Long non-coding RNA HOTAIR promotes UVB-induced
apoptosis and in
fl
ammatory injury by up-regulation of
PKR in keratinocytes
Guo Liu and Wenhao Zhang
Department of Burns and Plastic Surgery, Jining No.1 People’s Hospital, Jining, Shandong, China
Abstract
Excessive exposure to ultraviolet (UV) rays can cause damage of the skin and may induce cancer, immunosuppression, photo-aging, and inflammation. The long non-coding RNA (lncRNA) HOX antisense intergenic RNA (HOTAIR) is involved in multiple human biological processes. However, its role in UVB-induced keratinocyte injury is unclear. This study was performed to investigate the effects of HOTAIR in UVB-induced apoptosis and inflammatory injury in human keratinocytes (HaCaT cells). Quantitative real-time polymerase chain reaction was performed to analyze the expression levels of HOTAIR, PKR, TNF-a, and IL-6. Cell viability was measured using trypan blue exclusion method and cell apoptosis usingflow cytometry and western blot. ELISA was used to measure the concentrations of TNF-aand IL-6. Western blot was used to measure the expression of PKR, apoptosis-related proteins, and PI3K/AKT and NF-kB pathway proteins. UVB induced HaCaT cell injury by inhibiting cell viability and promoting cell apoptosis and expressions of IL-6 and TNF-a. UVB also promoted the expression of HOTAIR. HOTAIR suppression increased cell viability and decreased apoptosis and expression of inflammatory factors in UVB-treated cells. HOTAIR also promoted the expression of PKR. Overexpression of HOTAIR decreased cell viability and increased cell apoptosis and expression of inflammatory factors in UVB-treated cells by upregulating PKR. Overexpression of PKR decreased cell viability and promoted cell apoptosis in UVB-treated cells. Overexpression of PKR activated PI3K/AKT and NF-kB pathways. Ourfindings identified an essential role of HOTAIR in promoting UVB-induced apoptosis and inflammatory injury by up-regulating PKR in keratinocytes. Key words: HOTAIR; UVB-induced apoptosis; Inflammatory injury; PKR; Keratinocytes
Introduction
Human skin is repeatedly exposed to chronic ultravio-let (UV) irradiation; however, excessive exposure to UV can cause either acute or chronic skin injury (1). Short-term exposure to ultraviolet B (UVB) can cause sunburn and erythema, while long-term exposure induces various cellular responses, such as inflammation, aging, and skin cancer (2,3). UV exposure induces apoptosis of keratino-cytes. Additionally, insufficient repair of UV-induced DNA damage may initiate apoptosis in sunburn cells (4).
During inflammation, pro-inflammatory mediators, including cytokines, chemokines, and prostaglandins are produced, mostly by keratinocytes at the irradiated site (5). It was also found that excessive UVB irradiation increased the production of reactive oxygen species, influenced the cell metabolic pathway, and decreased the activity of superoxide dismutase (3). Therefore, it is of great importance to study the damage and regulation of UVB on skin keratinocytes for the clinical treatment of skin injury and skin cancer.
Long chain non-coding RNAs (lncRNAs) are a class of non-protein coding RNAs with more than 200 nucleotides in length, and with a role in regulating protein transcrip-tions (6). At present, research on lncRNAs is still in the beginning stage. lncRNAs exert critical functions in adult tissue stem cells, including skin, brain, and muscle, as well as in developmental patterning and pluripotency (7). A previous study showed that lncRNA-p21 was upregu-lated in UVB-treated human keratinocytes (HaCaT cells) and played a significant role in cell apoptosis (8).
HOX antisense intergenic RNA (HOTAIR) is a recently discovered lncRNA that plays a critical role in gene regula-tion and chromatin dynamics. HOTAIR has been shown to be dysregulated in a variety of cancers (9). Rinn et al. (10) introduced HOTAIR as a spliced and polyadenylated RNA with 2158 nucleotides and 6 exons. In addition, studies showed that HOTAIR could induce the expression of TNF-a in lipopolysaccharide-induced myocardial cell inflammatory injury (11). HOTAIR is considered as a promoter
Correspondence: Wenhao Zhang:<[email protected]>
Received September 16, 2017 | Accepted April 9, 2018
Brazilian Journal of Medical and Biological Research (2018) 51(8): e6896, http://dx.doi.org/10.1590/1414-431X20186896
ISSN 1414-431X Research Article
of inflammatory response (12). By studying mechanisms of HOTAIR in cell injury, here, we identified a role for double-stranded RNA-dependent protein kinase (PKR) in inflammasome activation. PKR has been identified as a necessary protein kinase for the activation of one or several types of inflammasomes (13). PKR deficiency could inhibit the secretion of IL-1b, IL-18, and high mobility group box 1 (HMGB1) inEscherichia coli-induced peritonitis (13). PKR upregulation was shown to be related to IL-24-induced apoptosis (14). However, little is known about the effects of HOTAIR and PKR on UVB-induced keratinocyte injury. In the present study, we explored the effects of HOTAIR on UVB-induced apoptosis and pro-inflammatory response in keratinocytes and analyzed the role of PKR in the action of HOTAIR.
Material and Methods
Cell culture and UVB treatment
Immortalized human keratinocytes, HaCaT cells, were purchased from the Chinese Academy of Sciences (China). The cells were cultured in DMEM/F12 (Gibco, USA), sup-plemented with 10% fetal bovine serum at 37°C under an atmosphere of 5% CO2and 95% air. HaCaT cells were plated onto culture dishes. The cells were then irradiated, covered with a thin layer of phosphate buffer saline (PBS), and exposed to UVB (30 mJ/cm2, 280
–320 nm) for 8, 16, and 24 h from a bank of lamps (Spectronics Corp., USA) that were placed at a distance of 25 cm. The irradiance of the lamps was calculated by a calibrated photometer (Spectronics Corp.). Cells with no UVB treatment were considered as the control. The cells were supplied with fresh culture medium after exposure to UVB and incubated for subsequent studies.
Viability assay
For the viability assay, 105cells per well were plated in 24-well plates until attachment. Cells were then trypsinized and stained with trypan blue dye (Beyotime Biotechnology, China); viable cells were counted using a cell-counting chamber (Thermo Fisher Scientific, USA).
Apoptosis assay
Flow cytometry analysis was performed to identify and quantify the apoptotic cells. The assay was done using Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, China) according to the protocol. The cells (105 cells/well) were seeded in 6-well plates. The cells were washed twice with cold PBS and then resuspended in 195mL binding buffer and 5mL Annexin V-FITC solution. PI (10 mL) was added to the mixture, which was then co-incubated for 30 min at room temperature in the dark. Finally, flow cytometry (Beckman Coulter, USA) was conducted to differentiate apoptotic cells (Annexin-V positive and PI-negative) from necrotic cells (Annexin-V and PI-positive).
siRNAs transfection
Cells transfected with si-PKR and the negative con-trols (NC) were synthesized by GenePharma Co. (China). Cell transfection was conducted using Lipofectamine 3000 reagent (Invitrogen, USA) following the manufacturer’s protocol.
Transfection and generation of stably transfected cell lines
Short-hairpin RNA directed against HOTAIR was ligated into the pcDNA3.1 plasmid (GenePharma, China) and was referred to as sh-HOTAIR. The sh-HOTAIR sequences were as follows: sense, 50-GATCCGCCACAT GAACGCCCAGAGATTTTCAAGAGAAATCTCTGGGCG TTCATGTGGTTTTTTG-30, and anti-sense, 50-AATTCAA AAAACCACATGAACGCCCAGAGATTTCTCTTGAAAAT CTCTGGGCGTTCATGTGGCG-30. The plasmid carrying non-targeting sequence was used as NC of sh-HOTAIR and was referred to as sh-NC. Plasmids overexpressing HOTAIR were ligated into the pcDNA3.1 and referred to as pc-HOTAIR. The full-length PKR sequences were constructed into pcDNA3.1 plasmids (GenePharma) to analyze the functions of PKR, and were referred to as pc-PKR. PKR siRNA was obtained from Bioneer (Daejeon Korea) with a sequence of 50-CGUUGCUUAUGAAUG GUCU-30. Lipofectamine 3000 reagent (Life Technologies Corporation, USA) was used for cell transfection accord-ing to the manufacturer’s instructions. The stably trans-fected cells were selected by the culture medium containing 0.5 mg/mL G418 (Sigma-Aldrich, USA). After approximately 4 weeks, G418-resistant cell clones were established.
Enzyme-linked immunosorbent assay (ELISA) After different treatments of HaCaT cells, the culture supernatant was collected from 24-well plates and infl am-matory cytokines were quantified using specific ELISA kits for tests of TNF-a(#DTA00C) and IL-6 (#D6050) con-centrations according to the protocol supplied by the manufacturer (R&D Systems, UK) and normalized to cell protein concentrations.
Quantitative real-time polymerase chain reaction (qRT-PCR)
for 35 cycles (denaturation at 94°C for 45 s, annealing at 55°C for 45 s, extension at 72°C for 90 s). The specific HOTAIR primers were: forward, 50-CAGTGGGGAACTCT GACTCG-30; reverse, 50-GTGCCTGGTGCTCTCTTACC-30.
The specific PKR primers were: forward, 50-CAGAAT TGACGGAAAGACTTAC GTT-30; reverse, 50-CATGA TCAAGTTTTGCCAATGC-30. HOTAIR levels were nor-malized to GAPDH: forward, 50-GTC AACGGATTTG Figure 1.UVB induced HaCaT cell injury;A, Cell viability assay revealed UVB inhibited HaCaT cell viability;B, Apoptosis assay revealed UVB promoted HaCaT cell apoptosis;C, Western blotting analysis revealed increased expression of Bax, decreased expression of Bcl-2, and activation of caspase-3 and caspase-9;D, ELISA results demonstrated that UVB promoted the expression of TNF-a;E, ELISA results demonstrated that UVB promoted the expression of IL-6;F, Western blotting analysis revealed overexpression of TNF-aand IL-6 in UVB-induced cells. Data are reported as means±SD. **Po0.01, ***Po0.001 compared to control (ANOVA). UVB: ultraviolet B; ELISA: enzyme-linked immunosorbent assay; TNF-a: tumor necrosis factor-a; IL-6: interleukin-6.
GTCTGTATT-30; reverse, 50-AGTCTTCTGGGTGGCAG TGAT-30.
Western blot
The proteins used for western blotting were extracted using RIPA lysis buffer (Beyotime Biotechnology) supple-mented with protease inhibitors (Roche, Switzerland). The proteins were quantified using BCAtProtein Assay Kit (Pierce, USA). Bio-Rad Bis-Tris Gel system (Hercules, USA) was used to establish western blotting system according to the manufacturer’s instructions. Samples were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (PVDF, GE Health-care, Germany). Primary antibodies were prepared in 5% blocking buffer (5% non-fat milk in TBS-T (20 mmol/L Tris-HCl (pH 7.6), 137 mmol/L NaCl, 0.1% Tween20) at a dilution of 1:1.000. The used primary antibodies were listed as follows: Bcl-2 (ab32124), Bax (ab32503), pro-caspase-3 (ab44976), cleaved-caspase-3 (ab32042), pro-caspase-9 (ab32539), cleaved-caspase-9 (ab32539), IL-6 (ab6672), TNF-a (ab1793), PKR (ab32506), PI3K (ab86714), p-PI3K (ab182651), AKT (ab32505), p-AKT (ab131443), p65 (ab16502), p-p65 (ab86299), IkBa (ab32518), p-IkBa (ab32518), and GAPDH (ab9485, Abcam, USA). The membrane was incubated with primary antibody at 4°C overnight, followed by washing and incu-bation with goat anti-rabbit (ab205718) or goat anti-mouse IgG (ab6789) marked by horseradish peroxidase for 1 h at room temperature. After rinsing, the PVDF membrane carrying the blots and antibodies were transferred onto Bio-Rad ChemiDoctXRS system, and 200mL Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA) was added to cover the membrane surface. The signals were captured and the intensity of the bands was quantified using Image LabtSoftware (Bio-Rad, Hercules, USA).
Statistical analyses
All experiments were done in triplicate. The results of multiple experiments are reported as means±SD. Statistical analyses were performed using SPSS 19.0 statistical software (USA). P values were calculated using one-way analysis of variance. Po0.05 was considered to be statistically significant.
Results
UVB induced HaCaT cell injury
Exposure of HaCaT cells to UVB decreased cell viability (Figure 1A) and increased apoptosis (Figure 1B) in a time-dependent manner. Western blot analysis for apoptosis (Figure 1C) also showed similar results, with increase in the expressions of pro-apoptotic proteins (Bax, cleaved caspase 3 and 9) and decrease in the expression of anti-apoptotic protein (Bcl-2).
The results of ELISA showed significantly increased expressions of TNF-a (Po0.001, Figure 1D) and IL-6
(Po0.001, Figure 1E) in UVB-treated HaCaT cells com-pared to non-irradiated control cells. Similar results were obtained by western blotting analysis (Figure 1F). The results indicated that UVB induced cell inflammatory injury in HaCaT cells.
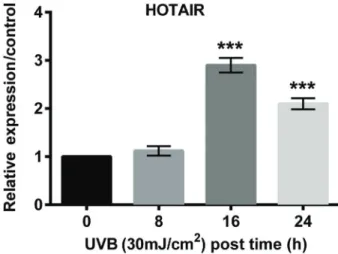
UVB up-regulated the expression of HOTAIR
The results of quantitative RT-PCR showed that HOTAIR was highly expressed at 16 h of UVB exposure (Figure 2). Therefore, subsequent experiments were conducted with a UVB treatment time of 16 h.
Effect of HOTAIR on UVB-induced cell injury
The results of RT-PCR (Figure 3A) showed significantly increased expression of HOTAIR in pc-HOTAIR group compared to pc-DNA3.1 (Po0.001), and significantly decreased expressions of HOTAIR in sh-HOTAIR#1 and #2 group compared to sh-NC (Po0.05 and Po0.01). We then measured the effects of altered expression of HOTAIR on cell viability, apoptosis, and pro-inflammatory cytokine concentration in UVB-treated cells. Overexpres-sion of HOTAIR significantly decreased cell viability (Po0.05; Figure 3B), increased apoptosis (Po0.01; Figure 3C), inhibited Bcl-2 expression (Po0.01; Figure 3D), altered apoptosis-associated factors (Figure 3E), and increased the expressions of TNF-a (Po0.05; Figure 3F and H) and IL-6 (Po0.05; Figure 3G and H) compared with the pcDNA3.1 group in UVB-treated cells. In contrast, suppression of HOTAIR expression using sh-HOTAIR#1 and #2 significantly increased cell viability (both Po0.05; Figure 3B), decreased apoptosis (both Po0.05; Figure 3C), and increased Bcl-2 expression (Po0.05; Figure 3D). No significant difference was found between sh-HOTAIR#1
and #2 transfection group. Due to the higher transfection efficiency of sh-HOTAIR#2, it was used for subsequent studies. We also found that inhibiting HOTAIR expression decreased the expressions of Bax, cleaved caspase-3, and cleaved caspase-9 (Figure 3E). Inhibiting HOTAIR expression reduced TNF-a (Po0.05; Figure 3F and 3H) and IL-6 (Po0.01; Figure 3G and H) compared with sh-NC
in UVB-treated cells. The results indicated that suppres-sion of HOTAIR alleviated UVB-induced cell injury and overexpression of HOTAIR further promoted UVB-induced cell injury.
The effects of HOTAIR on HaCaT cells with no UVB treatment were also analyzed in this study (Supplemen-tary Figure S1). Inhibiting HOTAIR increased cell viability, Figure 3.Effect of HOTAIR on UVB-induced cell injury;A, qRT-PCR revealed overexpression and suppression of HOTAIR in HaCaT cells after transfection assay;B, Cell viability assay revealed suppression of HOTAIR alleviated UVB-induced low cell viability and overexpression of HOTAIR further promoted UVB-induced inhibition of cell viability; C, Apoptosis assay revealed suppression of HOTAIR alleviated UVB-induced high cell apoptosis and overexpression of HOTAIR further promoted UVB-induced high cell apoptosis.
D, qRT-PCR analysis showed that mRNA expression of Bcl-2 was increased after HOTAIR was silenced and decreased after HOTAIR was overexpressed.E, Western blotting analysis showed increased expression of Bax and decreased expression of Bcl-2 in HOTAIR overexpressed cells; F, ELISA results showed suppression of HOTAIR alleviated UVB-induced high expression of TNF-a and overexpression of HOTAIR further promoted UVB-induced high expression of TNF-a;G, ELISA results showed suppression of HOTAIR alleviated UVB-induced high expression of IL-6 and overexpression of HOTAIR further promoted UVB-induced high expression of IL-6;
H, Western blotting analysis showed increased expression of TNF-aand IL-6 with HOTAIR overexpression. Data are reported as means±SD. *Po0.05; **Po0.01; ***Po0.001 (ANOVA). ns: P40.05. UVB treatment time was 16 h. HOTAIR: HOX antisense intergenic RNA; UVB: ultraviolet B; qRT-PCR: quantitative real-time polymerase chain reaction; TNF-a: tumor necrosis factor-a; IL-6: interleukin-6.
but had no significant effect on apoptosis or expression of pro-inflammatory factors. Enhancing HOTAIR expression inhibited viability, promoted apoptosis, and elevated expres-sions of TNF-aand IL-6. Thus, we speculated that HOTAIR might be an important regulator in HaCaT cells.
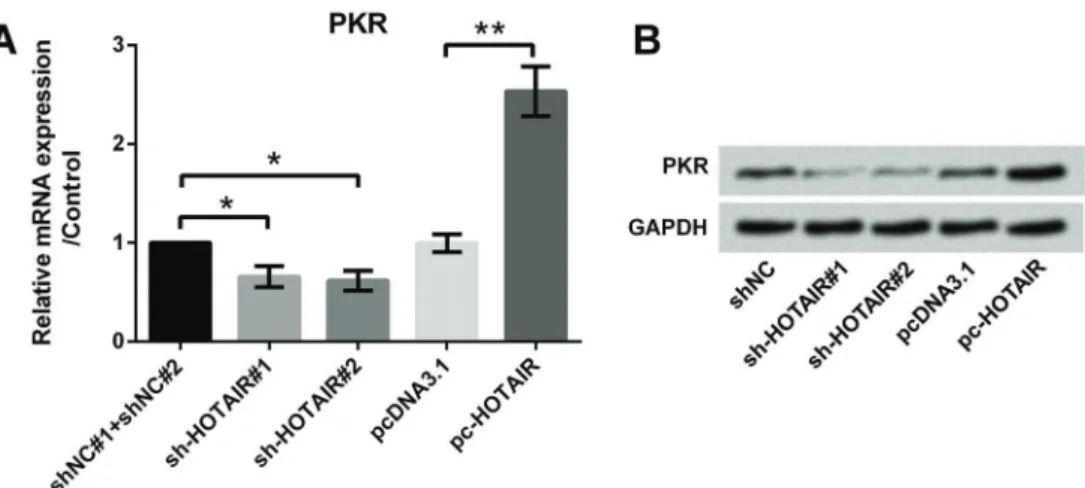
HOTAIR promoted the expression of RNA-dependent protein kinase (PKR)
Quantitative RT-PCR was performed to evaluate the effect of HOTAIR on the expression of PKR (Figure 4A). Results showed that overexpression of HOTAIR significantly promoted the expression of PKR (Po0.01), while suppres-sion of HOTAIR using sh-HOTAIR#1 and #2 decreased the expression of PKR (both Po0.05). Similar results were obtained by western blotting analysis (Figure 4B).
Effect of PKR on UVB-induced cell injury
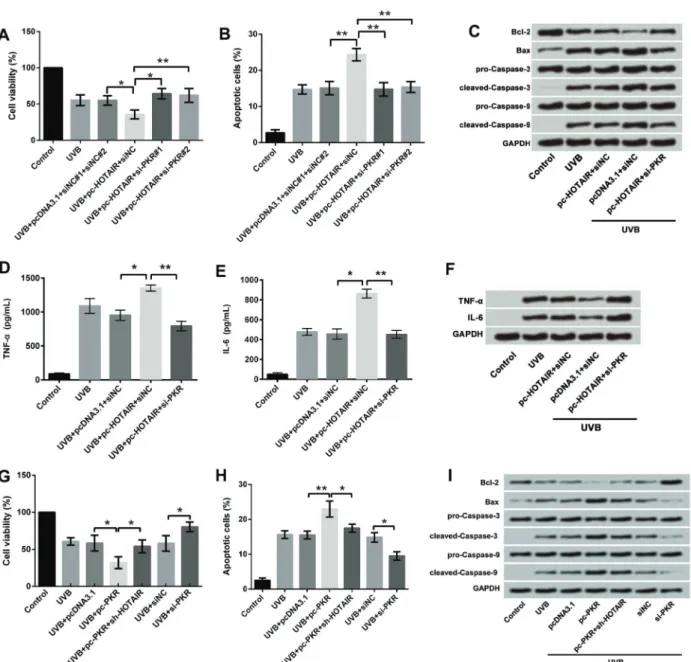
Next, we investigated the effect of PKR on UVB-induced cell injury. Designed si-PKR#1 and 2 were transfected into cells and it was found that PKR expressions in both mRNA and protein were inhibited (both Po0.01, Figure 5A and B). The full-length PKR sequences were constructed into pcDNA3.1 plasmids (pc-PKR), and, as expected, pc-PKR significantly increased the mRNA expression (Po0.01; Figure 5C) and protein expression (Figure 5D) of PKR. We then measured the effects of altered expres-sion of PKR on cell viability and apoptosis in UVB-treated cells. Overexpression of PKR significantly decreased cell viability (Po0.05; Figure 5E) and increased apoptosis (Po0.01; Figure 5F and G), compared with pcDNA3.1 group in UVB-treated cells. In contrast, suppression of PKR expression significantly increased cell viability (Po0.05; Figure 5E) and decreased apoptosis (Po0.05; Figure 5F and G), compared with sh-NC in UVB-treated cells.
Effect of HOTAIR and PKR on UVB-induced cell injury We then measured the effects of altered expressions of PKR and HOTAIR in combination on cell viability, apoptosis, and pro-inflammatory cytokine concentration in UVB-treated cells. For this, UVB-treated cells were transfected with pcDNA3.1+siNC, pc-HOTAIR+siNC, and pc-HOTAIR+si-PKR. As presented previously, over-expression of HOTAIR significantly decreased cell via-bility (Po0.05; Figure 6A), increased apoptosis (Po0.01; Figure 6B and C), and increased the concentrations and protein expression of TNF-a (Po0.05; Figure 6D and F) and IL-6 (Po0.05; Figure 6E and F) compared with pcDNA3.1 in UVB-treated cells. However, suppression of PKR expression reversed these effects by significantly increasing cell viability (Po0.01; Figure 6A), decreasing apoptosis (Po0.01; Figure 6B and C), and increasing the expressions of TNF-a(Po0.01; Figure 6D and F) and IL-6 (Po0.01; Figure 6E and F), compared with pc-HOTAIR in UVB-treated cells. These results suggested that over-expression of HOTAIR promoted UVB-induced cell injury by upregulation of PKR.
As described previously, PKR overexpression inhibited cell growth and promoted cell inflammation. These effects were also impaired by HOTAIR silence (Figure 6G, H, and I), which indicated that growth and inflammation injury of UVB-treated cells were affected by interaction between HOTAIR and PKR.
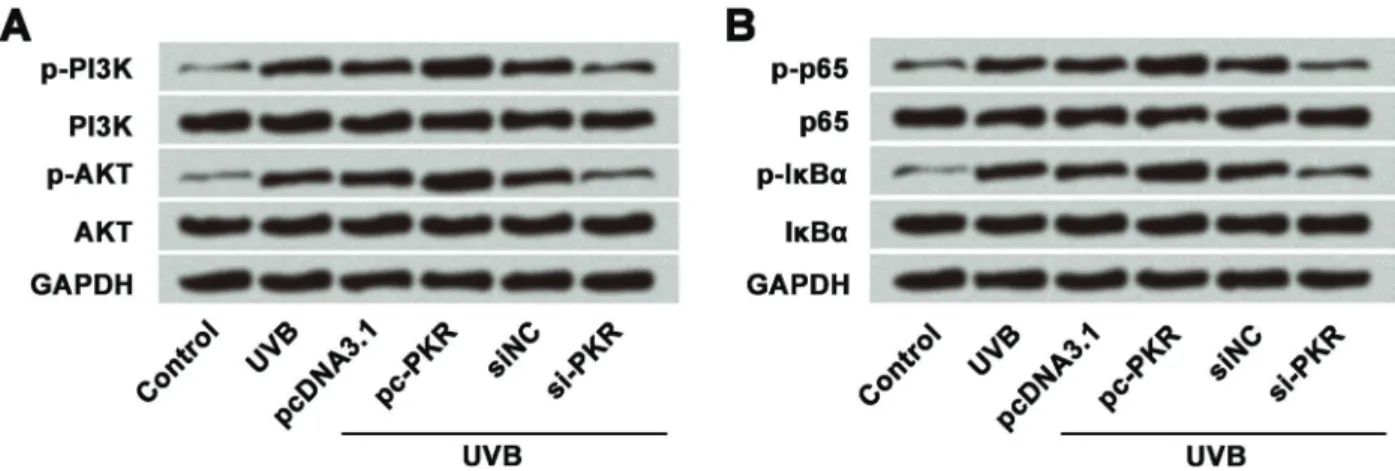
Effect of PKR on PI3K/AKT and NF-kB signaling pathways
Lastly, we measured effects of PKR on PI3K/AKT and NF-kB signaling pathway-related proteins (PI3K, AKT, p-65, and IkBa) in UVB-treated cells using western blot (Figure 7A and B). Overexpression of PKR increased the
Figure 4.Effect of HOTAIR on PKR expression;A, qRT-PCR revealed overexpression of HOTAIR promoted the expression of PKR;
B, Western blotting analysis revealed overexpression of HOTAIR promoted the expression of PKR. Data are reported as means±SD.
expression of phosphorylated PI3K, AKT, p65, and IkBa, whereas suppression of PKR expression showed the opposite effects. These results suggested that overex-pression of PKR activated PI3K/AKT and NF-kB pathways in UVB-injured HaCaT cells.
Discussion
In the present study, we explored the effects of HOTAIR on UVB-induced keratinocyte injury. The results
showed that UVB induced HaCaT cell injury and upregulated the expression of HOTAIR. Overexpression of HOTAIR further promoted the UVB-induced cell injury and suppression of HOTAIR reduced the cell damage. Further studies showed that HOTAIR upregulated the expression of PKR, activating the PI3K/AKT and NF-kB pathway.
Repeated UVB exposure results in damage to skin cells and induction of carcinogenesis (15,16), which requires the rapid removal of irreparably damaged cells after Figure 5.Effect of PKR on UVB-induced cell injury;AqRT-PCR revealed downregulation of PKR in HaCaT cells;B,Western blotting analysis revealed downregulation of PKR in HaCaT cells;C, qRT-PCR revealed overexpression of PKR in HaCaT cells;D, Western blotting analysis revealed overexpression of PKR in HaCaT cells;E, Cell viability assay revealed overexpression of PKR promoted UVB-induced cell viability inhibition, and the effects were reduced by suppression of PKR;F, Apoptosis assay revealed overexpression of PKR promoted UVB-induced cell apoptosis, and the effects were reduced by suppression of PKR;G, Western blotting analysis revealed overexpression of PKR promoted UVB-induced alterations in apoptosis-associated factors, and the effects were reduced by suppression of PKR. Data are reported as means±SD. *Po0.05; **Po0.01 (ANOVA). UVB treatment time was 16 h. PKR: RNA-dependent protein kinase; UVB: ultraviolet B; qRT-PCR: quantitative real-time polymerase chain reaction.
exposure to UV irradiation and condemned to cell apoptosis (17). It is important to maintain the balance of proliferation, stratification, differentiation, and apoptosis of skin cells for
epidermal homeostasis (18); DNA damage repair and induction of apoptosis are main cellular mechanisms for this balance.
Figure 6.Effect of HOTAIR and PKR on UVB-induced cell injury;A, Cell viability assay demonstrated overexpression of HOTAIR promoted UVB-induced cell viability inhibition by upregulation of PKR, and suppression of PKR reversed the results;B,Apoptosis assay showed overexpression of HOTAIR promoted UVB-induced cell apoptosis by upregulation of PKR, and suppression of PKR reversed the results;C,Western blotting analysis revealed overexpression of HOTAIR promoted the effects of UVB on apoptosis-related factors by upregulation of PKR, and suppression of PKR reversed the results;DandE, ELISA revealed overexpression of HOTAIR promoted UVB-induced high expression of TNF-aand IL-6 by upregulation of PKR, and suppression of PKR reversed the results;F, Western blotting analysis showed overexpression of HOTAIR promoted UVB-induced high expression of TNF-aand IL-6 by upregulation of PKR, and suppression of PKR reversed the results; G, Inhibiting expression of HOTAIR reversed the viability-inhibitory effect of PKR;
As an important epigenetic regulation mechanism of gene expression under environmental stress, lncRNAs have been found to play important roles in environmental response (19). Several techniques have been applied to study the molecular mechanisms underlying UVB-induced photo damage and skin carcinogenesis, and lncRNAs have been shown to be involved in this process (8). However, the specific functions of HOTAIR and the underlying mechanisms are still unknown in UVB-induced skin injury. The importance of lncRNAs has been impli-cated in many different contexts. HOTAIR interacts with Polycomb Repressive Complex 2 (PRC2) and is neces-sary for PRC2 occupancy and histone H3 lysine-27 trimethylation of different genes in different chromosomes. PRC2 is a histone methyltransferase that implements epigenetic silencing during different processes, including cancer development (20).
Our study revealed that overexpression of HOTAIR promoted UVB-induced apoptosis and inflammatory injury by up-regulation of PKR in keratinocytes. Several studies have shown antiviral defense mechanism through PKR in keratinocytes (21). Also, the association of HOTAIR and PKR has been demonstrated in several different cancers (22–24). However, no study has evaluated the association of HOTAIR and PKR in UVB-induced cell injury. Our study showed that overexpression of HOTAIR significantly promoted the expression of PKR, while suppression of HOTAIR inhibited the expression of PKR.
Several protein kinases are involved in the UV-induced signal transduction (25). To our knowledge, this is thefirst study that demonstrated the role of PKR in UVB-induced skin injury. However, the role of PKR in activation of infl am-masome has been confirmed. Lu et al. (13) demonstrated that PKR activity was integral to assembly and activation of inflammasome and that it mediated inflammatory response by activating caspase-1, cleavage IL-1b, and releasing HMGB1. PKR was involved in inflammation and
immune dysfunction through regulating mitogen-activated protein kinases (MAPKs), interferon regulatory factor 3 (IRF3), NF-kB, apoptosis, and autophagy pathways (26). These studies indicated the pro-inflammation effect of PKR, which were consistent with our study.
There are several pathways involved in UVB-induced skin injury as solar UVB radiation induces erythema, sunburn, hyperplasia, proliferation, inflammation, oxida-tive stress, DNA damage, p53 mutations, immunosup-pression, and alterations in PI3K/AKT and NF-kB cell survival signaling pathways eventually leading to skin cancer (27–31). UVB-induced inflammatory responses include increased production of pro-inflammatory cyto-kines such as TNFa, IL-1b, and IL-6 (32,33). UVB radia-tion activates PI3K/AKT pathway in cultured cells as well as in both mouse and human skin (34–36). In UVB-exposed skin, PI3K/AKT signaling increases the survival response of keratinocytes and facilitates tumorigenesis (37). Also, NF-kB regulates production of inflammatory mediators and induces transcription of pro-inflammatory genes. NF-kB also contributes to the regulation of cell proliferation and survival. Previous studies have shown that UVB radiation activates NF-kB signaling in keratino-cytes and mouse skin (38–40). Our study showed that overexpression of PKR activated PI3K/AKT and NF-kB pathways.
In conclusion, our studyfindings suggested that over-expression of HOTAIR promoted UVB-induced cell injury in HaCaT cells. Overexpression of HOTAIR upregulated the expression of PKR, leading to the activation of PI3K/ AKT and NF-kB pathways. These results indicated that HOTAIR promoted UVB-induced apoptosis and infl amma-tory injury by up-regulation of PKR in keratinocytes.
Supplementary material
Click here to view [pdf].
Figure 7.Western blotting analysis of PKR on PI3K/AKT and NF-kB signaling pathways, which suggested that overexpression of PKR activatedA, PI3K/AKT pathway andB, NF-kB pathway. UVB treatment time was 16 h. UVB: ultraviolet B; PKR: RNA-dependent protein kinase; PI3K: phosphatidylinositol-3 kinase.
References
1. Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M, et al. UV-induced skin damage.Toxicology2003; 189: 21–39, doi: 10.1016/S0300-483X(03)00150-1.
2. Li W, Wu YF, Xu RH, Lu H, Hu C, Qian H. miR-1246 releases RTKN2-dependent resistance to UVB-induced apoptosis in HaCaT cells. Mol Cell Biochem 2014; 394: 299–306, doi: 10.1007/s11010-014-2108-1.
3. Hou W, Gao W, Wang D, Liu Q, Zheng S, Wang Y. The protecting effect of deoxyschisandrin and schisandrin B on HaCaT Cells against UVB-Induced damage.PLoS One
2015; 10: e0127177, doi: 10.1371/journal.pone.0127177. 4. Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala
SLeffell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion.J Investig Dermatol Symp Proc1996; 1: 136–142.
5. Ryser S, Schuppli M, Gauthier B, Hernandez DR, Roye O, Hohl D, et al. UVB-induced skin inflammation and cutaneous tissue injury is dependent on the MHC class I-like protein, CD1d.J Invest Dermatol2014; 134: 192–202, doi: 10.1038/ jid.2013.300.
6. Da Sacco L, Baldassarre A, Masotti A. Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int J Mol Sci 2012; 13: 97–114, doi:
10.3390/ijms13010097.
7. Flynn RA, Chang HY. Long Noncoding RNAs in cell-fate programming and reprogramming.Cell Stem Cell2014; 14: 752–761, doi: 10.1016/j.stem.2014.05.014.
8. Hall JR, Messenger ZJ, Tam HW, Phillips SL, Recio L, Smart RC. Long noncoding RNA lincRNA-p21 is the major mediator of UVB-induced and p53-dependent apoptosis in keratinocytes.Cell Death Dis2015; 6: e1700, doi: 10.1038/ cddis.2015.67.
9. Bhan A, Mandal SS. LncRNA HOTAIR: A master regulator of chromatin dynamics and cancer.Biochim Biophys Acta
2015; 1856: 151–164. doi: 10.1016/j.bbcan.2015.07.001. 10. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann
SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs.
Cell2007; 129: 1311–1323, doi: 10.1016/j.cell.2007.05.022.
11. Wu H, Liu J, Li W, Liu G, Li Z. LncRNA-HOTAIR promotes TNF-alpha production in cardiomyocytes of LPS-induced sepsis mice by activating NF-kappaB pathway. Biochem Biophys Res Commun2016; 471: 240–246, doi: 10.1016/
j.bbrc.2016.01.117.
12. Carrion K, Dyo J, Patel V, Sasik R, Mohamed SA, Hardiman G, et al. The long non-coding HOTAIR is modulated by cyclic stretch and WNT/beta-CATENIN in human aortic valve cells and is a novel repressor of calcification genes.PLoS One
2014; 9: e96577, doi: 10.1371/journal.pone.0096577. 13. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P,
et al. Novel role of PKR in inflammasome activation and HMGB1 release.Nature2012; 488: 670–674, doi: 10.1038/
nature11290.
14. Pataer A, Vorburger SA, Barber GN, Chada S, Mhashilkar AM, Zou-Yang H, et al. Adenoviral transfer of the melanoma differentiation-associated gene 7 (mda7) induces apoptosis of lung cancer cells via up-regulation of the double-stranded RNA-dependent protein kinase (PKR). Cancer Res2002; 62: 2239–2243.
15. Soehnge H, Ouhtit A, Ananthaswamy ON. Mechanisms of induction of skin cancer by UV radiation.Front Biosci1997; 2: d538–551, doi: 10.2741/A211.
16. Yoshikawa T, Rae V, Bruins-Slot W, Van den Berg JW, Taylor JR, Streilein JW. Susceptibility to effects of UVB radiation on induction of contact hypersensitivity as a risk factor for skin cancer in humans.J Invest Dermatol1990; 95: 530–536, doi: 10.1111/1523-1747.ep12504877.
17. Metcalfe A, Streuli C. Epithelial apoptosis.Bioessays1997; 19: 711–720, doi: 10.1002/bies.950190812.
18. Bernerd F, Sarasin A, Magnaldo T. Galectin-7 overexpression is associated with the apoptotic process in UVB-induced sunburn keratinocytes.Proc Natl Acad Sci USA1999; 96: 11329–11334, doi: 10.1073/pnas.96.20.11329.
19. Xu Q, Song Z, Zhu C, Tao C, Kang L, Liu W, et al. Systematic comparison of lncRNAs with protein coding mRNAs in population expression and their response to environmental change.BMC Plant Biol2017; 17: 42, doi: 10.1186/s12870-017-0984-8.
20. Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscu-ous RNA binding by Polycomb repressive complex 2.
Nat Struct Mol Biol 2013; 20: 1250–1257, doi: 10.1038/
nsmb.2679.
21. Kalali BN, Kollisch G, Mages J, Muller T, Bauer S, Wagner H, et al. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling.J Immunol
2008; 181: 2694–2704, doi: 10.4049/jimmunol.181.4.2694.
22. Heinicke LA, Wong CJ, Lary J, Nallagatla SR, Diegelman-Parente A, Zheng X, et al. RNA dimerization promotes PKR dimerization and activation.J Mol Biol2009; 390: 319–338,
doi: 10.1016/j.jmb.2009.05.005.
23. Lee KS, Park JL, Lee K, Richardson LE, Johnson BH, Lee HS, et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget 2014; 5: 3944–3955. doi: 10.18632/ oncotarget.2047.
24. Wang H, Vardy LA, Tan CP, Loo JM, Guo K, Li J, et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer cell 2010; 18: 52–62, doi:
10.1016/j.ccr.2010.04.028.
25. Bode AM, Dong Z. Mitogen-activated protein kinase activa-tion in UV-induced signal transducactiva-tion. Sci STKE 2003; 2003: RE2. doi: 10.1126/stke.2003.167.re2.
26. Kang R, Tang D. PKR-dependent inflammatory signals.
Sci Signal2012; 5: pe47, doi: 10.1126/scisignal.2003511. 27. Afaq F, Adhami VM, Mukhtar H. Photochemoprevention of
ultraviolet B signaling and photocarcinogenesis.Mutat Res
2005; 571: 153–173, doi: 10.1016/j.mrfmmm.2004.07.019.
28. Adhami VM, Syed DN, Khan N, Afaq F. Phytochemicals for prevention of solar ultraviolet radiation-induced damages.
Photochem Photobiol 2008; 84: 489–500, doi: 10.1111/
j.1751-1097.2007.00293.x.
29. Afaq F, Katiyar SK. Polyphenols: skin photoprotection and inhibition of photocarcinogenesis.Mini Rev Med Chem2011; 11: 1200–1215. doi: 10.2174/138955711804586739.
30. Norval M, Halliday GM. The consequences of UV-induced immunosuppression for human health.Photochem Photobiol
31. Timares L, Katiyar SK, Elmets CA. DNA damage, apoptosis and langerhans cells--Activators of UV-induced immune tolerance. Photochem Photobiol 2008; 84: 422–436, doi:
10.1111/j.1751-1097.2007.00284.x.
32. Bashir MM, Sharma MR, Werth VP. UVB and proinfl amma-tory cytokines synergistically activate TNF-alpha produc-tion in keratinocytes through enhanced gene transcripproduc-tion.
J Invest Dermatol2009; 129: 994–1001, doi: 10.1038/jid. 2008.332.
33. Vaid M, Sharma SD, Katiyar SK. Honokiol, a phytochemical from the Magnolia plant, inhibits photocarcinogenesis by targeting UVB-induced inflammatory mediators and cell cycle regulators: development of topical formulation.
Carcinogenesis2010; 31: 2004–2011, doi: 10.1093/carcin/
bgq186.
34. Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling.Nat Rev Cancer2004; 4: 23–35, doi: 10.1038/nrc1253.
35. Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidyli-nositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model.
Mol Cancer Res2005; 3: 90–99, doi: 10.1158/1541-7786.
MCR-04-0065.
36. Tsoyi K, Park HB, Kim YM, Chung JI, Shin SC, Lee WS, et al. Anthocyanins from black soybean seed coats inhibit
UVB-induced inflammatory cylooxygenase-2 gene expres-sion and PGE2 production through regulation of the nuclear factor-kappaB and phosphatidylinositol 3-kinase/Akt path-way.J Agric Food Chem2008; 56: 8969–8974, doi: 10.1021/
jf801345c.
37. Pal HC, Athar M, Elmets CA, Afaq F. Fisetin inhibits UVB-induced cutaneous inflammation and activation of PI3K/ AKT/NFkappaB signaling pathways in SKH-1 hairless mice.
Photochem Photobiol 2015; 91: 225–234, doi: 10.1111/ php.12337.
38. Afaq F, Adhami VM, Ahmad N, Mukhtar H. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea Constituent (-)-epigallocatechin-3-gallate.Oncogene 2003; 22: 1035–1044, doi: 10.1038/sj.onc.1206206.
39. Afaq F, Ahmad N, Mukhtar H. Suppression of UVB-induced phosphorylation of mitogen-activated protein kinases and nuclear factor kappa B by green tea polyphenol in SKH-1 hairless mice.Oncogene2003; 22: 9254–9264, doi: 10.1038/
sj.onc.1207035.
40. Sharma SD, Meeran SM, Katiyar SK. Dietary grape seed proanthocyanidins inhibit UVB-induced oxidative stress and activation of mitogen-activated protein kinases and nuclear factor-kappaB signaling in in vivo SKH-1 hairless mice.
Mol Cancer Ther 2007; 6: 995–1005, doi: 10.1158/1535-7163.MCT-06-0661.