UNIVERSIDADE FEDERAL DE OURO PRETO INSTITUTO DE CIÊNCIAS EXATAS E BIOLÓGICAS
DEPARTAMENTO DE CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS
ANÁLISE DA EXPRESSÃO DAS ENZIMAS
DESUBIQUITINADORAS 5, 14 E 16 DURANTE O CICLO
DE VIDA DO
Schistosoma mansoni
.
AUTORA:HELAINE GRAZIELE SANTOS VIEIRA
ORIENTADORA: PROFª. DRª. RENATA GUERRA DE SÁ CO-ORIENTADOR: PROF. DR. ÉLIO HIDEO BABÁ
Dissertação submetida ao programa de Pós-Graduação do Departamento de Ciências Biológicas da Universidade Federal de Ouro Preto, como parte integrante dos requisitos para obtenção do título de Mestre em Ciências Biológicas, área de concentração: Biologia Molecular.
“
!
"
" #
"
%
"
&
"
)
Agradeço a Deus, pela minha vida e por me dar dia após dia fé e perseverança pra conseguir enfrentar as grandes tempestades.
Ao Prof. Dr. Élio Hideo Babá pela acolhida no LBBM, pela co-orientação neste trabalho, por todos estes anos de boa convivência e pelos grandes ensinamentos que levarei comigo pra sempre.
À Prof. Dra. Renata Guerra de Sá, pela orientação e pelo apoio constante durante a realização deste trabalho.
Aos meus pais, Liberticina e Oliver, aos meus irmãos e demais familiares que mesmo longe sempre me apoiaram, e através de suas orações para que meus experimentos dessem certo, contribuíram para a concretização deste trabalho.
Aos meus pais de bancada e queridos amigos Carmem Regina Nery e Silva e
Wander Jeremias de Jesus.
Ao mestrando e grande amigo Matheus Gomes de Souza, pessoa de um coração enorme, que teve grande participação neste trabalho auxiliando na parte experimental, sempre me incentivando e procurando soluções para os problemas que surgiam, faltam me palavras pra lhe agradecer.
Aos professores do Laboratório de Bioquímica e Biologia Molecular (LBBM), Profª Dra. Maria Lúcia Pedrosa sempre atenciosa e cativante, Prof. Dr. Elísio Alberto Evangelista, pela alegria, pelo apoio e incentivo.
Aos colegas e amigos do LBBM: Cássio, Nilza, Maísa, Robertinha, Letícia, Nayara, Natália, Leonardo, Roberta, Roenick, Tiago, Eneida. Aos ex-LBBM e aqueles que deixaram sua marca registrada por aqui Ângelo, Gustavo, Rafaela, Militão, Carla, Guilherme, Amanda, Ângela, Cláudia, Nancy, Juliana, Júlio, Keila, Levindo, Olavo, Hiroko, Fabiana; todos vocês contribuíram muito para a minha formação científica, seja ajudando no dia a dia de bancada seja oferecendo apoio e carinho.
Ao Ezequiel, técnico de nosso laboratório e por quem tenho enorme carinho, agradeço pela boa convivência durante estes anos, por sua alegria e pela amizade sincera.
Ao Dr. Omar dos Santos Carvalho, do Laboratório de Malacologia e ao Dr. Rodrigo Correia Oliveira do Laboratório de Imunologia do CPqRR/Fiocruz-BH, por gentilmente fornecerem os materiais biológicos (cercárias e vermes adultos) utilizados neste trabalho. A Dra. Liana, Delza, Dílci e Sueleny, responsáveis pela manutenção do ciclo do S. mansoni, agradeço pela atenção, pelo carinho, presteza e pelos ensinamentos.
Ao Prof. Dr. Luís Carlos Crocco e a Profª Dra. Simone pelo acesso ao Laboratório de Imuno Parasitologia, contribuindo muito para a realização deste trabalho. E a todos os colegas do LIP especialmente Eduardo, Leandro, Djalma, Roberta, Liz, Tiago, Marcos, Rafaela e Jamile.
Aos professores do NUPEB, pelo conhecimento fornecido e pela receptividade em seus laboratórios permitindo a realização deste trabalho.
A todos os colegas da pós-graduação Geórgia, Nilza, Cássio, Maísa, Fernanda, Thiago, Alessandra, Ana Maria, Juliana, Joelma, Anderson, Emerson, Rafaela, Gil, Helen, Analina, Alex e àqueles não listados aqui.
Aos colegas dos Laboratórios do NUPEB, pela solidariedade mútua e amistosa convivência. Um especial agradecimento a Zezé, Maristela e aos colegas do Laboratório de Patologia Clínica.
À Maria Aparecida (Cida) secretária do NUPEB, por sua alegria e prestatividade.
$
*
+$
,
+$
)
+)
)
+&
+)
+- .
011.1 - Esquistossomose humana 02
1.2 - Ciclo biológico do parasito 03
1.3 - Patologia e controle da esquistossomose 06
1.4 - Genoma e transcriptoma de S. mansoni 07
1.5 - Modificação pós-traducional dependente de ubiquitina 09
1.6 – Desubiquitinação 13
1.7 - Modificação pós-traducional dependente de ubiquitina em S. mansoni 16
/ . '
182.1 – Objetivos Específicos 19
0 . 1
12
203.1 - Obtenção das formas evolutivas do S. mansoni (vermes adultos, cercárias
e ovos)
21
3.2 - Transformação mecânica de cercárias em esquistossômulos in vitro 22
3.3 - Extração de DNA genômico 24
3.4 - Extração de RNA total 25
3.5 - Oligonucleotídeos iniciadores específicos para os genes DUB5, DUB14 e DUB16 (deubiquitinating enzyme)
27
3.6 - RT-PCR (Reverse- Transcription Polimerase Chain Reaction) 29
3.8 - Obtenção de células competentes para transformação 30 3.9 - Transformação de bactérias E. coli DH5 α 31
3.10 - Mini-preparação dos plasmídios recombinantes 32
3.11 – Sequenciamento 33
3.12 - Análise computacional das seqüências 34
3.13 - Análise da expressão de DUB 5, DUB 14 e DUB 16 durante as fases de
S. mansoni
34
3 . &
364.1 - Análise computacional das seqüências de cDNA codificadoras para as enzimas desubiquitinadoras
37
4.1.1 - Enzima desubiquitinadora 5 37
4.1.2 - Enzima desubiquitinadora 14 42
4.1.3 - Enzima desubiquitinadora 16 45
4.2 - Análise semi-quantitativa do padrão de expressão gênica das enzimas desubiquitinadoras durante o ciclo evolutivo do S. mansoni
48
4.2.1 - DUB5 48
4.2.2 - DUB14 50
4.2.3 - DUB16 52
4.2.4 - Padrão de expressão das DUB’s durante o desenvolvimento de S.
mansoni
54
4.3 - Padrão de expressão gênica das subunidades RPN9 e COP9S3 do proteassoma 26S durante o ciclo evolutivo do S. mansoni
55
4 .
575 . 6
7
688 . &
9
:
70$
Tabela 1 - Composição do meio 169 24
Tabela 2 - Oligonucleotídeos iniciadores gerados de DUB5, DUB14 e DUB16 28
Tabela 3 - Oligonucleotídeos utilizados como controle neste trabalho 35
$
,
Figura 1 - Ciclo de vida do S. mansoni apresentado em diagrama esquemático 05
Figura 2 - Estágios de desenvolvimento de S. mansoni 06
Figura 3 – Algumas das várias funções das modificações dependentes de ubiquitina 10
Figura 4 – Esquema representativo do proteassoma associado aos seus reguladores 12
Figura 5 – Funções das enzimas desubiquitinadoras 14
Figura 6 - DNA genômico de cercária de S. mansoni 25
Figura 7 – Extração de RNA total das fases evolutivas de S. mansoni 27
Figura 8 – PCR das colônias 32
Figura 9 – Mini-preparação 33
Figura 10 – Seqüência da ORF predita pelo programa ORF finder de SmDUB5 38 Figura 11 - Representação esquemática da localização dos domínios conservados em
SmDUB5
39
Figura 12 – Alinhamento da seqüência SmDUB5 com seqüências ortólogas de USP5 41 Figura 13 – Seqüência da ORF predita pelo programa ORF finder de SmDUB14 42 Figura 14 – Representação esquemática da localização dos domínios conservados em
SmDUB14
43
Figura 15 – Alinhamento da seqüência SmDUB14 com seqüências ortólogas de
USP14
44
Figura 16 – Seqüência da ORF predita pelo programa ORF finder de SmDUB16 45 Figura 17 - Representação esquemática da localização dos domínios conservados em
SmDUB16
46
Figura 18 – Alinhamento da seqüência SmDUB16 com seqüências ortólogas de
USP16
47
Figura 19 – Amplificação docDNA correspondente ao gene SmDUB5 na fase de
verme adulto de S. mansoni
48
Figura 20 – Padrão de expressão do gene SmDUB5 49
Figura 21 – Amplificação docDNA correspondente ao gene SmDUB14 na fase de
verme adulto de S. mansoni
50
Figura 23 – Amplificação docDNA correspondente ao gene SmDUB16 na fase de
verme adulto de S. mansoni
52
Figura 24 – Padrão de expressão do gene SmDUB16 53
Figura 25 – Gráfico comparativa da expressão das DUB’s nas fases Ce (cercárias), V.A (vermes adultos) e Ovos
54
Figura 26 - Gráfico comparativo da expressão das DUB’s durante o estágio larval 54
Figura 27 – Padrão de expressão das subunidades COP9S3 e RPN9 do proteassoma 26S
$
)
g Microgramas
g/mL Microgramas por mililitros
L Microlitros
M Micromolar
ATP Adenosina trifosfato
BLAST Basic Local Alignment Search Tool
cDNA DNA complementar
cm2 Centímetros quadrados
COP9S3 Constitutive photomorphogenic 9 subunidade 3
D.O Densidade óptica
DNA Ácido desoxiribonucléico
dNTP Deoxinucleosídeo trifosfato (N = A, C, G, ou T)
DTT Ditiotreitol
DUB Deubiquitin enzyme
EDTA Ácido etilenodiaminotetracético
g Gramas
HEPES 2-4-(2-Hidroxietil)-piperazinil-(1) ácido etanosulfônico IPTG Isopropil -D-galactosídeo
Kb Kilobases
kDa KiloDaltons
M Molar
Mb Megabases
mg Miligramas
mg/mL Miligramas por mililitros
Mg+2 Magnésio
mL Mililitros
mM Milimolar
MOPS [Ácido 3-(N-morfolino) propanosulfônico]
mRNA Ácido ribonucléico mensageiro
NCBI National Center for Biotechnology Information
ng Nanogramas
nm Nanômetros
ºC Grau celsius ou centígrados
pb Pares de bases
PCR Reação em cadeia da polimerase
pH Potencial hidrogeniônico
PM Peso molecular
pmol Picomoles
RNA Ácido ribonucléico
RNAi RNA interference
RNAse Ribonuclease
SDS Dodecil sulfato de sódio
SmDUB Enzima desubiquitinadora de Shistosoma mansoni
Taq Thermus aquaticus
Tris Tris-hidroximetilaminometano
Ub Ubiquitina
Volts Voltagem
)
) )
9
)
Alanina Ala A
Arginina Arg R
Asparagina Asn N
Ácido aspártico Asp D
Ácido glutâmico Glu E
Cisteína Cys C
Glicina Gly G
Glutamina Gln Q
Histidina His H
Isoleucina Ile I
Leucina Leu L
Lisina Lys K
Metionina Met M
Fenilanina Phe F
Prolina Pro P
Serina Ser S
Tirosina Tyr Y
Treonina Thr T
Triptofano Trp W
&
O Schistosoma mansoni apresenta um ciclo biológico bastante peculiar, no qual
diversas alterações bioquímicas e morfológicas ocorrem no verme. Estas mudanças são adaptações evolutivas encontradas pelo parasito para sobreviver nos distintos ambientes. Acredita-se que estas adaptações são promovidas por um conjunto de genes expressos coordenadamente nos seis estágios de vida do parasito, sendo de extrema importância a identificação destes genes para elabor vacinas e novas drogas. Neste contexto, as vias de modificações pós-traducional de proteínas como a ubiquitina devem ser atuantes em vários processos celulares no parasito. A ubiquitina se liga covalentemente a proteína alvo, através de uma ligação isopeptídica entre o resíduo de glicina (Gly 76) da ubiquitina, localizado na região C-terminal e o resíduo de lisina (Lys) do grupo amino da proteína a ser ubiquitinada. Esse processo é reversível, sendo regulado pelas enzimas chamadas desubiquitinadoras (DUBs). As DUBs são cisteíno proteases que clivam a ligação isopeptídica existente entre as ubiquitinas que formam a cadeia poliubiquitinada, mantendo o pool de Ub intracelular livre e processam a pré-proteína dando origem a forma madura da Ub. No presente trabalho, objetivamos validar as análises in silico das DUB’s 5, 14 e 16 em S. mansoni e avaliar o padrão de
expressão destas enzimas em cercárias, esquistossômulos obtidos in vitro nos tempos
de 3 horas, 18,5 horas; 24 horas, 3 dias, em verme adulto e ovos. A expressão comparada das DUB’s 5, 14 e 16 mostra que elas são mais expressas nas fases cercária, verme adulto e ovo, do que nos diferentes estágios de esquistossômulos estudados, confirmando resultados encontrados anteriormente em que foi verificada a atividade DUB total nas fases de esquistossômulos menor comparada às demais fases. Este padrão de expressão também corrobora com os resultados obtido por nosso grupo de pesquisa onde foi observada a presença de conjugados ubiquitinados em todas as fases de S. mansoni, com uma abundância predominante durante as diferentes culturas de
)
Schistosoma mansoni presents a peculiar life cycle, in which several
morphologic and biochemical alterations occur in different phases of development. These changes are evolutive adaptations found by the parasite to survive in distinct environments. These adaptations are promoted by a set of genes expressed coordinately in the six life periods of the parasite, being a target to elaboration of vaccines and new drugs. In this context, the post-transductional modifications of proteins, as ubiquitination, must be operating in some crucial cellular processes in the parasite. Ubiquitin (Ub) binds to target protein covalently, through an isopeptide linkage between the C-terminal glicina residue (Gly 76) of Ub and the amino group of Lysine (Lys). The reversible process of ubiquitination is the deubiquitination. DUBs (deubiquitin enzymes) are cystein proteases that specifically cleave off Ub from Ub-protein conjugates, Ub precursors and Ub adducts. In the present work, our objective was to validate the in silico analyses of DUB's 5, 14 and 16 in S. mansoni and evaluate
the expression patterns of these enzymes in cercariae, in vitro schistosomulum obtained
by times of 3 hours, 18,5 hours; 24 hours, 3 days, in adult worms and eggs. The comparative expression patterns of DUB' s 5, 14 and 16 shown that they are more expressed in cercariae, adult worm and egg phases, and in basal level in different periods of in vitro cultivate schistosomulum. Our results corroborate the low total DUB
activity in phases of schistosomulum when compared with the other phases. This expression pattern also corroborates with the results obtained by our group, where the presence of ubiquitin conjugates was observed in all the phases of S. mansoni, with a
1.1) Esquistossomose humana
A esquistossomose é uma importante doença parasitária humana que afeta mais de 200 milhões de indivíduos distribuídos em 76 países tropicais e subtropicais em desenvolvimento. É a segunda moléstia tropical de maior prevalência, causadora de severa morbidade e mortalidade tornando-se assim um problema de saúde pública. Estima-se que 500 a 650 milhões de pessoas estão em risco de infecção pela esquistossomose (WHO, 2006).
De acordo com os primeiros relatos sobre a doença, a esquistossomose teve sua provável origem nas bacias de dois importantes rios, o Nilo na África e o Yangtze na Ásia. Tudo indica que sua história é bastante antiga já que existem relatos de ovos de
Schistosoma sp em vísceras de múmias egípcias de 3.500 a.C. Dos vários tipos de
esquistossomoses conhecidas, aquela que mais se disseminou, atingindo inclusive o continente americano, foi a mansônica (Kloos, H. & David, R., 2002).
Três espécies contribuem para a maioria das esquistossomoses em humanos:
Schistosoma mansoni, que vive principalmente nas vênulas que drenam o intestino
grosso; S. japonicum, que é encontrado principalmente nas vênulas do intestino delgado
e S. haematobium que vive nas vênulas da bexiga urinária. Outras espécies como S.
intercalatum, S. mekongi e S. malayensis também podem infectar humanos (Hickman,
C.P.J. et al., 2004).
A distinção entre as diferentes espécies de Schistosoma pode ser feita através da
observação do formato dos ovos, do número de lobos testiculares dos vermes adultos machos, do comprimento relativo do útero e dos vitelógenos nas fêmeas.
O agente etiológico da esquistossomose no Brasil é o S. mansoni, localmente
conhecida como barriga d’água ou bilharziose em diversos países do mundo. O termo bilharziose foi uma homenagem ao patologista Theodor Bilharz, que em 1852 ao realizar uma autópsia observou a presença destes parasitos no sistema venoso porta intra-hepático.
O S. mansoni é um trematódeo digenético (Trematoda, Digenea) pertencente à
família Schistosomatidae, parasitando não somente o homem mas também alguns
1.2) Ciclo biológico do parasito
Os ovos de S. mansoni são excretados nas fezes de portadores da
esquistossomose, sendo que cada ovo contém uma larva ciliada chamada miracídio. Em contato com a água os ovos eclodem, através de estímulos como luz intensa, temperatura e oxigenação, liberando assim os miracídios. Estes procuram pelo hospedeiro intermediário do parasito, o molusco de água doce do gênero Biomphalaria.
As larvas, que têm sexo definido, penetram ativamente a hemocele do caramujo, multiplicam-se assexuadamente tornando-se esporocistos mães e posteriormente, surge uma segunda geração denominada esporocistos filhos. O hepatopâncreas do caramujo fica repleto de esporocistos filhos que se diferenciam por expansão clonal dando origem à terceira geração, as larvas agora denominadas cercárias (Cunha, A.S., 1970).
Através de estímulos como luz e calor, as cercárias deixam o caramujo entre a quarta e sexta semana após a infecção, permanecendo no meio aquático até encontrarem seu hospedeiro definitivo. Respondendo a sinais químicos, as cercárias penetram ativamente na pele humana através de ações mecânica e enzimática, perdendo suas caudas bifurcadas durante este processo. Após os primeiros 15 minutos de infecção no hospedeiro definitivo, já podem ser observadas alterações fisiológicas nas cercárias, que a partir deste momento são denominadas esquistossômulos. A passagem das cercárias pela epiderme e derme do hospedeiro pode durar de 24 a 72 horas aproximadamente (Cunha, A.S., 1970).
Após este período os esquistossômulos iniciam a migração através do sistema sanguíneo ou linfático e por volta do quarto dia eles chegam aos pulmões. Nos pulmões o número de esquistossômulos atigem o pico máximo entre os dias 7 e 9 após infecção. Durante o processo de migração, os esquistossômulos sofrem apenas um elongamento aumentando assim seu volume corporal, não havendo sinais de divisão celular durante esta fase (Clegg, J.A., 1965).
hepáticos onde, evidenciando o assincronismo, indivíduos jovens são encontrados ao lado de indivíduos adultos durante vários dias. A partir da terceira semana inicia-se a migração dos esquistossômulos para as veias mesentéricas, onde os parasitos já na fase adulta atigem a maturidade. Macho e fêmea se acasalam e as fêmeas realizam a postura de seus ovos completando assim o ciclo evolutivo (Cunha, A.S., 1970). A Figura 1 ilustra o ciclo biológico completo do parasito e a Figura 2 a passagem entre as fases cercária/verme adulto dividida em estágios.
A fêmea de S. mansoni permanece alojada no canal ginecóforo do macho. O
casal pode viver por muitos anos pareado no sistema porta intra-hepático de seu hospedeiro vertebrado. Este pareamento desencadeia uma cascata de eventos que promovem um bom desenvolvimento do ovário e das glândulas vitelogênicas nas fêmeas, de forma a garantir a manutenção do ciclo de vida do parasito (Cunha, A.S., 1970 and Shaw, M.K., 1987).
A longevidade de um Schistosoma adulto é em torno de 3 a 5 anos podendo se
estender por mais de 30 anos. Os casais de vermes adultos podem produzir cerca de 300 a 1.000 ovos por dia. O potencial teórico de reprodução por casal de parasitos é acima de 600 bilhões de Schistosomas (Gryseels, B. et al., 2006).
Ocorre uma divisão de trabalho entre o casal, onde a fêmea de S. mansoni
concentra toda sua energia metabólica em torno da reprodução, formando e propagando seus ovos com eficiência. O macho em contato com a fêmea promove o desenvolvimento do ovário e das glândulas vitelogênicas, ele fornece à fêmea proteção, alimento e esperma para a fertilização dos oócitos, além de outros fatores químicos envolvidos na maturação do sistema reprodutor feminino (Fitzpatrick, J.M. et al.,
2006).
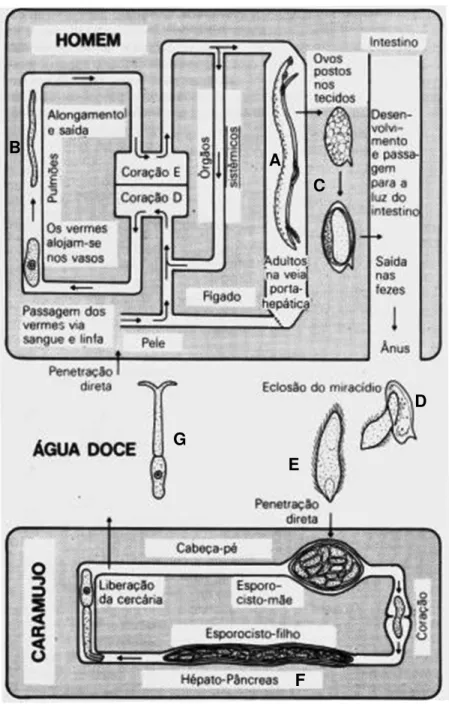
Figura 1 - Ciclo de vida do S. mansoni apresentado em diagrama esquemático - O parasito coexiste em três ambientes distintos: o interior do hospedeiro vertebrado, o ambiente aquático e o interior do hospedeiro invertebrado (moluscos do gênero Biomphalaria). No homem formas larvais (esquistossômulos B) dão origem a parasitos sexualmente maduros (verme adulto A), os quais se acasalam e produzem ovos (C) que são liberados no ambiente aquático. Os ovos eclodem (D) liberando os miracídios infectantes (E) que penetram no caramujo dando origem a numerosos esporocistos (F). Os esporocistos geram cercárias (G) que saem do caramujo e nadam ativamente na água a procura do hospedeiro vertebrado. Adaptado de : Introdução à Parasitologia. Wilson, R. A, 1980. pp13.
B
G
E C
D
1.3) Patologia e controle da esquistossomose
Os sintomas da esquistossomose em sua fase aguda se manifestam através de pequenas urticárias na região de penetração das cercárias, onde linfócitos e macrófagos são reunidos como resposta imune primária à invasão do parasito. Dependendo do número de parasitos e da sensibilidade do hospedeiro vertebrado, este pode desenvolver a forma toxêmica da doença, que é um processo de hipersensibilidade sistêmica contra a migração dos esquistossômulos, caracterizada por febre, fadiga, mialgia, linfadenopatia, tosse não produtiva, eosinofilia entre outros sintomas, que podem desaparecer em poucas semanas (Gryseels et al., 2006).
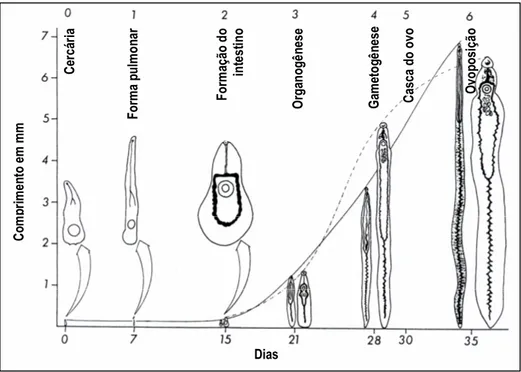
Figura 2 - Estágios de desenvolvimento de S. mansoni. O esquema ilustra o desenvolvimento do parasito em
As principais lesões na infecção crônica não são devidas aos vermes adultos e sim aos seus ovos. Somente cerca de 50% dos ovos liberados pela fêmea são eliminados nas fezes, os demais permanecem retidos nas paredes do intestino e no parênquima hepático do hospedeiro (Warren, K.S. et al., 1967). Os ovos secretam
enzimas proteolíticas que provocam reações inflamatórias eosinófilicas e granulomatosas. Na fase crônica pode ocorrer perda de peso, diarréia, dipnéia, dores abdominais difusa, hipertensão portal e hepatoesplenomegalia. A severidade dos sintomas está relacionada com a intensidade da infecção, o estado nutricional, a idade, a carga parasitária, a linhagem do parasito, e com a resposta imune do indivíduo (Gryseels et al., 2006).
O tratamento da doença é realizado com praziquantel, um fármaco muito utilizado no combate a esquistossomose que, embora seja seguro e efetivo, não oferece proteção contra reinfecções em áreas endêmicas (Ismail M et al., 2002). Muitas cepas de S. mansoni apresentam resistência ao tratamento com este fármaco, porém os
mecanismos utilizados pelo parasito neste processo ainda não estão bem esclarecidos (Ismail M. e cols., 1999).
Várias medidas de saúde pública têm sido adotadas no controle de doenças parasitárias tais como: melhoramento de moradias, higiene sanitária e controle dos vetores. Entretanto, estas abordagens para o controle da esquistossomose têm se mostrado em muitos casos insuficientes ou pouco efetivas, sendo assim de grande importância o desenvolvimento de vacinas e novos quimioterápicos.
1.4) Genoma e transcriptoma de S. mansoni
Atualmente o genoma de S.mansoni está estimado em aproximadamente 300
Mb, organizado em oito pares de cromossomos sendo sete pares autossômicos e um par de cromossomos sexuais. Os machos são homogaméticos (ZZ) e a fêmea heterogamética (ZW). Uma característica do genoma do parasito é o elevado conteúdo de DNA repetitivo (40 a 60%), elementos gênicos móveis, microsatélites e minisatélites, SNPs (Single nucleotide polymorphisms) e íntrons que podem variar em
Em 1994 teve início o Projeto Genoma do Schistosoma, criado através de um
consórcio internacional, tendo como alvo as espécies S. mansoni e S. japonicum, que
foram analisadas separadamente. A iniciativa para a criação deste projeto partiu da Organização Mundial de Saúde (WHO) juntamente com outras agências de fomento, com o intuito de promover o desenvolvimento de novas drogas e quimioterápicos (Lo Verde, P.T. et al., 2004).
Os resultados deste trabalho propiciaram o desenvolvimento de bibliotecas de cDNA, construção de bibliotecas de cromossomos artificiais de bactérias e leveduras, mapas físicos parciais dos cromossomos, e a geração de ESTs (Expressed Sequence Tags) de Schistosoma. As primeiras ESTs foram depositadas por Franco et al 1995,
sendo sequenciadas 607 ESTs de uma biblioteca de cDNA de vermes adultos, onde foram identificados 154 novos genes, dando início a era de descoberta de genes do parasito (Lo Verde, P.T. et al., 2004).
Paralelamente ao sequenciamento de ESTs em larga escala, diversos trabalhos foram publicados descrevendo genes importantes para a biologia do parasito, porém um grande avanço foi alcançado com a liberação do resultado do projeto Transcriptoma do
S. mansoni realizado no Brasil. Este projeto contou com a participação de um consórcio
de laboratórios conhecido como a rede ONSA (Organization of Nucleotide Sequence
and Analysis), que foi financiado pela FAPESP (Fundação de Amparo a Pesquisa do
Estado de SãoPaulo).
Cerca de 163.586 ESTs de S. mansoni foram seqüenciadas, cobrindo 92% do
transcriptoma, destas 151.684 foram originadas de minibibliotecas ORESTES (Open reading frames ESTs) e 11.902 seqüências de bibliotecas normalizadas de vermes adultos. O número de genes preditos foi de aproximadamente 14.000 genes, sendo 50% expressos na fase de verme adulto e 1.000 genes com um padrão de expressão estágio-específica (Verjovski-Almeida, S. et al., 2003). Atualmente, estima-se a existência de
~17.500 genes no genoma do S. mansoni, muitos destes apresentando modulação de
função através de trans-splincing ou splincing alternativo (Brindley, P.J., 2005 e Wilson, R.A. et al., 2006).
O outro projeto, o Transcriptoma do S. japonicum, foi realizado na China e teve
de S. japonicum. Esse conjunto de EST’s foi agrupado em 13.131 contigs, sendo que
cada contig é um provável representante gênico do parasito (Hu, W. et al., 2004).
A análise do transcriptoma de S. mansoni e S. japonicum, através da
classificação funcional dos transcritos pelo Gene Ontology (GO), confirma e sugere a presença de genes associados a eucariotos e processos específicos dos metazoários, como diferenciação eixo anterior-posterior, dorsoventralidade, epitélio, processo neural, motilidade, diferenciação sexual e maturação, longevidade, parasitismo, evasão imune e resposta a stress (Verjovski-Almeida, S. et al., 2003).
Hoje, na era pós genômica, novas ferramentas são utilizadas para validar a presença e quantificar a abundância dos transcritos sequenciados das diferentes fases do
S. mansoni, possibilitando assim a aplicação do genoma funcional. Experimentos tais
como expressão gênica diferencial por "microarrray”, manipulação do genoma silenciando genes utilizando RNAi e estudo do proteoma através da separação de proteínas por eletroforese bidimensional, vêm sendo realizados. Estas inovações possibilitaram a identificação e caracterização dos genes estágios específicos, bem como a constatação do envolvimento das vias de modificação pós-traducional durante um ciclo de vida completo do parasito (Dillon, G.P. et al., 2006).
1.5) Modificação pós-traducional dependente de ubiquitina
Muitos processos biológicos são controlados pelas modificações pós-traducionais dependentes de ubiquitina e proteínas similares a ubiquitina, dentre eles: proteólise intracelular, controle do ciclo celular, desenvolvimento embrionário, resposta a estresse, reparo do DNA, resposta imune, transdução de sinal, biogênese de organelas, regulação transcricional, endocitose e endereçamento de proteínas (Quesada, V., 2004).
A ubiquitina é uma proteína de 76 resíduos de aminoácidos, presente no citosol, núcleo e em diversos compartimentos da célula, sendo bastante conservada entre os organismos eucariotos. Ela se conjuga covalentemente a proteína alvo, através de uma ligação isopeptídica entre o resíduo de glicina (Gly 76) da ubiquitina, localizado na região C-terminal e o resíduo de lisina (Lys) do grupo amino da proteína a ser ubiquitinada (Kim, J.H. et al., 2003). Os sinais para a ubiquitinação nas proteínas alvo
seqüências de aminoácidos na região N-terminal necessárias para direcionar a sua ubiquitinação. Uma outra forma de sinalização ocorre por meio da fosforilação regulando a ubiquitinação das proteínas alvo (Wilkinson, K.D., 2000).
O processo de ubiquitinação de proteínas é catalizado por três enzimas: enzima ativadora de ubiquitina (E1), enzima conjugadora de ubiquitina (E2) e enzima ligase (E3). A ubiquitina é ativada pela E1, por uma ligação ATP dependente, formando um intermediário tio-éster, sendo em seguida transferida ao grupo tio-éster de uma E2 conjugada. Essas E2 transferem a ubiquitina a uma enzima ligase (E3) que se liga a E2 e ao substrato, como ilustrado na Figura 3 (Pickart, C.M., 2001).
Citosol
Núcleo
Ativação Transcricional
Lisossomo Peptídeos
Proteas-soma 26S
Enzimas Desubiquiti-nadora
Proteína alvo
Fusão de Membrana
Endosso- mo precose
CM
Proteína X
d
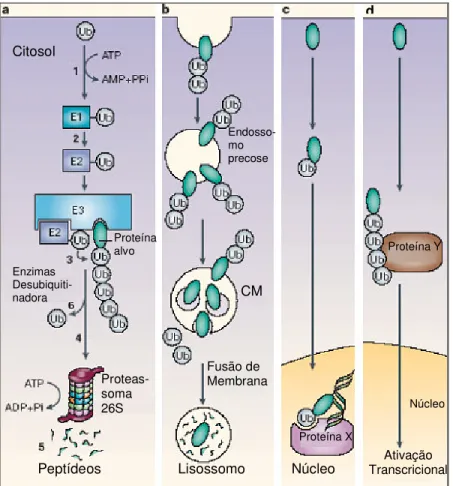
Figura 3 – Algumas das várias funções das modificações dependentes de ubiquitina: (a) Uma molécula de ubiquitina livre é ativada pela enzima ativadora de ubiquitina E1 por um mecanismo ATP dependente – passo (1). Em seguida a ubiquitina é transferida para uma enzima de conjugação E2 - passo (2). O conjugado E2-Ubiquitina e a proteína substrato associam-se especificamente a uma enzima de ligação E3 e a ubiquitina é então transferida para a proteína substrato passo (3). Sucessivas conjugações de moléculas de ubiquitina na proteína alvo geram uma cadeia poliubiquitinada que funciona como sinal para a proteína ser degradada no proteassoma 26S – passo (4). O substrato é degradado em pequenos peptídeos - passo (5) e monômeros de ubiquitina livre são gerados pela atividade das enzimas desubiquitinadoras (DUB) – passo (6). (b) Proteínas de membrana marcadas por diubiquitinação para a degradação no lisossomo. (c) Monoubiquitinação ou uma simples modificação por uma ubiquitina pode marcar proteínas para diferentes destinações subcelulares. (d) A geração de uma cadeia de poliubiquitina pode ativar a regulação transcricional direta ou indiretamente. CM - corpo muiltivesicular; Pi – fosfato inorgânico; PPi - pirofosfato inorgânico; Ub - ubiquitina. Figura adaptada de Ciechanover, A., 2005
As reações de ligação entre as enzimas e a ubiquitina envolvem a glicina do grupo carboxil terminal da ubiquitina e o resíduo de cisteína das enzimas envolvidas neste processo.
Um substrato alvo pode ser modificado pela adição de uma, duas ou mais ubiquitinas. Substratos com quatro ou mais moléculas de ubiquitina ligados no resíduo de lisina 29 ou lisina 48 são direcionados para a proteólise dependente do proteassoma 26S. A mono e poliubiquitinação no resíduo de lisina 63 está envolvida no processo de degradação lisossomal, endocitose, reparo de DNA, ativação de quinases e herança mitocondrial (Kim, J.H. et al., 2003). Numerosas permeases, transportadores,
receptores, histonas e fatores de transcrição são regulados pela monoubiquitinação via Lys 63 (Kim, J.H. et al., 2003).
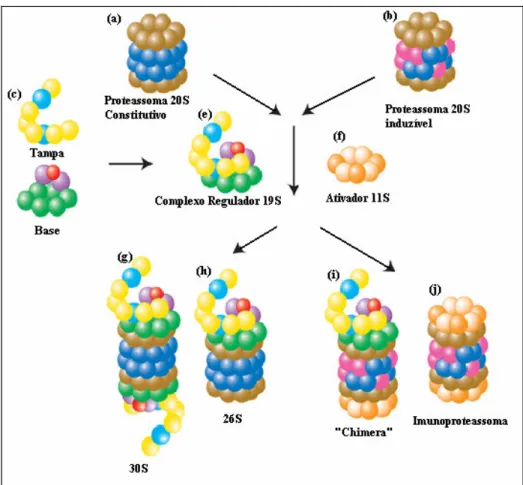
O proteassoma 26S é um complexo proteolítico de 2.000 a 3.000 kDa, constituído por múltiplas proteases diferentes que se associam formando um cilindro contendo um poro central por onde as proteínas passam para serem degradadas (Figura 4). Ele consiste de uma porção proteolítica central de coeficiente de sedimentação 20S, e em suas terminações está associada uma ou duas partículas regulatórias 19S (Ciechanover, A., 2005).
A estrutura cilíndrica central é formada por quatro anéis heptaméricos, dois anéis periféricos compostos por sete subunidades alfa, e dois aneis centrais compostos por sete subunidades beta. O proteassoma é o sistema de proteólise mais importante em eucariotos sendo responsável pela remoção de diversas proteínas regulatórias envolvidas em funções celulares essenciais (Ciechanover, A., 2005; Jackson, P. et al.,
2000; Tanaka, K. et al., 1998).
As diferentes montagens do proteassoma 20S associado aos reguladores PA28 (Proteassome activator 28), PA700 ou ao PA200 permitem a degradação de
importantes polipeptídeos. PA700 ou 19S é o único ativador que estimula a degradação de proteínas poliubiquitinadas e este mecanismo é dependente de ATP. O complexo regulatório 19S é constituído de 17 subunidades ATPásicas e não ATPásicas que se dividem em dois sub-complexo chamado de tampa e base.
sete subunidades. Ahn, J.Y. et al. (1995) verificaram que PA28 é induzido por
interferon (IFN) , sugerindo que este regulador tem importante papel no sistema imune de vertebrados. A Figura 4 ilustra o proteassoma associado aos reguladores 19S e 11S.
PA200 é constituído de uma cadeia simples de proteínas com aproximadamente 200kDa, ele é o mais recente ativador descoberto, suas propriedades biológicas indicam seu envolvimento no processo de reparo de DNA (Rechsteiner, M. & Hill, C.P., 2005).
Outro regulador multifuncional do proteassoma é o COP9 signalossoma (CSN), um complexo constituído por oito subunidades protéicas que são conservadas entre os organismos. A função bioquímica do COP9 signalossoma não está clara, mas alguns dados apontam para uma cooperação entre este complexo e a proteólise mediada pelo proteassoma 26S, implicando na sua participação em processo de desenvolvimento (Bech-Otschir, D. et al., 2002). Somando-se a estas evidências, Freilich, S. et al., (1999)
demonstraram que o COP9 é essencial para o desenvolvimento de D. melanogaster.
1.6) Desubiquitinação
A ubiquitinação de proteínas é um processo reversível, sendo as enzimas desubiquitinadoras responsáveis pela remoção de ubiquitina dos conjugados ubiquitinados, em um mecanismo denominado desubiquitinação. Assim como fosforilar e defosforilar, a ubiquitinação e a desubiquitinação são de extrema importância na regulação das vias dependentes de ubiquitina, que está relacionada a numerosos processos celulares.
No princípio da década de oitenta foi caracterizada a primeira atividade desubiquitinadora, quando pôde ser verificado que a enzima denominada isopeptidase era capaz de clivar as ligações isopeptídicas de ubiquitina de histona H2A (Matsui, S. et
al., 1982). Desde então um vasto número de DUB’s vêem sendo identificadas, porém
muito pouco é conhecido sobre o mecanismo de ação destas enzimas.
As enzimas desubiquitinadoras (DUBs) estão envolvidas no processamento de ubiquitina, reconhecendo a ligação peptídica entre os resíduos de glicina e metionina da cadeia de poliubiquitina. A molécula de ubiquitina é codificada como um transcrito policistrônico (poliubiquitina) ou em fusão com subunidades ribossomais L40 e S27a. Estes mRNAs são traduzidos e processados gerando a ubiquitina livre (Hershko, A. & Ciechanover, A., 1998).
As DUBs regulam de forma negativa a degradação de proteínas poliubiquitinadas, evitando a proteólise via proteassoma ou vacuolar, aumentando assim a meia-vida destas proteínas. Elas são responsáveis também pela retirada das cadeias de ubiquitina ancoradas ao proteassoma 26S, que podem competir com substratos ubiquitinados pelos sítios de ligação à ubiquitina, além de estarem envolvidas na remoção de ubiquitina de proteínas que regulam o ciclo celular durante a mitose (Kim, J.H. et al., 2003; Amerik A.Y. and Hoshstrasser M., 2004). As funções catalíticas das
Figura 5 – Funções das enzimas desubiquitinadoras: (1) Processamento dos precursores de Ub. (2) Edição ou recuperação de conjugados ubiquitinados. (3) Reciclagem da Ub ou oligômeros de Ub dos conjugados proteína/substrato para a degradação. (4) Separação dos oligômeros de ubiquitina em monômeros. Figura adaptada de Wilkinson, K.D., 2000.
As enzimas desubiquitinadoras são organizadas em 5 grupos de cisteíno-proteases: UCH (ubiquitin C-terminal hydrolase), USP (ubiquitin-specific protease), OTU (Ovarian Tumor), MJD (Machado-Joseph diseases protease), o quinto grupo envolve as metaloproteases JAMM (JAB1/MPN/Mov34, metaloenzima) (Nijman, S.M.B. et al., 2005).
As proteases específicas de ubiquitina (USP), também denominadas proteases processadoras de ubiquitina (UBP) ou ubiquitinas carboxil terminal hidrolases 2, apresentam tamanhos variados e grande complexidade estrutural. Cerca de 90 proteases constituem este grupo, sendo proteínas de alto peso molecular, 50-300 kDa, que contém várias regiões conservadas em suas seqüências de aminoácidos, incluindo aquelas em torno dos resíduos Cys, His e Asp que formam a tríade catalítica característica de USP (Wing, S.S., 2003 e Hu, M. et al., 2002).
As USPs exibem extensões N-terminal e C-terminal que parecem ter importante papel na determinação da localização celular e na especificidade/reconhecimento dessas enzimas pelo substrato. Estas proteases atuam em múltiplos níveis na via da ubiquitina, sendo responsáveis pela geração de ubiquitina livre através da hidrólise das cadeias não ancoradas de poliubiquitina e de proteínas precursores de ubiquitina. As UBPs também
4
Ribossomo
1
estão correlacionadas com a estabilidade de fatores transcricionais entre outras funções (Kim, J.H. et al., 2003 e Quesada, V. et al., 2004).
Ubiquitina carboxil-terminal hidrolase (UCH) é o segundo grupo de cisteíno-proteases composto de proteínas relativamente pequenas, entre 20 a 30 kDa, com algumas exceções. UCHs têm um domínio catalítico de aproximadamente 230 aminoácidos, que é estruturalmente caracterizado pela presença de uma tríade catalítica apresentando os resíduos conservados Cys, His e Asp (Kim, J.H. et al., 2003).
As UCHs atuam principalmente na reciclagem de ubiquitina quando esta é inapropriadamente conjugada (como exemplo, glutationa, poliaminas). Elas também estão envolvidas no processamento da síntese de ubiquitina, que é traduzida ou como precursor de poliubiquitina ou fundida ao precursor de proteína ribossomal.
Alguns estudos sugerem um papel para as UCHs em processos específicos regulados por ubiquitina. Mutações em UCH-L1 (UCH expressa especificamente em neurônios), foram descritas em dois irmãos com doença de Parkinson (PD). UCH-L1 mutada tem sua atividade DUB reduzida e um polimorfismo neste gene tem sido relacionado à redução no risco de PD. Nem todos os estudos têm encontrado uma relação estrita entre a atividade UCH-L1 e PD (Nijman, S.M.B. et al., 2005).
A ataxin-3, uma proteína com domínio MJD (Doença Machado-Joseph),
representa o terceiro grupo de cisteíno-proteases. Instabilidade de um nucleotídeo nas repetições CAG no gene da ataxin-3 leva a uma condição neurológica hereditária
conhecida como ataxia espinocerebelar tipo 3 ou doença de Machado Joseph. A
ataxin-3 se associa ao proteassoma, participando da via ubiquitina-proteassoma, e apresenta
propriedades típicas das DUBs (Nijman, S.M.B. et al., 2005 e Amerik, A.Y. &
Hochstrasser, M., 2004). Alguns experimentos indicam que a função normal da ataxin-3
está envolvida na regulação transcricional, mas se a atividade desta DUB tem um papel neste processo não se sabe ao certo.
As proteases OTU (ovarian tumor) compreendem um grupo pontual de
cisteíno-proteases que são homólogas ao gene do tumor ovariano de Drosophila. Otubain-1 e
otubain-2 foram as duas primeiras proteínas OTU onde a atividade DUB foi verificada in vitro. Cézanne, outra proteína contendo o domínio OTU, regula negativamente o
NF-kB clivando cadeias de poliubiquitina in vitro (Balakirev, M.Y., et al., 2003 e Evans,
O grupo das metaloproteases JAMM/MNP+ pode ser representado por um constituinte da tampa do proteassoma 26S, a subunidade Rpn11, que é de extrema importância para a degradação de substratos poliubiquitinados. Outra proteína com motivo JAMM/MPN+ foi recentemente encontrada, a AMSH (molécula associada com o domínio SH3 de STAM), que também tem atividade desubiquitinadora (Amerik, A.Y. & Hochstrasser, M., 2004).
1.7) Modificação pós-traducional dependente de ubiquitina em S. mansoni
Em 2000 foi realizada a primeira caracterização de cDNA codificando para poliubiquitina em S. mansoni, e foi verificada a expressão do gene SmUbi(Schistosoma
mansoni poliubiquitina) em todas as formas de desenvolvimento do parasito (Guerra-Sá,
R., 2000). Além disso, Castro-Borges, W. (2005), utilizando a técnica de Westen Blot em gel de proteína bi-dimensional com extratos totais de cercárias, esquistossômulos de 3horas, 3 dias e vermes adultos, confirmou a presença de conjugados ubiquitinados em todas as fases analisadas. Nas fases larvais, a quantidade de conjugados ubiquitinados detectados foi consideravelmente superior em relação a fase adulta, sugerindo que a via de degradação dependente de ubiquitina e proteassoma exercem papel importante nos processos de remodelagem do S. mansoni.
Tendo como bases as evidências anteriores foi constatada a atividade proteolítica endógena dependente de ubiquitina e do proteassoma 26S em vermes adultos de S.
mansoni. Experimentos realizados na presença do MG132, inibidor da atividade
peptidásica do proteassoma, mostraram 90% de inibição da proteólise exógena e 80% da endógena (Guerra-Sá, R. et al., 2005).
Infecção experimental de camundongos Balb-C com cercárias previamente incubadas com MG132, mostrou uma redução significativa no número dos esquistossômulos, na carga parasitária e no número de ovos presente nas fezes desses animais. Sugerindo assim que a inibição transitória do proteassoma de cercárias compromete o desenvolvimento do parasito no hospedeiro vertebrado. O uso deste inibidor em parasitos adultos mantidos in vitro também mostrou uma alteração no
Analisando os resultados de atividade proteolítica endógena e exógena dependente do proteassoma 20S e 26S de vermes adultos e cercárias, pôde ser observado que a taxa de proteólise é aproximadamente 50% menor em cercária quando comparado a vermes adultos (Guerra-Sá, R., et al., 2005).
Com relação a atividade desubiquitinadora em S. mansoni, Andreolli, A.B.P.
(2005), através de análises in silico, identificou cerca de 32 enzimas desubiquitinadoras,
destas 25 pertencem a família USP e 7 à família UCH. Para a realização desta análise foram utilizados dados gerados no projeto de sequenciamento do Transcriptoma de S.
mansoni.
Considerando a presença de conjugados ubiquitinados em todas as fases de S.
mansoni, a quantidade relevante destes conjugados durante a fase de esquistossômulos e
'
Analisar a expressão das enzimas desubiquitinadoras 5, -14 e -16 durante o ciclo do parasito S. mansoni enfatizando o estágio de transformação de cercária em verme
adulto.
/ - '
1) Obter cercária, esquistossômulos de 3 horas, 18,5 horas, 24 horas, 3 dias de cultivo
in vitro, verme adulto e ovos.
2) Obter in silico o cDNA completo para as enzimas desubiquitinadoras 5, 14 e 16 de
verme adulto e sequenciar estes genes.
1 12
21
22
3.1) Obtenção das formas evolutivas do S. mansoni (vermes adultos, cercárias e
ovos)
O ciclo biológico do S. mansoni, cepa LE é rotineiramente mantido no
Moluscário "Lobato Paraense" do Centro de Pesquisa René Rachou, Fundação Oswaldo Cruz (Fiocruz) em Belo Horizonte/MG.
Os vermes adultos foram perfundidos do sistema porta-hepático de camundongos da linhagem Swiss Webster após 50 dias de infecção com aproximadamente 100 cercárias inoculadas na via subcutânea, conforme descrito por Smithers S.R. & Terry R.J., (1965).
Os ovos do S. mansoni presentes nos fígados de 30 camundongos infectados
foram obtidos após perfusão dos vermes adultos. Os fígados foram picados e colocados em um béquer contendo solução salina 1,7% e deixados a 4ºC por 24 horas. Em seguida o material foi incubado a 37ºC em banho-maria por 2 horas e homogeneizado, com o auxílio de um liquidificador na rotação máxima por 5 minutos. A velocidade do liquidificador foi reduzida para a rotação média e nova homogeneização foi realizada por mais 3 minutos. O homogenato foi lavado através da emissão de jatos de salina 1,7% sob a amostra, utilizando para isto um borrifador. Após a lavagem a mistura foi passada em duas peneiras próprias para separar os ovos do “macerado” de fígado. Depois desta etapa a amostra foi acondicionada em 3 cálices de vidro de 500 mL, e incubada novamente a 4ºC por uma hora para a sedimentação dos ovos. Passada a etapa de sedimentação, as fibras foram removidas por aspiração, os ovos ressuspendidos em salina 1,7% e distribuídos em tubos Falcon de 50 mL. Vários processos de lavagens dos ovos com salina 1,7% foram realizados, seguidos de centrifugação a 4ºC a 1200 x g por 3 minutos e posterior aspiração do sobrenadante.
Os ovos maduros foram separados dos ovos imaturos e dos hepatócitos através de um gradientes descontínuos de PercollTM (Amersham Biosciences) preparado de
23
descartado e os ovos maduros submetidos a sucessivas lavagens com meio RPMI 1640 (INVITROGEN) seguido de centrifugações por 30 segundos a 1200 x g até completa remoção do Percoll. Os ovos purificados foram estocados a -70°C até o momento do
uso.
As cercárias foram obtidas de caramujos B. glabrata infectados através de um
processo de eliminação induzido artificialmente. Em um béquer com 200 mL de água declorada aquecida a 28ºC foram colocados 50 caramujos e estes expostos à luz branca fria em uma estufa a 25ºC. Depois de duas horas de exposição dos moluscos a luz, as cercárias eliminadas foram recolhidas. Após centrifugação por 10 minutos a 12.000 x g, uma alíquota das cercárias foi armazenada a -70ºC até o momento de uso.
3.2) Transformação mecânica de cercárias em esquistossômulos in vitro
Aproximadamente 100.000 a 150.000 cercárias foram obtidas segundo o procedimento anteriormente descrito. Processos de lavagens com água declorada foram realizados para a limpeza dos resíduos dos caramujos. As cercárias foram transferidas juntamente com a água declorada para um béquer previamente gelado e após duas horas de incubação em banho de gelo obteve-se um sedimento. O sobrenadante foi removido e descartado com o auxílio de uma pipeta e o sedimento de cercárias transferido para 4 tubos do tipo Falcon de 50 mL. Em cada tubo foi adicionado 25 mL de água declorada e o material foi novamente sedimentado por 15 minutos em banho de gelo. O sobrenadante foi retirado com o auxílio de uma pipeta e o procedimento repetido por mais duas vezes ou até o sedimento de cercárias ficar completamente sem resíduos. Na última lavagem foi adicionado meio de cultura RPMI 1640 (INVITROGEN). Os sedimentos de cercárias contidos nos tubos tipo Falcon de 50 mL foram concentrados em um único tubo e o sobrenadante retirado deixando apenas um volume final de 5 mL.
O processo de transformação de cercárias em esquistossômulo foi realizado utilizando o método descrito por Ramalho-Pinto F.J. et al., (1974). As cercárias,
24
esquistossômulos, concentraram-se ao fundo do tubo tipo Falcon e as caudas suspensas no sobrenadante foram descartadas.
Os esquistossômulos foram transferidos para um frasco de cultura de 75 cm2 e a
ele acrescentado 30 mL de meio de cultura RPMI suplementado com 100 U/mL de penicilina e 100 g/mL de estreptomicina. Em seguida a cultura foi incubada em estufa de CO2 a 5% a 37°C por 3 horas, tempo necessário para completar a transformação.
Após esse período, 28 mL de RPMI contendo caudas cercarianas foram retirados e descartados. Os outros 2 mL foram transferidos para dois tubos de poliestireno estéreis (tipo eppendorf). Os esquistossômulos foram submetidos a sucessivas lavagens (10 a 12) com meio RPMI, seguidas por sedimentação com intervalos de 4 minutos e remoção das caudas remanescentes suspensas no sobrenadante. Posteriormente todo o sobrenadante foi removido e o pellet contendo os corpos cercarianos(neste momento os
parasitos foram considerados larvas de 3 horas), sem as caudas, foi estocado em tubo de poliestireno estéril (tipo eppendorf) e armazenado a -70°C até o momento do uso.
25
Tabela 1- Composição do meio 169
Componentes Concentração
Meio líquido BME (Eagle) __
Hidrolisado de lactoalbumina 0,1%
Glicose 0,1%
Hipoxantina 5 X 10-7M
Serotonina 1 X 10-6M
Hidrocortisona 1 X 10-6M
Triiodotironina (T3) 210-7M
Meio Mínimo Vitamina 0,5%
Meio Schneider 5%
3.3) Extração de DNA genômico
Para a extração do DNA genômico de S. mansoni foram utilizadas cerca de
100.000 cercárias. O protocolo adotado foi descrito por Costa P.I. (Tese de doutorado, 1997), apresentando neste trabalho algumas modificações.
As cercárias armazenadas previamente a -70ºC foram ressuspensas em 500 µL
de tampão de extração (Tris 50 mM , EDTA 1 mM, pH 7,5 e 1% de N-Laurilsarcosina). A amostra foi incubada a 37ºC por 16 horas contendo 50 µL de proteinase K 20
mg/mL. Posteriormente foram adicionados 250 µL de NaCl 5M à amostra e outra
incubação a 65ºC foi realizada por 10 minutos. Para a desproteinização da amostra foi adicionado 100 µL de brometo de cetiltrimetilamônio (CTAB)/NaCl 10%, após 20
26
adicionado 60 µL de água milli-Q estéril. O DNA genômico foi incubado durante uma
noite a 37° C e na manhã seguinte uma alíquota de 5 L, analisada em gel de agarose
0,5% em eletroforese a 90 volts. A Figura 6 mostra uma preparação típica de DNA genômico obtido utilizando o protocolo descrito acima.
A quantificação da amostra foi obtida por espectrofotometria com leitura em densidade ótica de 260 e 280 nm. A pureza da preparação de DNA genômico foi estimada utilizando a relação A260 /A280 nm.
3.4) Extração de RNA total
Após a obtenção do material biológico (vermes adultos, cercárias, ovos e esquistossômulos de 3 horas, 18,5 horas, 24 horas e 3 dias) o RNA total foi extraído, utilizando Trizol (Invitrogen), como descrito abaixo.
Aproximadamente 100 mg de vermes adultos foram homogeneizados em vortex, na rotação máxima, contendo 1,0 mL de Trizol LS (Invitrogen) e incubados a temperatura ambiente por 60 minutos. A cada 15 minutos novas homogeneizações em vortex foram realizadas até a completa solubilização dos vermes. Para que a amostra não apresentasse contaminação com DNA genômico, a mistura foi passada 10 vezes em uma seringa descartável (10 mL/25 x 8) com agulha, para fragmentar o DNA, facilitando assim sua separação do RNA durante os procedimentos de extração. Em seguida, foram adicionados 200 µL de clorofórmio à amostra e realizada uma agitação
moderada com o auxílio de vortex durante 1 minuto. Posteriormente, uma incubação Figura 6 - DNA genômico de cercária de S. mansoni
27
por 20 minutos a temperatura ambiente foi realizada e a amostra foi centrifugada a 12.000 x g durante 10 minutos.
Após centrifugação três fases foram observadas, a fase aquosa foi transferida para um tubo de poliestireno estéril (tipo eppendorf) e a ela adicionados 500 µL de
isopropanol para a precipitação do RNA total. O tubo foi invertido por três vezes cuidadosamente, incubado a temperatura ambiente por 30 minutos e centrifugado a 12.000 x g durante 10 minutos. O sobrenadante foi descartado e o precipitado lavado com etanol 70% gelado seguido de nova centrifugação por 10 minutos a 12.000 x g. Novamente o sobrenadante foi descartado e o RNA foi seco a temperatura ambiente em fluxo laminarpor 15 minutos. O RNA total foi ressuspenso em 30 µL de água tratada
com dietilpirocarbonato (DEPC) e incubado por 10 minutos a 56° C.
Para a extração de RNA total das fases larvais do ciclo do S. mansoni, foram
utilizadas cerca de 150.000 cercárias, 100.000 esquistossômulos e 5.000 ovos. Na extração de RNA total de esquistossômulos utilizamos glicogênio a 40 g/mL para co-precipitar o RNA das amostras juntamente com o isopropanol. E na extração de RNA total de ovos após a adição do Trizol a mistura foi congelada em nitrogênio líquido e levada ao vortex por três vezes para romper as cascas dos ovos e garantir uma boa extração do RNA.
A qualidade do RNA total foi avaliada em gel de agarose-formaldeído 1%. Para a preparação do gel, inicialmente 600 mg de agarose foram dissolvidos em 40,0 mL de água DEPC autoclavada. Em outro recipiente, foram adicionados: 12,8 mL de água DEPC, 6,0 mL de MOPS 10X concentrado (MOPS, acetato de sódio diidratado, EDTA tetrassódico, água DEPC completando para um volume final de 500 mL), 1,2 mL de formaldeído e 0,5 L de brometo de etídio. A solução foi misturada à agarose e deixada em repouso até completa solidificação. As amostras foram previamente desnaturadas em tampão (formamida, formaldeído, MOPS 1X, azul de bromofenol, água DEPC e brometo de etídio), incubadas por 15 minutos a 65°C, deixadas em banho de gelo
durante 3 minutos e aplicadas no gel. As condições de realização da eletroforese foram: 80 volts em tampão de corrida (MOPS 1X) por aproximadamente 75 minutos. A Figura 7 mostra uma preparação típica de RNA total, obtido como descrito acima.
28
3.5) Oligonucleotídeos iniciadores específicos para os genes DUB5, DUB14 e DUB16 (deubiquitinating enzyme)
Os oligonucleotídeos iniciadores específicos para os genes DUB5, DUB14 e DUB16 (deubiquitinating enzyme), foram idealizados no programa GenneRunning. As
seqüências utilizadas são originadas do banco de dados da FAPESP (Fundação de Apoio à Pesquisa do Estado de São Paulo), que foi criado através do consórcio de sequenciamento do transcriptoma de S. mansoni.
Durante a análise das seqüências optamos por desenhar iniciadores, cujos produtos, cobrissem a maior parte das seqüências das enzimas desubiquitinadoras em estudo. Na Tabela 2 estão listados os iniciadores gerados pelo programa Genne Running e seus respectivos número de acesso (Contig).
Tabela 2 - Oligonucleotídeos iniciadores gerados de DUB5, DUB14 e DUB16
Enzima Contig F (direto) R (Reverso)
Produto Esperado
(nt)
DUB 5 607508.1 (F) 5'ACAAGCACACAAGAGCAG3'
(R) 5'ATCTTCCACTGATGCTGG3' 550
DUB 14 609068.1 (F) 5'GCTTATTCCACGGAAGAC3’
(R) 5'GTTGCAATTCAGGCGTAC3' 844
DUB 16 608180.1 (F) 5'GGCTGTTTAGATCAGTGC3'
(R) 5'GGGAACAGAACAGAAAGG3' 882
Figura 7 – Extração de RNA total das fases evolutivas de S. mansoni. Gel de agarose/formaldeído 1% contendo em cada canaleta aproximadamente 5 g de RNA total. (E) abreviatura de esquistossômulos e (h) horas.
!
"#
29
Para validar os iniciadores gerados, PCR’s (Polimerase Chain Reaction) foram
realizadas para amplificar os possíveis genes das enzimas desubiquitinadoras 5, 14 e 16. O DNA genômico de cercária foi utilizado como DNA molde para a reação.
Cada reação de amplificação foi realizada em tubo ependorff de 200µl,
contendo aproximadamente 350 ng de DNA genômico, 10 mM de cada dNTP’s (dATP, dCTP, dTTP, e dGTP - Promega), 50 mM de MgCl2, 5 picomoles de cada
oligonucleotídeo (direto e reverso), 2,5 unidade de Taq DNA polimerase (Invitrogen) e 1X do tampão PCR 10X concentrado sem Mg+2; completando o volume da reação com água milli-Q para 50 µl.
Depois de vários testes as temperaturas de anelamento das DUB’s foram padronizadas em 49ºC para DUB 5 e DUB 16, e 47ºC para DUB 14. A seguinte ciclagem foi utilizada nas reações de amplificação: após uma desnaturação inicial de 2 minutos a 95ºC um programa de 35 ciclos foi estabelecido, com desnaturação a 95ºC por 1 minuto, anelamento a 49ºC (DUB 5 e 16) ou 47ºC (DUB 14) por 1 minuto, extensão por 90 segundos a 72ºC e uma elongação final de 6 minutos a 72ºC.
Os produtos das PCR’s foram aplicados em gel de agarose 1,2% (90 volts – 75 minutos), a temperatura ambiente, juntamente com o padrão de peso molecular 100pb DNA Ladder (Invitrogen). O gel foi corado com brometo de etídio, observado ao transiluminador (Vilber Lourmat) e fotografado.
3.6) RT-PCR (Reverse- Transcription Polimerase Chain Reaction)
Para a confirmação dos genes preditos DUB 5, DUB 14 e DUB 16, através do sequenciamento, foi obtido o cDNA a partir de mRNA do verme adulto de S. mansoni.
As técnicas utilizadas para a obtenção deste cDNA estão detalhadas a seguir.
A partir do RNA total obtido de verme adulto foi sintetizada uma fita simples de cDNA pela atividade da enzima transcriptase reversa(Thermoscript RT- PCR System
-Invitrogen), seguindo as recomendações dadas pelo fabricante. Inicialmente foram utilizados aproximadamente 5 µg de RNA total, 50 pmol de oligodT, 10 mM de dNTPs
e água livre de RNAse para um volume final de 10 µL. A desnaturação do RNA foi
30
durante 5 minutos, seguida de banho de gelo por 1 minuto. Em seguida, foi adicionado o tampão da enzima, 0,1 M de DTT (ditiltreitol) e 15 unidades da enzima transcriptase reversa. Posteriormente, essa mistura foi incubada em termociclador a 37°C durante 60
minutos, seguida de outra incubação a 85°C por 5 minutos. Finalizando a reação foi
acrescentado 1,0 µL de RNAse H e a amostra incubada por 20 minutos a 37ºC para a
digestão do RNA utilizado como molde. A amostra foi estocada a -20°C até o momento
do uso.
No intuito de obter o cDNA fita dupla, amplificações foram realizadas utilizando 2µL do cDNA de verme adulto de S. mansoni, combinados com os
oligonucleotídeos específicos para a amplificação dos genes DUB 5, DUB14 e DUB 16. Nestas reações de amplificações foram utilizados os parâmetros previamente estabelecidos para os genes de DUB, como descrito no item 3.5.
3.7) Purificação e clonagem dos produtos das PCR’s
Os produtos das PCR’s gene específicas foram purificados através da adição de 1/10 de acetato de sódio 3M pH 7.0 por tubo e dois volumes de etanol absoluto, sendo em seguida incubados a -20ºC por 30 minutos. Após centrifugação a temperatura ambiente por 10 minutos a 12.000 g, o sobrenadante foi descartado e o pellet lavado com 500µl de etanol 70%. Depois de centrifugado nas mesmas condições que a etapa
anterior, o etanol 70% foi descartado e o pellet ressuspendido em 50 µl de água milli-Q
estéril.
As reações de ligação dos produtos de PCR purificados DUB5, DUB14 e DUB16 ao vetor pGEMT Easy (Promega) foram realizadas conforme orientações do
fabricante. Foram utilizados 3 µL do produto de PCR (150 ng/µL) por reação de
ligação que foi incubada por 16 horas a 4°C.
3.8) Obtenção de células competentes para transformação
As bactérias E.coli – DH5 α utilizadas em experimentos de transformação
foram tornadas competentes de acordo com o método Pipes (Hanahan, D., 1985). Bactérias E.coli – DH5 α não competentes, armazenadas a -70°C foram
31
seletivo (sem antibióticos) Luria-Bertani caldo (10 g de NaCl; 5 g de extrato de levedura; 10 g de tryptona). Este meio foi suplementado com 50 µL de MgSO4 1 M, 50
µL de MgCl2 1 M e incubado a 37°C por 16 horas sobre agitação orbital de 200 x g
(agitador - B. Braum Biotech International). Uma alíquota de 250 µL desta cultura foi
expandida em um tubo tipo Falcon contendo 50 mL de meio SOB (20 g triptona, 5 g extrato de levedura, 0,58 g NaCl, 0,2 g KCl pH 7,5, com KOH para 1 litro), onde foi adicionado 250 µL de MgSO4 1 M e 250 µL de MgCl2 1 M. A cultura de bactérias foi
incubada a 37°C sob agitação orbital de 270 x g, por 2 horas, ou até atingir uma leitura
em densidade ótica de 600 nanômetros (espectrofotômetro Metrolab 1700 U.V. visível). Ao atingir DO 600 entre 0,4 e 0,6 nm a cultura foi incubada em banho de gelo por 30 minutos e centrifugada a 4°C 3.000 x g por 10 minutos. O sobrenadante foi descartado
e o precipitado celular ressuspendido em 10 mL de tampão Pipes (CaCl2 60 mM, 100
mL de água destilada, 10 mM Pipes, 15% de Glicerol pH 7.0) gelado. A suspensão de células foi novamente centrifugada a 4°C 2.000 x g por 10 minutos e o sobrenadante
descartado, esta etapa foi repetida por mais duas vezes. As células foram ressuspendidas em mais uma vez em tampão Pipes e incubadas em banho de gelo por 30 minutos, após este intervalo outra centrifugação a 4°C 2.000 x g por 10 minutos foi
realizada. O precipitado bacteriano foi resuspendido com 1,2 mL de tampão Pipes, alíquotas de 200µl foram colocadas em tubo de poliestireno estéril (tipo eppendorf) e
mantidas no gelo para proceder à transformação.
3.9) Transformação de bactérias E. coli DH5 αααα
Os plasmídios recombinantes foram introduzidos em bactérias competentes da linhagem DH5α como descrito a seguir.
Aos tubos de poliestireno estéril (tipo eppendorf) contendo as células E.coli
DH5 α competentes foram colocados, em cada tubo, aproximado de 5 µL daconstrução
plasmidial pGEMT-Easy/DUB5, pGEMT-Easy/DUB14 e pGEMT-Easy/DUB16 e levados a banho de gelo por 30 minutos seguido por um choque térmico de 42°C por
exatamente 90 segundos e novamente incubados no gelo por 2 minutos. Foi adicionado 1 mL de Luria-Bertani caldo (LB caldo) por tubo e as amostras incubadas por 90 minutos a 37°C sob agitação orbital de 270 x g. Após a incubação 100µl da cultura foi
32
mg/mL e 40 mg/mL de Xgal (LB-XIA), de acordo com Maniatis. As placas foram incubadas a 37°C durante a noite e os clones recombinantes foram identificados pela
seleção azul/branca.
Controles negativos com o plasmídio sem o inserto foram preparados em paralelo para descartar a possibilidade de contaminação e um controle positivo que atesta a eficiência das células competentes foi realizado utilizando o plasmídio PUC 18. As colônias brancas que cresceram foram coletadas isoladamente e inoculadas em 1 mL de LB caldo contendo 100 µg/mL de ampicilina em tubos de poliestireno
estéreis (tipo eppendorf) e incubados a 37°C sob agitação orbital de 220 x g durante a
noite. Em seguida, após adição de 30% do volume de glicerol, foram armazenadas a -70°C.
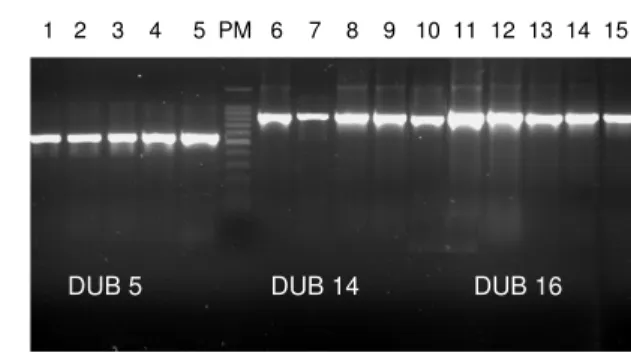
Para verificar a presença dos insertos nos transformantes, PCR das colônias foram realizadas utilizando como DNA molde cerca de 2 µl da cultura de cada clone
obtido. Os produtos das PCR’s foram aplicados em gel de agarose 1,2% corado com brometo de etídio que está representado na Figura 8.
3.10) Mini-preparação dos plasmídios recombinantes
Cerca de 50 L de cultura de cada clone recombinante obtidos como descrito anteriormente foram crescidos em 5 mL de LB/ampicilina, durante a noite sob agitação orbital a 37°C. Aproximadamente 3 mL foram retirados da cultura bacteriana de cada
clone, transferidos para tubos de poliestireno e centrifugados a 14000 x g a temperatura ambiente por 3 minutos. Os precipitados foram homogeneizados em 300 µL de
GTE/RNAse (500 mM TRIS-HCl; 10 mM EDTA; 100 µg de RNAse A; pH 8,0) e
DUB 5 DUB 14 DUB 16
Figura 8 – PCR das colônias. Nas canaletas de 1 a 5 foram aplicadas alíquotas dos produtos de PCR de 5 transformantes contendo o gene DUB 5. Nas canaletas 6 a 10 pode ser verificada a presença do gene DUB 14 nos transformantes e de 11 a 15 também há amplificações para o gene DUB 16. PM – 100pb (Invitrogen)