químicos carcinogénicos eficaz na indução experimental de neoplasias da bexiga.
Amostras de fígado foram recolhidas dos grupos experimentais controlo, BBN, cisplatina, DMAPT e com a co-administração cisplatina e DMAPT. Nas amostras, que tinham sido previamente congeladas, foram avaliados vários parâmetros de stresse oxidativo como marcadores de possível hepatotoxicidade: atividade da superóxido dismutase (SOD), catalase (CAT), glutatião-S-tranferase (GST), glutatião redutase (GR), glutationa peroxidase (GPx), o teor em tióis e a peroxidação lipídica. Também foi avaliada a atividade dos complexos mitocondriais (complexos I, II e IV) para determinar possível interferência na mitocôndria. Por último, foi avaliada a atividade do citocromo P450.
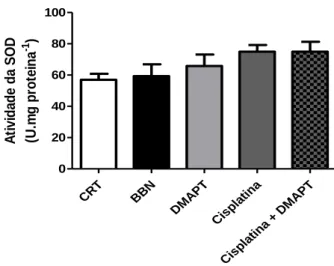
O grupo tratado com BBN e o grupo tratado com DMAPT não apresentaram diferenças em nenhum dos parâmetros avaliados quando comparados com o grupo controlo.
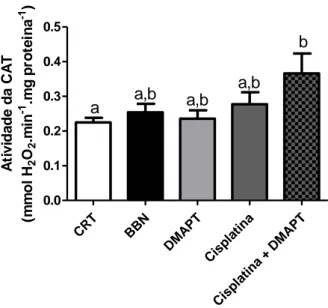
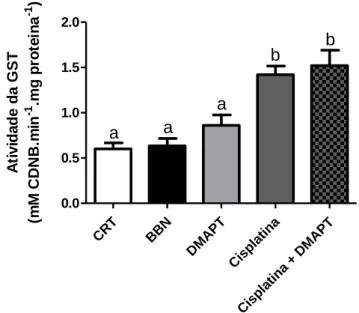
Os resultados obtidos para as atividades das enzimas antioxidantes mostraram que, relativamente ao grupo controlo, houve um aumento da atividade da CAT no grupo tratado com cisplatina + DMAPT; comparativamente ao grupo controlo e ao grupo BBN houve um aumento da atividade da GST e GR nos grupos da cisplatina e cisplatina + DMAPT. Os níveis de GSH encontraram-se aumentados no grupo da cisplatina relativamente ao grupo controlo. A
peroxidação lipídica também se encontrou aumentado no grupo da cisplatina relativamente ao grupo controlo, confirmando-se a indução de stresse oxidativo pela cisplatina.
Na atividade dos complexos mitocondriais e do citocromo P450 não foram observadas
alterações.
Neste trabalho foram pela primeira vez avaliados os efeitos secundários, particularmente alterações nos parâmetros de stresse oxidativo na co-administração de cisplatina e DMAPT.
Este estudo confirma o efeito hepatotóxico da cisplatina, já demonstrada em vários estudos anteriores e que a co-administração do DMAPT induz uma menor produção de ROS a nível hepático e em simultâneo mantem alguma atividade enzimática antioxidante.
Em suma, podemos assim concluir que a co-administração dos dois fármacos induz menos efeitos secundários com base na produção de ROS e que a atividade enzimática antioxidante pode representar um efeito benéfico a nível terapêutico.
Palavras-chave: Cancro da bexiga, cisplatina, dimetilamino-partenolídeo, espécies reativas de
of the most common urinary tract cancers in the world. In Europe, bladder cancer represents 7% of all cancers, and about 2 to 5% of all malignant neoplasm. Cisplatin is one of the oldest compounds used in the treatment of bladder cancer. However, despite its efficacy, it causes a lot of side effects, such as cancer cells’ drug resistance and hepatotoxicity. Several studies have shown that the side effects are associated with the increased number of reactive oxygen species (ROS) in tissues as well as cell resistance. It has recently been discovered that dimethylaminoparthenolide (DMAPT), extracted from the medicinal plant Tanacetum
parthenium, has promising antitumor properties. Thus, there has been an increasing interest in
these two drugs’ association (cisplatin and DMAPT) in the treatment of bladder cancer. In this context, this work’s main motivation was to determine whether the co-administration of cisplatin and DMAPT does or does not cause a higher level of toxicity compared with the administration of cisplatin, in bladder cancer treatment in mice. For this cancer model was used for its induction N-butyl-N- (4-hydroxybutyl) nitrosamine (BBN), one of the most widely used carcinogenic chemical compounds in experimental induction of bladder neoplasms.
Liver samples were collected from the control, BBN, cisplatin treatment, DMAPT treatment and co-administration of cisplatin and DMAPT treatment groups. To achieve the objective proposed in the samples that had been previously frozen, were evaluated several parameters of oxidative stress as possible hepatotoxicity markers: Activity of superoxide dismutase (SOD), catalase (CAT), glutathione-S-transferase (GST), glutathione reductase (GR), glutathione peroxidase (GPx), thiol content and lipid peroxidation. The activity of mitochondrial complexes (complexes I, II and IV) it was also evaluated to determine possible mitochondrial interference and the cytochrome P450 activity.
The group treated with BBN and the group treated with DMAPT did not show any difference in all parameters evaluated when compared with the control group.
The results obtained for the activities of antioxidant enzymes showed that there was an increase in CAT activity in the group treated with cisplatin + DMAPT, when compared with the control group. Compared to the control group and the BBN group there was an increase in GST and GR activity in the cisplatin and cisplatin + DMAPT groups. GSH levels were
was also increased in the cisplatin group when compared to the control group, confirming the induction of oxidative stress by cisplatin.
In the activity of the mitochondrial complexes and in the cytochrome P450 activity it was not observed significant differences.
It was evaluated for the first time in this work the side effects, particularly, the changes in the oxidative parameters of the co-administration of cisplatin and DMAPT.
This study confirms the hepatotoxic effect of cisplatin, already demonstrated in several previous studies, and that the co-administration of DMAPT induces a lower production of ROS at the liver and at the same time maintains some antioxidant enzymatic activity.
Summing up, it was concluded that the co-administration of the two drugs induces fewer side effects based on ROS production and that the antioxidant enzyme activity may represent a beneficial therapeutic effect.
Keywords: Bladder cancer, cisplatin, dimethylamino-parthenolide, reactive oxygen species,
ÍNDICE
AGRADECIMENTOS ... I RESUMO ... V ABSTRACT ... VII ÍNDICE ... IX ÍNDICE DE FIGURAS ... XIII ÍNDICE DE TABELAS ... XIII ABREVIATURAS ... XV
CAPÍTULO I - INTRODUÇÃO ...1
1. O CANCRO ...3
1.1. Biologia do cancro ...3
1.2. O cancro da bexiga ...4
1.3. Indução experimental do cancro da bexiga ...5
2. TERAPIA DO CANCRO ...6
2.1. Quimioterapia ...6
2.2. Cisplatina ...8
2.3. Dimetilaminopartenolídeo ...8
2.4. Potencial utilização de cisplatina e dimetilaminopartenolídeo em associação ... 10
3. FÍGADO ... 11
4.A MITOCÔNDRIA ... 12
4.1. Estrutura ... 12
4.2. Mitocôndria como alvo da ação tóxica de xenobióticos ... 13
5.STRESSE OXIDATIVO ... 15
5.1. Formação de Radicais Livres ... 15
5.2. Defesas antioxidantes ... 17
CAPÍTULO II - OBJETIVOS ... 21
CAPÍTULO III - MATERIAL E MÉTODOS ... 25
1.1. Homogeneização do Fígado para as determinações das enzimas do stresse
oxidativo ... 27
1.2. Quantificação da proteína... 28
2. AVALIAÇÃO DOS MARCADORES DE STRESSE OXIDATIVO ... 29
2.1. Superóxido dismutase (SOD) ... 29
2.2. Catalase (CAT) ... 29
2.3. Glutationa Peroxidase (GPx) ... 30
2.4. Glutatião S-Transferase (GST) ... 30
2.5. Glutatião Redutase (GR) ... 30
2.6. Determinação da peroxidação lipídica avaliada pelo método dos TBARS ... 31
2.7. Quantificação do conteúdo em glutatião ... 31
3. CITOCROMO P450(CYP1A) ... 32
3.1. Homogeneização do fígado para a determinação da atividade do CYP1A ... 32
3.2. Determinação da atividade da CYP1A ... 33
4. DETERMINAÇÃO DA ATIVIDADE DAS ENZIMAS MITOCONDRIAIS ... 33
4.1. NADH:ubiquinona oxidoredutase (complexo I) ... 33
4.2. Succinato-coenzima Q redutase (complexo II) ... 34
4.3. Citocromo c oxidase (complexo IV) ... 35
5. ANÁLISE ESTATÍSTICA ... 35
CAPÍTULO IV - RESULTADOS E DISCUSSÃO... 37
1. AVALIAÇÃO DAS ATIVIDADES ENZIMÁTICAS ... 39
1.1. Superóxido Dismutase (SOD) ... 39
1.2. Catalase (CAT) ... 40
1.3. Glutatião peroxidase (GPx) ... 42
1.4. Glutatião-S-Transferase (GST) ... 44
1.5. Glutatião Redutase (GR) ... 45
1.6. Glutatião nas formas reduzida (GSH) e oxidada (GSSG) ... 47
2. DETERMINAÇÃO DA PEROXIDAÇÃO LIPÍDICA ... 50
3. CITOCROMO P450(CYP1A) ... 52
3.1. Atividade da CYP1A ... 52
4.1. Avaliação do complexo mitocondrial I (NADH: ubiquinona oxidoredutase)
complexo II (Succinato-coenzima Q redutase) e IV (Citocromo c oxidase) ... 54
CAPÍTULO V - CONCLUSÕES FINAIS ... 57
CAPÍTULO VI - BIBLIOGRAFIA ... 63
ÍNDICE DE FIGURAS
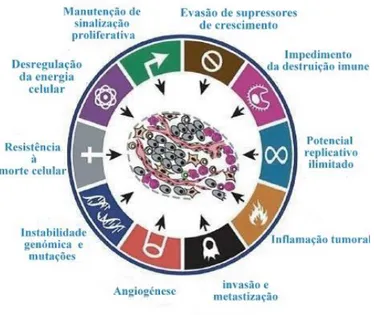
Figura 1- Caraterísticas que as células adquirem para se desenvolver, sobreviver e progredir..4
Figura 2- Uma visão geral das vias canónicas e não-canónicas levando à ativação do fator nuclear NF-ĸβ ...7
Figura 3- Estrutura química da Cisplatina. ...8
Figura 4- Estrutura do dimetilaminopartenolido. ...9
Figura 5 – Produção de ATP pela fosforilação de ADP pela ATP sintase ao longo da membrana mitocondrial interna.. ... 14
Figura 6– Ciclo redox da quinona como mecanismo para gerar ROS ... 16
Figura 7- Enzimas participantes na defesa antioxidante enzimática ... 19
Figura 8 – Atividade da superóxido dismutase (SOD) ... 39
Figura 9 - Atividade da catalase (CAT)... 42
Figura 10 - Atividade da glutationa peroxidase (GPx) ... 43
Figura 11 - Atividade do glutatião-S-Transferase (GST) ... 44
Figura 12 - Atividade do Glutatião Redutase (GR)... 46
Figura 13 – Concentração de GSH (A) e GSSG (B) e a razão entre as duas formas ... 48
Figura 14 - Peroxidação lipídica avaliada pelo método de TBARS. ... 51
Figura 15 - Atividade do citocromo P450 ... 53
Figura 16 - Atividade dos complexos mitocondriais I, II e IV. ... 55
ÍNDICE DE TABELAS
Tabela 1 - Quantificação da proteína pelo método do Biureto. ... 81Tabela 2 - Construção da reta padrão de MDA. ... 81
Tabela 3 - Construção da reta padrão de GSH. ... 82
Tabela 4 - Construção da reta padrão de GSSG. ... 82
ABREVIATURAS
ATP – Adenosina trifosfato
BAFF – Fator de ativação das células B
BBN – N-butil-N-(4-hidroxibutil)-nitrosamina CAT – Catalase CRT – Grupo controlo DCIP – 2,6-diclorofenol-indofenol DDT - Ditiotreitol DMAPT – Dimetilaminopartenolído DTNB – 5,5-ditio-bis-(2-nitrobenzóico)
FADH2 – Flavina adenina dinucleótido, forma reduzida
GPx – Glutationa peroxidase GR – Glutationa redutase GSH – Glutatião reduzido GSSG – Glutatião oxidado GST – Glutationa-S-Transferase H2O2 – Peróxido de hidrogénio
IKK – Complexo IκB cinase i.p. – Intraperitonial
LTβ – Linfotoxina β MDA - Malondialdeído mtDNA – DNA mitocondrial
NADH – Nicotinamida adenina dinucleótido, forma reduzida NEM – N-etilnaleimida
NF-ĸβ – Fator nuclear kappa-ativador
NIK – Via ativação da cinase indutora do NF-ĸβ O2 – Oxigénio molecular
O2.- - Radical anião superóxido
OPT – o-ftaldeído
PAHs – Hidrocarbonetos aromáticos policíclicos PCBs – Bifenilos policlorados
RNS – Espécies reativas de nitrogénio/azoto ROS – Espécies reativas de oxigénio
rpm – Rotações por minuto SOD – Superóxido dismutase TBA – Ácido tiobarbitúrico
TBARS – Substâncias reativas ao ácido tiobarbitúrico TCA – Ácido tricloroacético
TMPD – N’,N’,N’,N’ – tetrametil-1,4-benzenediamina di-hidrocloreto XO – Xantina oxidase
Capítulo I
INTRODUÇÃO
Efeito de dois Fármacos Antineoplásicos (Cisplatina e
Dimetilaminopartenolídeo) a nível dos Parâmetros de Stresse Oxidativo no
Fígado de Murganhos com Cancro da Bexiga Induzido com
1. O cancro
O cancro é uma das doenças com maior taxa de mortalidade. Anualmente, estima-se que sejam diagnosticados cerca de 14 milhões de novos casos [1]. A Organização Mundial de Saúde considera que em 2020 esta será a primeira causa de morte no Mundo. Para reduzir a mortalidade prematura provocada pelo cancro devem existir medidas que facilitem o diagnóstico precoce, o acesso a tratamento e a cuidados paliativos [1,2].
A quimioterapia é atualmente um dos métodos mais utilizados para combater o cancro. No entanto, apesar da sua eficácia, acarreta uma grande quantidade de efeitos secundários que necessitam ser conhecidos e eliminados [3].
1.1. Biologia do cancro
A origem do cancro, assim como os vários aspetos relacionados com a progressão e proliferação tumoral continuam por clarificar [1]. Existe uma grande necessidade de identificar os mecanismos moleculares e as vias de sinalização essenciais à formação e à sobrevivência das células cancerígenas. Este conhecimento é essencial para que se consigam elaborar terapias mais eficazes e com menos efeitos secundários [2, 4, 5].
As células tumorais apresentam variadas alterações a nível genético, bioenergético e histológico quando comparadas com células normais. Estas alterações podem ser promovidas pela ativação de oncogenes, por mutações em genes supressores tumorais, pela reprogramação metabólica ou por mudanças induzidas pelo tumor no respetivo microambiente [6]. Para que as células se tornem malignas, é necessário a acumulação gradual de várias mutações génicas, resultando na desregulação de múltiplas vias de sinalização, dando possibilidade às células de adquirirem capacidades específicas [7].
A capacidade que as células adquirem para conseguirem sobreviver, proliferar e metastizar foram resumidas em 2000 por Hanahan e Weinberg [8]. Estes dois investigadores descreveram algumas caraterísticas essenciais às células cancerígenas e que ditam o seu comportamento desregulado: autossuficiência em sinais de crescimento; insensibilidade a sinais antiproliferativos; capacidade de encontrar alternativa à apoptose; potencial replicativo ilimitado; ativação sustentada da angiogénese invasão e metastização de tecidos (Figura 1) [9].
Existem três palavras que descrevem as várias fases do cancro: eliminação, equilíbrio e inibição. A eliminação refere-se à perda de imunovigilância do cancro, onde as células do
imunitário e o tumor estabelecem um equilíbrio dinâmico que vai ajudá-lo na sua evolução. Por fim a inibição do sistema imunitário ou a resistência das células tumorais, desencadeando o seu desenvolvimento e proliferação [9, 10, 11].
Figura 1- Caraterísticas que as células adquirem para se desenvolver, sobreviver e progredir. Adaptado de [9].
1.2. O cancro da bexiga
Os primeiros arquivos históricos que sugerem a ocorrência de doenças da bexiga são os registos da civilização Egípcia a documentarem a ocorrência de urina de cor escura nas pessoas idosas, presumivelmente hematúria. O primeiro caso de cancro da bexiga foi descrito por Ferri em 1552 [12]. As taxas mais elevadas de cancro da bexiga são encontradas na Europa, América e no Norte de África [13]. O cancro na bexiga é considerado um dos cancros a nível do trato urinário, mais comuns a nível mundial, com uma maior incidência no sexo masculino do que no feminino [14, 15]. Na Europa, o aparecimento de cancro da bexiga representa 7% de todos os cancros, sendo que representa aproximadamente 2 a 5% de todas as neoplasias malignas [16]. No entanto, desde os anos 90, tem sido observado um declínio na taxa de mortalidade (particularmente nos homens), talvez devido à redução da prevalência do tabagismo nos países ocidentais e a menor exposições em zonas propícias a agentes que podem causar cancro da bexiga [17]. O fumo do tabaco e produtos químicos industriais específicos, como por exemplo os nitratos, representam os fatores de risco exógenos mais importantes associados ao cancro da bexiga [13, 18].
O sucesso do seu tratamento e prognóstico estão intimamente relacionados com o grau histopatológico e estado clínico do tumor [3]. O aparecimento deste tipo de cancro é maioritariamente provocado pelo tabaco, uma vez que este possui uma série de compostos cancerígenos, sendo um deles o N-butil-N-(4-hidroxibutil)nitrosamina (BBN) (produto derivado metabolicamente de um composto N-nitroso encontado no fumo do tabaco) que tem especificidade em provocar cancro na bexiga [3, 14].
1.3. Indução experimental do cancro da bexiga
O trato urinário inferior dos roedores possui estrutura e funções semelhantes ao trato urinário inferior dos Humanos [19]. A mucosa que recobre o trato urinário inferior está classificada como epitélio das células de transição ou urotélio. Na bexiga, o urotélio é constituído por três camadas de células: camada superficial, camada intermédia e camada basal [20, 21, 22]. Considerando que todo o trato urinário é revestido por urotélio seria de prever, teoricamente, que os ureteres e a uretra pudessem desenvolver lesões neoplásicas. Contudo, no rato e no murganho, as neoplasias nestas estruturas são raras, a rápida passagem da urina por elas, em contraste com o seu armazenamento na bexiga, justifica esta diferença [23].
Os modelos mais utilizados para o estudo das neoplasias da bexiga são os roedores [22]. Os roedores possuem certas vantagens na investigação médica: a sua fisiologia e genética são bem conhecidas; são animais económicos e fáceis de manter; quando expostos a compostos químicos carcinogénicos desenvolvem neoplasias num período de tempo, cerca de 8 a 12 meses e, tal como o Homem, são mamíferos [24].
A BBN é um dos compostos químicos carcinogénicos mais utilizados na indução experimental de neoplasias da bexiga, e o seu poder carcinogénico limita-se a este órgão, pois é neste local que se encontram as enzimas que provocam a sua ativação [25]. Por esta razão, muitos têm sido os estudos realizados em cancro da bexiga, induzido pela BBN, nomeadamente em roedores, uma vez que estes apresentam similaridade com a bexiga humana, como referido anteriormente [26, 27].
2. Terapia do cancro
2.1. Quimioterapia
A cirurgia, a radioterapia e a quimioterapia são atualmente os tratamentos mais eficazes e utilizados para combater o cancro [3]. Na quimioterapia está disponível um grande número de compostos, que podem ser divididos em várias categorias: agentes alquilantes (ex: ciclofosfamida, dimetilaminopartenolídeo (DMAPT); antibióticos com ação sobre ácidos nucléicos (ex: doxorrubicina); compostos platinados (ex: cisplatina); inibidores mitóticos (ex: vincristina); antimetabolitos (ex: 5-fluorouracil); modificadores da resposta biológica (ex: interferon) e compostos hormonais (ex: tamoxifeno) [28].
Os tratamentos para combater o cancro têm como objetivo a eliminação de células cancerígenas, sendo que cada tipo de cancro exige uma seleção prévia do fármaco a ser utilizado [3, 29]. No entanto, é frequente o aparecimento de efeitos secundários associados a estes tratamentos, com alteração do funcionamento das células normais e também resistência das células cancerígenas [29, 30].
Em muitos tipos de cancros, verificou-se que a resistência à quimioterapia é fortemente dependente da ativação do fator nuclear kappa-ativador (NF-ĸβ). A via NF-ĸβ e a sua regulação são altamente complexas, podendo levar à invasão do cancro de diversas formas, incluindo resistência à apoptose, alterações metabólicas e modulação do microambiente tumoral através da produção de mediadores inflamatórios [31].
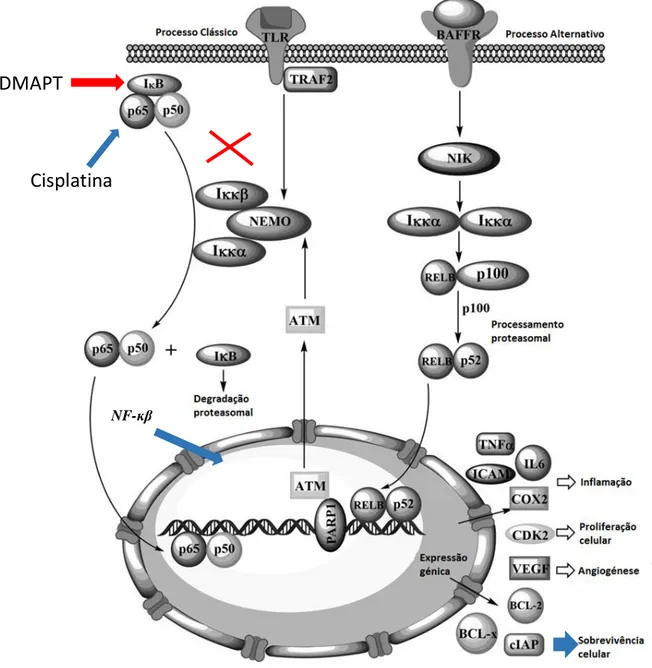
O NF-ĸβ pode ser ativado através de 2 processos principais, o clássico (canónico) e o alternativo (não-canónico) (Figura 2). No processo clássico a célula é ativada por uma variedade de mediadores do sistema imunitário inato e adaptativo, nomeadamente espécies reativas de oxigénio e proteínas virais. Apesar de todos estes estimuladores do NF-κB ligarem-se a diferentes recetores e sinalizarem diferentes proteínas adaptadoras, todos eles convergem na ativação do complexo IκB cinase (IKK), o qual consiste em 2 subunidades catalíticas, IKKα e IKKβ e a subunidade reguladora e estrutural IKKγ, também conhecida por modulador essencial do NF-κB. Uma vez ativo por fosforilação, o complexo IKK fosforila a subunidade IκBα da proteína IκB em 2 resíduos de serina na região N-terminal. Estes eventos de fosforilação marcam a proteína IκB para ubiquitinação pela ação da ubiquitina ligase e subsequente degradação proteolítica. Este processo leva à libertação do NF-ĸβ e à sua consequente translocação para o núcleo, onde regula a transcrição de vários genes, sendo um deles responsável pela sobrevivência/resistência celular. No processo alternativo, a célula é ativada
pelo fator de ativação das células B (BAFFR). Este processo prossegue via ativação da cinase indutora do NF-ĸβ (NIK) que ativa, por fosforilação, o dímero IKKα, o qual fosforila a subunidade p100, induzindo o seu processamento proteolítico a p52. A ativação deste processo leva à translocação nuclear do heterodímero p52/RelB e é particularmente importante na maturação das células B e na formação de órgãos linfóides [31, 32, 33].
Figura 2- Uma visão geral das vias canónicas e não-canónicas levando à ativação do fator nuclear NF-ĸβ.
Adaptado de [31].
Cisplatina
DMAPT
2.2. Cisplatina
A cisplatina ou cis-diaminodicloroplatina (II) (CDDP) é um dos compostos mais antigos a ser utilizado nos tratamentos de diferentes tipos de cancro, nomeadamente no cancro da bexiga. Este faz parte de uma classe de medicamentos antineoplásicos e citotóxicos que têm na sua constituição um átomo de platina (Pt) no centro da molécula, com uma coordenação planar a dois átomos de cloro (Cl) e duas moléculas de amoníaco (NH3), Figura 3 [30, 34, 35].
Figura 3- Estrutura química da Cisplatina.
A cisplatina é uma opção com boa eficácia clínica no tratamento de vários tipos de cancro, particularmente nos pulmões e aparelho urogenital [36]. Contudo, a dose administrada e os intervalos de tempo entre os ciclos de tratamento são uma limitação devido aos efeitos secundários que induz [37], entre eles nefrotoxicidade, neurotoxicidade e hepatotoxicidade [38]. Os efeitos tóxicos da cisplatina devem-se à sua atuação não seletiva sobre as células tumorais e ao seu efeito acumulativo em células e tecidos normais com consequente disfunção quando se atingem concentrações elevadas [39]. Vários estudos mostraram que os efeitos tóxicos estão associados a um aumento das espécies reativas de oxigénio (ROS) nos tecidos e que vários agentes antioxidantes reduzem esses efeitos sem alterar a eficácia terapêutica da cisplatina [40, 41].
2.3. Dimetilaminopartenolídeo
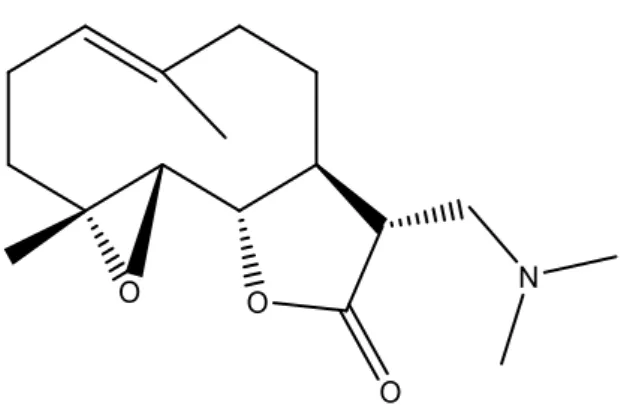
O dimetilaminopartenolídeo (DMAPT) representado na Figura 4 é um análogo do partenolídeo, um sesquiterpeno extraído da erva medicinal tanaceto (Tanacetum parthenium), solúvel em água. Este composto demonstrou em estudos preliminares uma eficácia promissora
nos tratamentos oncológicos, no entanto a ausência de possíveis efeitos secundários ainda não está completamente esclarecida [42].
Figura 4- Estrutura do dimetilaminopartenolido.
Os sesquiterpenos são uma abundante e diversificada classe de metabolitos secundários, pertencentes ao grupo dos terpenóides. Os seus quinze átomos de carbono podem ciclizar e sofrer transformações oxidativas, gerando-se assim diferentes grupos estruturais que são classificados de acordo com o seu esqueleto carbocíclico [43].
O partenolídeo foi dos primeiros compostos de pequeno tamanho a despertar o interesse da comunidade científica para o tratamento de cancro. Todavia, este composto apresentabaixa solubilidade em água o que influencia a sua biodisponibilidade, comprometendo assim a sua administração por via oral [44].
A introdução de um grupo dimetilamina deu origem ao análogo DMAPT. Este composto apresenta uma boa biodisponibilidade podendo, assim, ser administrado oralmente [44, 45]. Adicionalmente, de forma semelhante ao partenolídeo, o DMAPT parece apresentar baixa toxicidade, o que é uma caraterística importante entre os compostos quimioterapêuticos disponíveis [46]. Nas células tumorais as suas principais vias de atuação são a produção de ROS e através da ligação direta às subunidades do fator NF-ĸβ [31] que levará à apoptose das células tumorais [44].
De acordo com a literatura, a apoptose das células tumorais provocada pelo tratamento com o DMAPT deve-se à sua capacidade em induzir nas células um estado de stresse oxidativo. Este composto leva a uma produção de ROS seletivas, ou seja, estas apenas atuam nas células malignas e, deste modo, os seus efeitos tóxicos não prejudicam as células saudáveis [46].
2.4. Potencial utilização de cisplatina e dimetilaminopartenolídeo em associação
As células tumorais desenvolvem mecanismos de resistência a alguns fármacos, durante o tratamento com a cisplatina pela ativação do fator nuclear NF-ĸβ. O tratamento do cancro da bexiga, com a cisplatina, perde frequentemente a sua eficácia pelo simples facto das células tumorais se tornarem resistentes ao fármaco [30, 47]. Como referido anteriormente, a cisplatina induz a formação de ROS que levam à ativação do fator nuclear NF-ĸβ. Este está presente no citoplasma, na forma inativa, e após a sua estimulação, desloca-se para o núcleo, onde promove a ativação da transcrição de genes alvo que conferem a sobrevivência celular às células tumorais [48].
Por este motivo, têm sido realizados diversos estudos de modo a diminuir os efeitos secundários provocados por este fármaco, nomeadamente a associação de outros compostos/fármacos à cisplatina. Ozkol e colaboradores avaliaram a proteção de um composto com propriedades antioxidantes na antagonização dos efeitos secundários do tratamento com a cisplatina [49]. Yu e colaboradores utilizaram o biciclol em conjugação com a cisplatina no tratamento de murganhos portadores de tumores de hepatocarcinoma 22, de modo a verificar se o biciclol diminuía os efeitos secundários provocados pela cisplatina. Estes investigadores observaram que o biciclol pode proteger a membrana celular através da eliminação de radicais livres e redução da apoptose [50].
As principais vias de atuação do DMAPT nas células tumorais é através da produção de ROS e pela ligação direta às subunidades do fator NF-ĸβ e assim tem a capacidade de inibir a ativação do fator NF-ĸβ [44].
O facto de fármacos como o partenolídeo e o DMAPT atacarem principalmente uma parte do tumor, sugere que a sua ação será mais eficaz, no combate à doença, quando combinada com outro tratamento anticancerígeno [44]. Como o DMAPT é um inibidor do fator pela qual as células se tornam resistentes à cisplatina, poderá ser uma boa opção a conjugação dos dois fármacos.
3. Fígado
3.1. A sua importância
O fígado é um órgão fundamental para a sobrevivência porque é essencial para a coordenação do metabolismo, incluindo homeostase da glicose, metabolismo xenobiótico e desintoxicação [51, 52]. O fígado é também um importante local para a síntese de hormonas esteróides e degradação e síntese de proteínas plasmáticas. Ele fornece continuamente energia para todo o corpo, fazendo a gestão do suprimento sistémico de nutrientes. Além disso, o fígado é um mediador da imunidade inata sistémica e um importante local da regulação imunológica [3, 15, 25].
A sua localização estratégica, o fluxo sanguíneo e o papel proeminente no metabolismo dos xenobióticos tornam este órgão particularmente suscetível a lesões por produtos químicos aos quais estamos muitas vezes expostos. A maioria das lesões hepáticas induzidas por xenobióticos é iniciada pela conversão metabólica de substâncias químicas em espécies intermediárias reativas, como compostos eletrófilos ou radicais livres, que podem alterar a estrutura e a função das macromoléculas celulares. Muitas espécies intermediárias reativas podem induzir stresse oxidativo, que pode ser igualmente prejudicial à célula. Quando as defesas protetoras são subjugadas por excesso de substâncias tóxicas, os efeitos das espécies intermediárias reativas levam à desregulação das vias de sinalização celular e à disfunção de biomoléculas e eventual morte celular [15, 25].
A biotransformação é um mecanismo fundamental para a eliminação dos xenobióticos, tornando-os mais hidrofílicos facilitando, assim, a sua excreção e fazendo com que esses compostos não reajam com as estruturas celulares. As reações que ocorrem durante a biotransformação estão divididas em dois grupos: reações de fase I, onde ocorrem reações de oxidação, redução e hidrólise de maneira a tornar o composto mais solúvel e reações de fase II, ou reações de conjugação, que permitem que os produtos resultantes da fase I sejam excretados devido à sua conjugação com outros compostos [53].
São exemplo de xenobióticos os agentes antineoplásicos e estes são fármacos que na sua maioria originam danos a nível hepático.
3.2. Hepatotoxicidade
DMAPT) causam ao nível do fígado. Geralmente, estes compostos geram stresse oxidativo provocando danos nas células saudáveis, levando à sua toxicidade. Os efeitos tóxicos no fígado induzido por estes fármacos, por vezes não resulta do seu efeito direto, mas advém do processo de biotransformação com consequente formação de produtos tóxicos [54].
O fígado é um dos órgãos responsável pela ativação metabólica da BBN. Este composto pode ainda induzir stresse oxidativo uma vez que o metabolismo de eliminação das nitrosaminas ocorre no fígado, e por isso leva à formação de radicais livres. É evidente um estado de stresse oxidativo devido ao facto do BBN atacar a mitocôndria o que faz com que haja uma maior produção de radicais [55].
Durante o tratamento com a cisplatina, o fígado é um dos órgãos mais afetados, uma vez que é neste órgão que ocorre o metabolismo deste fármaco. A elevada quantidade de ROS, induzida por este composto e o consequente stresse oxidativo pode levar a danos no fígado. Por este motivo, a exposição deste órgão à cisplatina, pode levar a uma redução da quantidade de enzimas antioxidantes e um aumento da peroxidação lipídica, traduzido num aumento de malonildialdeído (MDA) [56, 44].
No entanto, a nível hepático existe uma elevada quantidade de mecanismos antioxidantes de maneira a controlar o stresse oxidativo. Por este motivo, a avaliação das enzimas que participam nestes mecanismos é fundamental para estudos/investigações onde se pretende avaliar os danos causados por determinado fármaco ou xenobiótico. Por esta razão, o desenvolvimento de novas soluções terapêuticas eficazes para tratar e reduzir os efeitos secundários provocados pelos tratamentos, constitui uma mais valia na área da biomedicina [57].
4. A mitocôndria
4.1.
EstruturaA mitocôndria é um organelo citoplasmático delimitado por duas membranas: uma externa e lisa, e outra interna formando múltiplas pregas designadas por cristas, que limita um espaço interno aquoso com estrutura gelatinosa devido à presença de múltiplas cópias da cadeia de DNA mitocondrial (mtDNA) e de inúmeras proteínas, designado por matriz mitocondrial [58]. Apesar dos manuais de bioquímica apresentarem, normalmente, a mitocôndria com uma forma oval, estudos de microscopia confocal mostraram que a mitocôndria é uma estrutura muito dinâmica que opera em estreita associação com o citoesqueleto e outros sistemas
membranares, tais como o retículo endoplasmático e o aparelho de Golgi, podendo por isso deslocar-se no espaço intracelular e apresentarem alterações morfológicas constantes [59, 60]. A dinâmica mitocondrial é bem evidenciada pelos processos de fissão (uma mitocôndria divide-se por divisão binária para originar duas) e de fusão (duas ou mais mitocôndrias unem-divide-se originando uma estrutura de maior dimensão) constantes, os quais são fundamentais não só para a manutenção funcional da rede mitocondrial como para garantir o funcionamento e homeostasia celular. A fusão permite a troca de conteúdos mitocondriais de tal modo que a percentagem de mitocôndrias com conteúdo defeituoso (e.g. mtDNA) possa permanecer mínimo, dado que quando o dano mitocondrial atinge um dado limiar as mitocôndrias “defeituosas” são degradadas por mitofagia. Por outro lado, a fissão permite concentrar numa das mitocôndrias “filhas” a maior parte dos componentes danificados da estrutura, permitindo o sequestro e eliminação de mitocôndrias com conteúdo mitocondrial danificado de forma irreversível sem que a dimensão da rede mitocondrial seja comprometida. O equilíbrio entre estes dois processos é muito sensível a várias condições fisiológicas do interior da célula, incluindo a concentração intracelular de iões (e.g. Ca+2) e o estado de oxidação-redução da
célula (e.g. stresse oxidativo) [59].
4.2. Mitocôndria como alvo da ação tóxica de xenobióticos
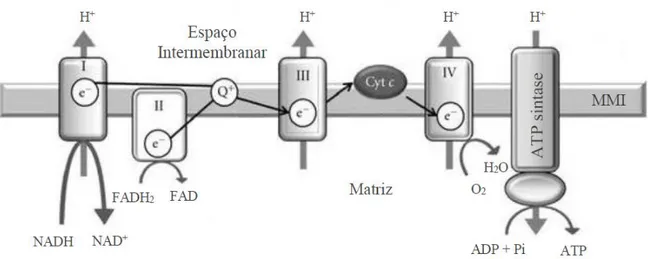
Na fosforilação oxidativa, a síntese de ATP ocorre em simultâneo com a oxidação do NADH e do succinato, substratos produzidos durante o ciclo de Krebs e a β-oxidação dos ácidos gordos. A oxidação do NADH (e do succinato) pelo oxigénio molecular ocorre em etapas sucessivas ao longo de um sistema de transporte de eletrões (impropriamente designado por cadeia respiratória) localizado na membrana mitocondrial interna. A enorme quantidade de energia envolvida neste processo é utilizada para o transporte vetorial de protões, da matriz para o espaço intermembranar. Este transporte vetorial de protões conduz à formação de uma força protomotriz, essencialmente na forma de potencial elétrico transmembranar, utilizado pelo complexo proteico F0F1-ATP-sintase, para a síntese de ATP [61].
O sistema de transporte de eletrões é formado por duas “lanchas” rápidas (elevada mobilidade lateral) que asseguram o transporte dos eletrões entre três complexos proteicos de elevada massa molecular e, consequentemente com reduzida mobilidade lateral (Figura 5). Assim a ubiquinona, uma quinona lipossolúvel que se desloca no interior da membrana lipídica,
citocromo c, uma pequena proteína extrínseca, recebe os eletrões do complexo III e transporta-os, deslocando-se na superfície externa da membrana interna da mitocôndria, para o complexo IV (citocromo-c-oxirredutase), que reduz o oxigénio molecular a água (respiração). Além deste percurso principal há ainda que considerar o abastecimento do sistema respiratório a partir do succinato, que ocorre ao nível do complexo II (succinato:ubiquinona-oxidorredutase), uma enzima membranar que faz parte do ciclo de Krebs. Esta enzima oxida o succinato a fumarato, ficando com o seu grupo prostético reduzido (FADH2 - acrónimo de flavin adenine
dinucleotide), o qual é regenerado pela transferência dos eletrões para a ubiquinona membranar,
os quais seguem depois o percurso indicado acima. Associado ao transporte de eletrões está o transporte vetorial de protões para o espaço intermembranar, contra o gradiente de concentração, ao nível dos complexos I, III e IV, que funcionam como bombas protónicas. Os protões são novamente arrastados eletroforeticamente para a matriz através da componente F0
(canal transmembranar de protões) da F0F1-ATP-sintase e aproveitados pela componente
catalítica F1 (localizada na matriz mitocondrial) para sintetizar ATP a partir de ADP e ácido
ortofosfórico [62, 63, 64].
Figura 5 – Produção de ATP pela fosforilação de ADP pela ATP sintase ao longo da membrana mitocondrial interna (MMI). (Fonte: Adaptado de http://www.hindawi.com/journals/jdr/2012/703538/ Acedido em 8/07/2017).
Existem xenobióticos que atuam diretamente nas mitocôndrias e causam a sua disfunção, quer seja nos seus componentes como nas vias metabólicas nos quais estão envolvidas. Uma das consequências é o excesso de produção de ROS, provocando, assim, um estado de stresse oxidativo [54, 65, 66].
5. Stresse oxidativo
O normal funcionamento das células de organismos aeróbios é acompanhado da produção de radicais livres: as espécies reativas de oxigénio (ROS) bem como as espécies reativas de azoto (RNS) [67]. Estes são normalmente removidos pelas defesas antioxidantes da célula que englobam enzimas e moléculas não enzimáticas. Para o normal funcionamento do organismo é essencial que haja a manutenção do equilíbrio entre a produção de radicais livres e defesas antioxidantes [68]. A perda do equilíbrio entre a formação de ROS e RNS e a capacidade das defesas antioxidantes para neutralizá-los é definido como stresse oxidativo [69, 70, 71]. As ROS são designados como moléculas que apresentam pelo menos um eletrão desemparelhado, portanto necessitam de ganhar estabilidade atacando moléculas vizinhas para obter outro eletrão, danificando assim a estrutura e função celular. A diminuição das defesas antioxidantes leva à formação de danos oxidativos, que estão envolvidos no aparecimento de várias patologias tais como o cancro [69].
5.1. Formação de Radicais Livres
Os radicais livres podem ter origem em fontes endógenas (Figura 6) ou exógenas. Como fontes endógenas encontramos: a cadeia transportadora de eletrões na mitocôndria, os peroxissomas, as NADP(H) oxidases, a xantina oxidase e o metabolismo do citocromo P450. No
que diz respeito à origem exógena, são de destacar os xenobióticos, agentes biológicos (bactérias, vírus), gases poluentes, as radiações UV e ionizantes. Mesmo que existam várias fontes de onde possam ser oriundos os radicais livres, a mitocôndria é a principal no organismo humano [70, 72].
Estas substâncias tanto podem ter um efeito benéfico como um efeito prejudicial. Em níveis apropriados, as ROS são necessárias e benéficas, podendo proteger as células contra infeções através da destruição de agentes patogénicos invasores em locais de inflamação, desempenhar um papel fisiológico como segundos mensageiros, participar na regulação da concentração de cálcio intracelular e funcionarem como moléculas de sinalização na proliferação celular e apoptose [69, 70, 73]. Podem também causar danos oxidativos nos lípidos que resulta em danos nas membranas de organelos celulares.
A exposição a xenobióticos pode levar a um aumento da produção de ROS. Um exemplo dessa produção são as quinonas que podem sofrer ciclos redox levando assim a uma maior
pela sua função antibacteriana em células fagocíticas. Esta enzima pode estar presente noutras células desempenhando atividades importantes de transdução de sinal [64].
Figura 6– Ciclo redox da quinona como mecanismo para gerar ROS. Adaptado de [64].
O O2 tende a receber eletrões, reagindo com iões dos metais de transição ou ao nível da
cadeia mitocondrial de eletrões. Nestas situações ocorre a formação direta do radical anião superóxido (O2∙-).
De acordo com estudos realizados por Haber e Fenton [64], o radical hidroxilo (HO.) poderia ser produzido a partir de uma interação entre o superóxido (O2) e o peróxido de
hidrogénio (H2O2). A reação de Haber-Weiss (Eq. (3)) podia ser um meio através do qual se
geram radicais mais tóxicos, partindo do superóxido, menos reativo, e do peróxido de hidrogénio que poderia ser gerado enzimaticamente. No entanto, reconheceu-se que esta reação é termodinamicamente desfavorável em sistemas biológicos, e exigiria algum tipo de catalisador para ocorrer. Por esta razão, surgiu a necessidade de um catalisador ião metálico, e foi demonstrado que a reação (Eq.(3)) pode ser dividida em duas reações químicas (Eqs. (1) e (2)). Esta reação pode ocorrer em células e é, portanto, uma possível fonte de stresse oxidativo. A reação é muito lenta, no entanto, é catalisada pelo ferro. O primeiro passo do ciclo catalítico envolve a redução do ião férrico ao ferroso. Embora outros iões de metais de transição sejam capazes de catalisar esta reação, a reação de Haber-Weiss catalisada por ferro, que utiliza a química de Fenton, é agora considerada o principal mecanismo pelo qual o radical hidroxilo altamente reativo é gerado em sistemas biológicos [74, 75, 76].
5.2. Defesas antioxidantes
Quando há formação de ROS, é necessário a existência de um mecanismo de defesa responsável pela redução dos efeitos do stresse oxidativo, mecanismo que é levado a cabo por antioxidantes (Figura 7). Portanto, um antioxidante é qualquer substância que vai ser responsável por retardar, prevenir ou eliminar os danos oxidativos [69, 70, 77].
Estas substâncias podem ser sintetizadas in vivo ou podem ser obtidas a partir da dieta localizando-se em diferentes tipos de células e podem ser classificados em defesas enzimáticas e não enzimáticas [68, 69].
Das defesas enzimáticas fazem parte as enzimas superóxido dismutase (SOD), catalase (CAT), glutationa peroxidase (GPx), glutationa redutase (GR). Também pode ser incluída neste grupo a glutatião-s-transferase (GST) porque, embora esteja classificada como uma enzima de fase II, utiliza uma molécula (GSH) muito importante das defesas antioxidantes. As defesas não enzimáticas são constituídas pela glutationa na forma reduzida (GSH), ácido úrico, bilirrubina, coenzima Q, ácido lipóico e melatonina que são endógenas, e por vitaminas tais como a vitamina C e vitamina E, β-caroteno, ácido cafeico, resveratrol e flavonoides como exógenos [70].
A SOD é a enzima responsável por catalisar a dismutação de dois radicais O2∙- em O2 e
H2O2 (Eq.4) [78].
Existem diversas isoformas desta enzima, diferindo entre si na natureza do metal do local ativo, na constituição dos aminoácidos e no número de subunidades e cofatores. Em humanos existem três formas, uma citoplasmática em que os metais são o cobre e o zinco (Cu/Zn-SOD), uma outra mitocondrial com manganês (Mn-SOD) e outra extracelular
(EC-Equação 4
Equação 1
Equação 2
SOD) [78, 79]. As bactérias contêm Mn-SOD e Fe-SOD e em alguns casos também Cu/Zn-SOD. Além destas existe uma outra presente nos procariotas que contem níquel (Ni-SOD) [79].
A CAT é uma enzima que está presente em células de plantas, animais e bactérias aeróbicas, sendo que na célula está localizada nos peroxissomas. Esta enzima é responsável pela conversão do H2O2 em O2 e H2O (Eq.5) [78, 79].
A Glutatião Peroxidase (GPx), uma enzima dependente de selénio, utiliza dois eletrões para reduzir o H2O2 e hidroperóxidos a compostos menos reativos como a água e álcoois. Esta
enzima atua em conjunto com o glutatião (GSH) para transformar o H2O2 e peróxidos orgânicos
(ROOH) em água ou álcoois, respetivamente, oxidando em simultâneo duas moléculas de GSH formando um dímero oxidado, designado por glutationa (GSSG) (Eq.6) [69, 78, 80].
A Glutatião-S-Transferase (GST), uma enzima incluída na fase II dos processos de destoxificação, é responsável pela conjugação do GSH a um elevado número de compostos, tanto hidrofílicos como lipofílicos, produzindo, de um modo geral, compostos menos reativos e mais hidrossóluveis que são mais fáceis de eliminar do organismo. No entanto, esta conjugação pode, por vezes, promover a formação de metabolitos que apesar de mais polares são mais tóxicos (Eq.7) [69].
A Glutatião Redutase (GR) é uma flavoenzima que utiliza o poder redutor do NADPH para regenerar o glutatião reduzido (GSH) a partir dos dímeros oxidados (GSSG). Trata-se de uma reação, essencialmente irreversível, fundamental para garantir uma razão GSH/GSSG elevada (normalmente superior à unidade), a qual é essencial para garantir o ambiente redutor necessário ao funcionamento das células (Eq.8) [69, 78].
Equação 5
Equação 6
O glutatião na forma reduzida (GSH) é um antioxidante intracelular que faz parte do grupo dos não enzimáticos. As principais funções deste antioxidante relativamente à proteção contra o stresse oxidativo são atuar como cofator de várias enzimas desintoxicantes tais como a GPx e a GST; participar no transporte de aminoácidos através da membrana; eliminar radicais hidroxilo e oxigénio singleto; ser capaz de regenerar outros antioxidantes como a vitamina E e C [78].
A capacidade do GSH para regenerar antioxidantes está relacionada com o estado redox da razão GSH/GSSG. O GSH é acumulado no interior das células e a proporção GSH/GSSG é uma medida do stresse oxidativo de um organismo. Uma concentração muito elevada de GSSG pode levar ao dano oxidativo de várias enzimas [68].
Figura 7- Enzimas participantes na defesa antioxidante enzimática. Adaptado de [77].
Capítulo II
OBJETIVOS
Efeito de dois Fármacos Antineoplásicos (Cisplatina e
Dimetilaminopartenolídeo) a nível dos Parâmetros de Stresse Oxidativo no
Fígado de Murganhos com Cancro da Bexiga Induzido com
O objetivo geral deste trabalho foi verificar se a co-administração de cisplatina e DMAPT não provoca um nível de hepatotoxicidade superior ao observado com a administração da cisplatina, no tratamento do cancro da bexiga de murganhos. Assim pretende-se avaliar vários parâmetros de stresse oxidativo como marcadores de possível hepatotoxicidade utilizando o tratamento com cisplatina, DMAPT e cisplatina + DMAPT.
Este trabalho é diferenciador relativamente à maioria dos realizados previamente com estes ou com compostos semelhantes devido à avaliação do stresse oxidativo ter sido realizada num modelo oncológico, neste caso induzido com a BBN.
Assim, este trabalho tem como objetivos específicos analisar as variações nos seguintes parâmetros:
• Atividade enzimática da superóxido dismutase (SOD), catalase (CAT), glutatião-s-tranferase (GST), glutatião redutase (GR) e glutationa peroxidase (GPx);
• Teor em tióis;
• Peroxidação lipídica (TBARs);
• Atividade das enzimas dos complexos mitocondriais (complexos I, II e IV);
Capítulo III
MATERIAL E MÉTODOS
Efeito de dois Fármacos Antineoplásicos (Cisplatina e
Dimetilaminopartenolídeo) a nível dos Parâmetros de Stresse Oxidativo no
Fígado de Murganhos com Cancro da Bexiga Induzido com
1. Modelo animal e amostragem
Este estudo foi realizado em amostras de fígado previamente congeladas a -80 ºC. As amostras foram recolhidas de murganhos do sexo feminino e em idade adulta, utilizados como modelo animal num ensaio experimental para o tratamento do cancro da bexiga.
Os animais foram mantidos durante o ensaio experimental no Biotério da UTAD e todos os procedimentos realizados de acordo com as normas legislativas em vigor, Portuguesas (Decreto-Lei nº 113/2013) e Europeias (Diretiva 2010/63/EU). O alojamento foi realizado em gaiolas coletivas, com cama, condições controladas de temperatura (22±2 ºC) e humidade relativa (50-60 %), ração e água ad libitum e num ciclo de 12h dia/noite.
O ensaio experimental foi constituído por cinco grupos de sete animais distribuídos aleatoriamente e tratados do seguinte modo:
Todos os grupos tratados foram induzidos com a BBN nas primeiras 12 semanas. A ultima administração da cisplatina foi feita 1 semana antes e o DMAPT 24hr antes do sacrifício, respetivamente. Após o sacrifício dos animais, as amostras de fígado foram rapidamente recolhidas e congeladas a -80 ºC até à sua utilização.
1.1. Homogeneização do Fígado para as determinações das enzimas do stresse oxidativo
Para a homogeneização do fígado as amostras foram descongeladas a 4 °C, pesadas e colocadas em copos com tampão fosfato (KH2PO4 50 mM) a pH 7,4. O tecido foi cortado em
Elvejhem e procedeu-se à sua homogeneização num homogeneizador mecânico a 500 rotações/minuto com a finalidade de libertar o conteúdo intracelular.
Os tubos foram colocados numa centrífuga (SIGMA 2-16K) devidamente equilibrados e procedeu-se à sua centrifugação a 1000 g, durante 15 minutos e a 4 °C. No final, os sobrenadantes foram transferidos para novos tubos de centrífuga e procedeu-se a uma nova centrifugação a 14000 g, durante 20 minutos e a 4 °C. O sobrenadante resultante foi transferido para eppendorfs para a quantificação da proteína, avaliação da atividade das enzimas do stresse oxidativo e posteriormente para a quantificação dos tióis. O pellet obtido em cada uma das centrifugações foi ressuspendido em 400 µL de tampão fosfato e foi transferido para eppendorfs e congelado para a determinação da peroxidação lipídica e atividade dos complexos mitocondriais. Todo o processo de obtenção de homogeneizado foi realizado tendo o cuidado de manter todos os acessórios de laboratório e as amostras em gelo granulado, mantendo a temperatura entre 0 e 4 °C.
1.2. Quantificação da proteína
Para quantificação da proteína seguiu-se o método colorimétrico do Biureto [81]. Este método baseia-se na reação do CuSO4 em solução alcalina (reagente de Biureto) com as
ligações amida das proteínas, originando um complexo de cor violeta. A densidade ótica deste complexo, isto é, a sua absorvância, é determinada a um comprimento de onda específico. Foi preparada uma série de padrões de BSA 0,4% (p/v) com as concentrações 0; 0,4 0,8; 1,6 e 2,4 mg de proteína, a que corresponderam os volumes 0, 100, 200, 400 e 600 μL e adicionaram-se 100 μL de dodecilsulfato de sódio (SDS) 10% (p/v) a cada tubo, o volume foi ajustado a 2000 μL com água desmineralizada e procedeu-se finalmente à adição de 2000 μL do reagente de biureto (CuSO4.H2O a 0,15% (p/v), tartarato de sódio e potássio (NaKC4H4O6.4H2O) a 0,6%
(p/v), NaOH a 3% (p/v) e KI a 0,1% (p/v)) perfazendo um volume final de 4 mL. Para as amostras o procedimento foi semelhante, mas com a adição de 50 μL do homogeneizado a quantificar. As absorvâncias dos padrões e das amostras foram lidas após 15 minutos de reação à temperatura de 30ºC, a 540 nm, contra um branco que continha todos os reagentes exceto a proteína. As leituras realizaram-se num espectrofotómetro UV-Vis – Varian Cary 50 (Tabela 1, Anexo A).
2. Avaliação dos marcadores de stresse oxidativo
Foram avaliadas a atividade das seguintes enzimas: superóxido dismutase (SOD), catalase (CAT), glutatião-s-tranferase (GST), glutatião redutase (GR) glutatião peroxidase (GPx), os níveis de tióis GSH e GSSG e a peroxidação lipídica pelo método de TBARS.
2.1. Superóxido dismutase (SOD)
A atividade da SOD (EC 1.15.1.1) foi determinada espetrofotometricamente utilizando o método descrito por Payá e colaboradores [82], com algumas alterações, aplicando o sistema xantina-xantina oxidase na presença de um cromóforo, o cloreto de azul de nitrotetrazólio (NBT).
Numa cuvete de 1 mL foram adicionados 965 μL de tampão (KH2PO4 50 mM e EDTA
1 mM a pH 7,4), 10 μL de NBT 10 mM, 10 μL de hipoxantina 10 mM e 10 μL do homogeneizado de fígado a analisar (10x diluído). A mistura foi incubada durante 2 minutos, e cerca 20 segundos antes de terminar esse tempo adicionaram-se 5 μL de xantina oxidase 5 U/mL, com agitação para garantir a homogeneização da mistura. A cinética da reação foi acompanhada a 560 nm durante 3 minutos, a 30˚C, num espectrofotómetro UV-Vis-Varian Cary 50. A atividade foi expressa em U min-1 mg proteína-1, sendo que uma unidade de
atividade (1U) corresponde a uma redução de 50% da atividade de um ensaio sem amostra (Branco).
2.2. Catalase (CAT)
A atividade da catalase (EC 1.11.16) foi lida num elétrodo de oxigénio do tipo Clark (Hansatech, sistema Oxygraph). O método baseia-se na determinação da velocidade inicial da produção de oxigénio resultante da decomposição do peróxido de hidrogénio pela enzima [83]. Na câmara de reação, termostatizada a 30ºC e sob agitação magnética constante colocaram-se 973 μL de tampão fosfato (KH2PO4 50 mM a pH 7,0) e 10 μL do homogeneizado de fígado
(600x diluído). Após 45 segundos de incubação a reação foi iniciada com a adição de 17 μL de peróxido de hidrogénio (H2O2) 10 mM. A cinética da reação foi acompanhada durante cerca de
2.3. Glutationa Peroxidase (GPx)
A atividade da glutationa peroxidase (GPx) foi determinada espectrofotometricamente utilizando o método descrito por Mannervik [84].
Na cuvete foi adicionado um volume de sobrenadante do fígado equivalente a 0,5 mg de proteína, 5 µL de GR (glutationa redutase), 10 µL de GSH (glutatião reduzido) a 100 mM e um volume de tampão (KH2PO4 100 mM e EDTA 1 mM a pH = 7,0), perfazendo um volume
final de 1 mL. Após incubação de 2 minutos adicionaram-se 10 µL de NADPH 10 mM, 30 segundos antes do final do tempo e agitou-se durantes alguns segundos. Após o registo de cerca de um minuto são adicionados 50 µL de H2O2 a 12 mM. A cinética da reação foi acompanhada
a 340 nm durante 3 minutos a 30˚C, num espectrofotómetro UV-Vis-Varian Cary 50. A atividade é expressa em µM NADPH.min-1 mg de proteína-1 recorrendo-se ao coeficiente de extinção molar do NADPH de 6220 M-1 cm-1.
2.4. Glutatião S-Transferase (GST)
A atividade da GST (EC 2.5.1.18) foi determinada espetrofotometricamente pelo método de Hatton [85]. Numa cuvette de 2 mL colocaram-se 1920 μL de tampão fosfato (KH2PO4 100 mM a pH 7,4), 20 μL de CDNB (1-cloro-2,4-dinitrobenzeno) 100 mM e 20 μL
da amostra a determinar (10x diluída). A mistura reacional foi incubada durante 2 minutos, a 30 °C, e a reação iniciou-se 1 minuto após o tempo de incubação com a adição de 40 μL GSH (glutatião reduzido) a 100 mM. A cinética da reação foi acompanhada a 340 nm ao longo de 3 minutos, num espectrofotómetro UV-Vis-Varian Cary 50. A atividade enzimática foi expressa em μM de CDNB conjugado min-1 mg proteína-1, recorrendo-se ao coeficiente de extinção
molar do CDNB de 9,6 x 103 M-1 cm-1.
2.5. Glutatião Redutase (GR)
A atividade da GR (EC 1.6.4.2) foi determinada espetrofotometricamente de acordo com o método de Carlberg e Mannervik [86]. Numa cuvete de 1 mL foram colocados 955 μL de tampão (KH2PO4 100 mM e EDTA 0,5 mM a pH 7,4) e 25 μL de homogeneizado de fígado.
A mistura foi incubada durante 2 minutos, e poucos segundos antes de terminar esse período adicionaram-se 10 μL de NADPH 10 mM seguido da agitação da cuvete. A reação foi iniciada após 30 segundos com adição de 10 μL de GSSG (glutatião na forma oxidada) 100 mM. A
cinética de reação foi acompanhada através do consumo do NADPH a 340 nm durante 3 minutos, a 30˚C, num espectrofotómetro UV-Vis-Varian Cary 50. A atividade foi expressa em μM de NADPH min-1.mg proteína-1, recorrendo-se ao coeficiente de extinção molar do NADPH
de 6220 M-1 cm-1.
2.6. Determinação da peroxidação lipídica avaliada pelo método dos TBARS
O método utilizado foi baseado no descrito por Ottolenghi [87]. O método de quantificação de substâncias reativas ao ácido tiobarbitúrico (TBARS) é, provavelmente, o método mais utilizado para a quantificação da peroxidação lipídica in vitro. Este método reflete a quantidade de malonildialdeído (MDA) formado, um produto final da peroxidação de ácidos gordos da membrana [88], e baseia-se na reação deste biomarcador de stresse oxidativo com o ácido tiobarbitúrico [89]. Da reação entre ambos resulta um composto de cor amarela, mensurável por análise espectrofotométrica, o qual é diretamente proporcional à concentração de MDA resultante da peroxidação lipídica. Apresenta como vantagens: o baixo custo e a facilidade de execução, é sensível para quantificação da peroxidação lipídica, porém, não é muito específico [90].
O meio de reação é constituído por 50 µL de amostra (pellet 1) e 100 µL de amostra (pellet 2), 550 µL e 500 µL de tampão – Tris-HCl (10 mM) em KCl (175 mM), respetivamente, e 100 µL de uma solução contendo 0,38% TBA (p/v), 37,5% TCA (v/v) e 0,015% BHT (p/v). Após agitação vigorosa, os tubos tapados com esferas de vidro, foram colocados num banho de água a ferver (100ºC) durante cerca de 15 minutos e arrefecidos por imersão em gelo. O seu conteúdo foi transferido para eppendorf’s que foram centrifugados a 3000 r.p.m durante 10 minutos para sedimentação de proteínas e membranas.
De cada eppendorf, foram retirados 200 µL de sobrenadante para uma microplaca de 96 poços e a absorvância foi lida a 530 nm num leitor de microplacas (Power Wave XS2 BioTek). O resultado foi expresso em µM MDA.mg proteína-1, utilizando o coeficiente de extinção molar
de 1,56 x 105M-1.cm-1 (Tabela 2, Anexo B).
2.7. Quantificação do conteúdo em glutatião
Para a quantificação do GSH retiraram-se 30 μL do homogeneizado obtido e adicionou-se um volume de tampão fosfato (KH2PO4 100 mM (pH 8,0) contendo EDTA a 5 mM, de modo
a obter um volume final de 2,3 mL. Seguidamente, adicionaram-se 200 μL da solução de o-ftaldeído (OPT) 1 mg/mL, contendo 200 μg deste composto. A mistura foi homogeneizada, incubada à temperatura de 30ºC e na ausência de luz, durante 15 minutos. Ao fim deste tempo a fluorescência foi lida a um comprimento de onda de emissão de 426 nm, e excitação a 339 nm num espetrofluorímetro Varian Cary Eclipse.
A concentração de GSH na amostra foi calculada por extrapolação com uma curva padrão cujas soluções padrão foram tratadas da mesma forma que as amostras (Tabela 3, Anexo C).
Para quantificar o GSSG retiraram-se 50 μL de homogeneizado aos quais se adicionou um volume de tampão de modo a que o volume final fosse 100 μL. De seguida adicionaram-se 40 μL de N-etilmaleimida (NEM) a 40 mM, homogeneizou-se e incubou-se à temperatura ambiente durante 30 minutos. Ao fim deste período adicionaram-se 1,66 mL de NaOH 100 mM, 500 μL de tampão para um volume final de 2,3 mL e 200 μL da solução de OPT a 1 mg/mL contendo 200 μg deste composto. Homogeneizou-se novamente e incubou-se à temperatura ambiente na ausência de luz durante 15 minutos. Seguidamente leu-se a fluorescência emitida a 426 nm, com excitação a 339 nm, num espetrofluorímetro Varian Cary Eclipse.
A concentração de GSSG na amostra foi calculada por extrapolação com uma curva padrão cujas soluções padrão tiveram o mesmo tratamento das amostras (Tabela 4, AnexoD).
3. Citocromo P450 (CYP1A)
3.1. Homogeneização do fígado para a determinação da atividade do CYP1A
A atividade do CYP1A foi determinada a partir de uma fração rica em microssomas adaptada do método descrito por Völkl e Fahimi [92]. Os fígados, armazenados a -80 °C, foram descongelados e colocados numa solução tampão de homogeneização (KH2PO4 0,1 M e KCl
0,1 M a pH 7,6) a 4 °C. De seguida, cada fígado foi cortado em pequenas porções, lavado em tampão e a solução foi transferida para um Potter-Elvejhem e homogeneizada num homogeneizador mecânico a 700 rotações/minuto. O homogeneizado foi transferido para tubos de centrífuga e procedeu-se à sua centrifugação a 25300 g durante 20 min. Após a centrifugação o precipitado foi descartado e procedeu-se a uma nova centrifugação do sobrenadante, desta
vez a 30000 g durante 20 minutos. Ambas as centrifugações foram realizadas numa centrífuga Sigma 3-16K refrigerada a 4° C. Após a centrifugação os precipitados foram descartados e os sobrenadantes ricos em microssomas recolhidos para determinação da atividade do CYP1A.
3.2. Determinação da atividade da CYP1A
A determinação da atividade da CYP1A foi realizada em eppendorfs de 2 mL. Adicionaram-se 500 μL de amostra a 470 μL de uma solução tampão contendo KH2PO4 100
mM, KCl 150 mM, EDTA 1 mM e DDT 1 mM, seguidamente adicionaram-se 5 μL de etoxiresorufina 400 μM e 25 μL de NADPH 20 mM, iniciando-se a reação. Os eppendorfs foram imediatamente colocados num banho a 25 °C durante 30 minutos ao fim dos quais se adicionou 1,0 mL de metanol arrefecido com o objetivo de parar a reação. Os eppendorfs foram seguidamente transferidos para uma centrifugadora, refrigerada a 4 °C, e centrifugados a 7500
g durante 10 minutos. Finalmente procedeu-se à leitura de cada amostra. A produção de
resorufina é medida no espetrofluorímetro a 530 nm e 585 nm, os comprimentos de onda de excitação e emissão, respetivamente. A atividade catalítica da enzima reflete a quantidade de enzima presente e é expressa em concentração de resorufina produzida min-1.mg de proteína-1. A determinação da atividade da CYP1A nas amostras foi calculada por extrapolação com uma curva padrão cujas soluções padrão foram preparadas apenas com tampão e resorufina (Tabela 5, AnexoD).
4. Determinação da atividade das enzimas mitocondriais
Os ensaios de bioenergética mitocondrial foram realizados com as frações mitocondriais isoladas dos fígados dos ratos utilizados neste trabalho. Antes da realização dos ensaios, cada fração mitocondrial foi submetida a 3 ciclos de congelamento/descongelamento para aumentar o acesso do substrato ao local ativo da respetiva enzima.
4.1. NADH:ubiquinona oxidoredutase (complexo I)
A NADH desidrogenase (E.C.1.6.5.3.), também conhecida por NADH-ubiquinona oxidoredutase ou vulgarmente, complexo I, é uma enzima localizada na membrana interna da mitocôndria que catalisa a oxidação do NADH com a redução da ubiquinona a ubiquinol,
![Figura 7- Enzimas participantes na defesa antioxidante enzimática. Adaptado de [77].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15837359.1083858/41.892.219.715.556.920/figura-enzimas-participantes-na-defesa-antioxidante-enzimática-adaptado.webp)
![Figura 10 - Atividade da glutationa peroxidase (GPx), apresentada em unidades: [NADPH] μM min -1 mg proteína -](https://thumb-eu.123doks.com/thumbv2/123dok_br/15837359.1083858/65.892.268.626.428.772/figura-atividade-glutationa-peroxidase-apresentada-unidades-nadph-proteína.webp)
![Figura 12 - Atividade do Glutatião Redutase (GR), apresentada em unidades: [NADPH] μM min -1 mg proteína -1 em sobrenadante de sobrenadante de homogeneizados de fígado de murganhos submetidos à exposição do BBN, DMAPT e cisplatina](https://thumb-eu.123doks.com/thumbv2/123dok_br/15837359.1083858/68.892.261.624.479.816/atividade-glutatião-apresentada-sobrenadante-sobrenadante-homogeneizados-submetidos-exposição.webp)
