Ce document a été réalisé avec LATEX2ε et thèses-IRINversion 0.92 classe de l'association LOGIN des jeunes chercheurs en informatique, Université de Nantes. Merci aux membres de l'Institut d'électronique et d'informatique Gaspard-Monge de l'Université de Marne-la-Vallée de m'avoir permis, en m'accueillant pour la rentrée 2006, de revenir en région parisienne.
Introduction
La deuxième partie de ce manuscrit concerne nos travaux visant à comparer les structures des molécules d'ARN. Dans le chapitre 3, nous donnons un aperçu des résultats existants concernant quatre problèmes impliquant les structures des molécules d'ARN.
P ARTIE I
Concepts généraux
C HAPITRE 1
Notions de génétique
La génétique du XIX eme ` siècle à nos jours
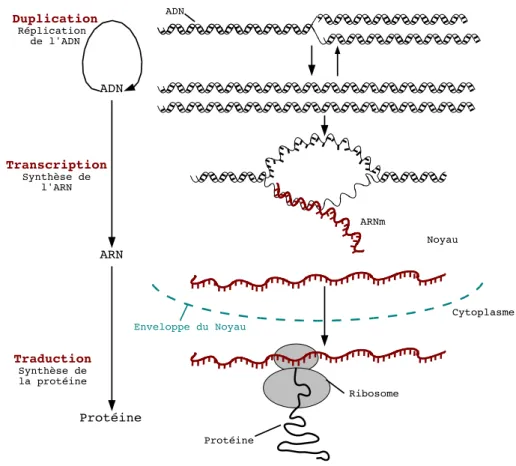
C'est en 1957 que Crick et le physicien nucléaire George Gamov définissent le dogme central de la biologie moléculaire. cf. Ces concepts seront présentés dans leur ordre d'utilisation dans le déploiement des mécanismes du dogme central de la biologie moléculaire.

Le dogme central de la biologie moléculaire
- Les gènes
- L’Acide DésoxyriboNucléique
- L’Acide RiboNucléique
- Les protéines
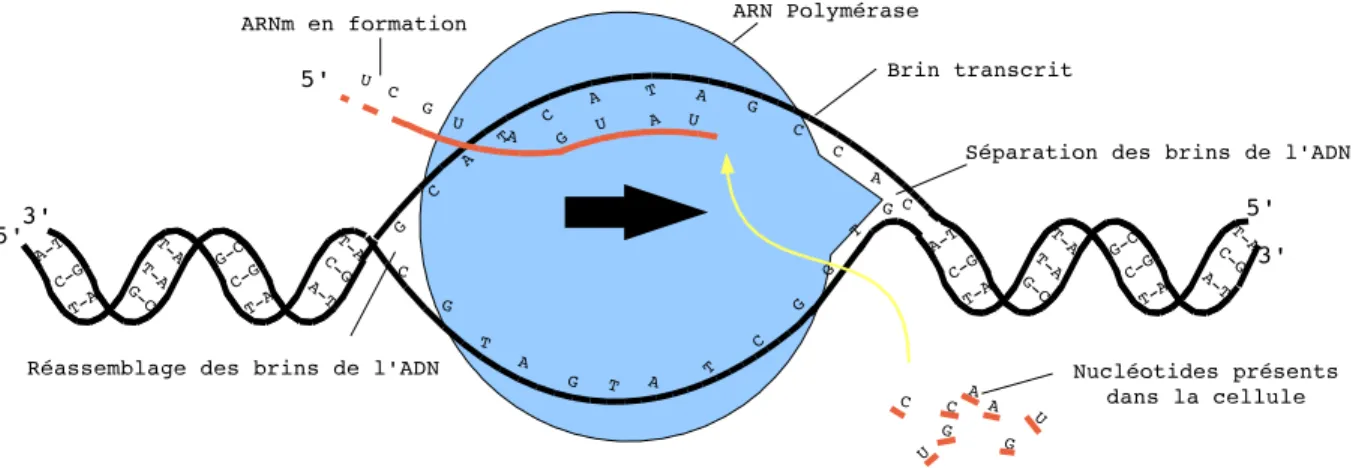
Deuxièmement, l'ADN polymérase va attacher les nucléotides disponibles dans le noyau de la cellule aux bases complémentaires d'un des brins libérés. Cette structure est chargée de l'organisation des éléments de la structure secondaire formant entre eux un tout compact.
C HAPITRE 2
Complexité et algorithmique
- Différences entre problème et algorithme
- Classification des algorithmes
- Classification des problèmes
- Classes P et NP
- Les problèmes NP-complets
- Contourner la NP-complétude
- Branch-and-Bound
- Heuristique
- Approximation
- Complexité paramétrée
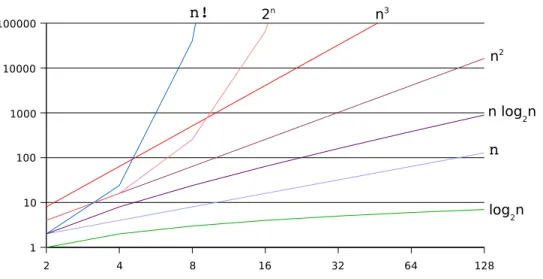
La complexité d'un algorithme est une mesure du nombre d'opérations atomiques (c'est-à-dire indivisibles) qu'il effectue sur les entrées du problème. La complexité du problème correspond à la complexité de l’algorithme connu1, qui est le plus efficace pour le résoudre.

P ARTIE II
Comparaison de structures de molécules d’ARN
C HAPITRE 3
Présentation générale
Les séquences arc-annotées
- Notations et modélisation de molécules d’ARN
- Quelques problèmes utilisant les séquences arc-annotées
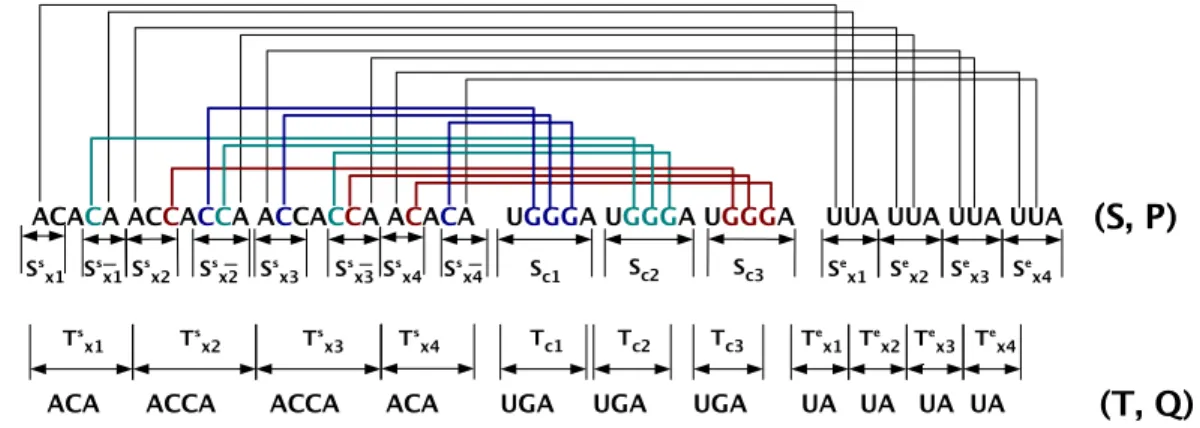
Notez que dans cette extension, les opérations d'édition d'un caractère libre ne peuvent pas être appliquées à un caractère marqué. 2Dans [75], les opérations d'insertion d'un arc et de marquage de deux caractères ne sont pas explicitement définies, mais sont prises en compte.

EDIT
- Les 2-intervalles
- Notations et modélisation de molécules d’ARN
- Quelques problèmes utilisant les 2-intervalles
Par cette définition, si un ensemble de deux intervalles D est R-similaire, D est un ensemble de deux intervalles disjoints. Le support d'un ensemble de 2 intervalles D, noté Support(D), est l'ensemble de tous les intervalles impliqués dans D. Toute structure secondaire sans pseudo-nœud peut être modélisée à l'aide d'un ensemble de 2 intervalles {<, @} similaires qui respectent le niveau de complexité du support DISJOINT.
Tout modèle à 2 intervalles peut être associé à un ensemble de 2 intervalles respectant un modèle d'équation exact.

C HAPITRE 4
Introduction
Un ensemble indépendant de sommets d'un graphe G = (V, E) est un ensemble V0 ⊆ V de sommets de V tel qu'il n'existe aucune arête dans E reliant deux sommets de V0.
NP-complétude du sous-problème L-EDIT( NESTED , NES -
TED )
Conclusion et perspectives
Malheureusement, l’utilisation de ces nouvelles opérations d’édition pour la comparaison de structures secondaires rend le problème L-EDIT(NESTED,NESTED) NP-complet. Malgré la difficulté, en général, du problème du calcul de la distance d'édition entre deux structures secondaires de molécules d'ARN, il existe des algorithmes précis et efficaces pour résoudre ce type de problème dans des classes d'instances plus spécifiques. La figure 4.5 montre le calcul de la distance de modification entre deux arbres représentant les structures secondaires des molécules d'ARN.
La polynomialité de cet algorithme est rendue possible par le fait que nous ne prenons pas en compte certains cas pouvant être inclus dans le calcul de la distance de contrôle comme Lin et al.

C HAPITRE 5
Le problème A RC -P RESERVING
S UBSEQUENCE
- Introduction
- NP-complétude du sous-problème APS( CROSSING , PLAIN )
- Raffinement du problème APS
- De nouveaux niveaux de complexité
- Résultats triviaux
- Polynomialité de certains sous-problèmes de APS
- Conclusion et perspectives
Dans cette section, nous montrons que le sous-problème APS(CROSSING,PLAIN) est NP-complet. Selon notre modèle, ce résultat implique que le sous-problème APS({<,@},{<,@}) peut être résolu par un algorithme de complexité temporelle O(nm). Il existe un algorithme de complexité temporelle O(nm2) pour résoudre le sous-problème APS({./},{./}).
Il existe un algorithme de complexité temporelle O(nm2) pour résoudre le sous-problème APS({./},∅).
C HAPITRE 6
La recherche de motifs de 2-intervalles
Introduction
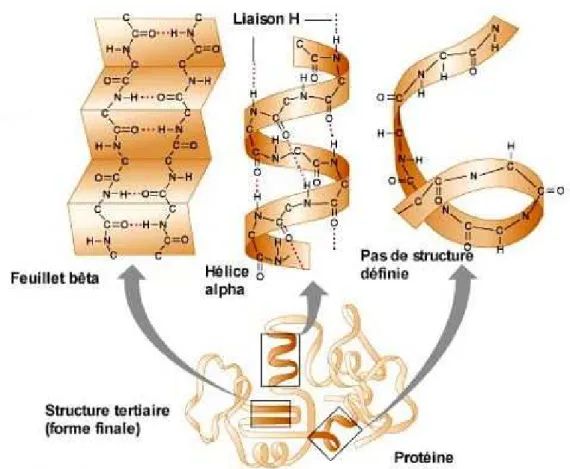
L’élément le plus à gauche (resp. le plus à droite) d’un ensemble d’intervalles 2-disjoints D désigne le 2-intervalle Di ∈ D tel que Left(Di) Un graphe de trapèze est un graphe d'intersection d'un ensemble fini de trapèzes entre deux droites parallèles. Nous avons donné un algorithme optimal de complexité temporelle O(nlogn) pour le sous-problème 2-IP(UNLIMITED,{@}), qui améliore la meilleure complexité existante pour ce problème. Enfin, nous avons prouvé que le sous-problème 2-IP(UNITARY,{<, ./}) est NP-complet, et de plus, nous avons montré que ce sous-problème dans sa généralité appartient à la classe FPT pour un paramètre fortement associé à l'intersection structure de l’ensemble des 2 intervalles. Étant donné le tableau 6.1, nous supposons qu'un algorithme polynomial existe pour ce problème. Par contre, vous savez si le sous-problème 2-IP(n1,{<, ./})∀n1∈ {UNLIMITED, UNITARY, DISJOINT} est dans la classe FPT pour le paramètre Depth(D) (en fait, rappelez-vous que FCcrossing ( D) + 1 ≥ Depth (D)) reste un problème ouvert. Données : un graphe de structure induiteGΓavecnsommets, un ensemble de fonctions de similaritéF ={f1,. fn}et une bonne dichotomieP ={Et,Eb}des arêtes de GΓ. Ensuite, nous prouverons que le problème MRSO est NP-complet même si le nombre minimum de pages du graphe de structure induit est de deux. Le problème MRSO est NP-complet si le graphe de structure induit a un nombre minimum de pages égal à 2. Étant donné un ensemble de n fonctions de similarité F, une séquence d'ARNm S de longueur 3n et un graphe structurel Γ avec V(Γ) = {u1, u2,. P ARTIE III Calcul de distances intergénomiques en présence de gènes dupliqués Un intervalle commun entre P1 et P2 est une séquence consécutive de gènes de P1 qui, étant donné le signe et l'ordre, est également une séquence consécutive de gènes de P2. Dans ce manuscrit nous nous intéressons aux problèmes de calcul du nombre d'inversions, de points d'arrêt, d'intervalles communs et d'intervalles conservés entre deux permutations. Dans le cas des points d'arrêt, un algorithme naïf parcourant les deux permutations a une complexité temporelle quadratique, par contre si l'on considère que l'une des deux permutations est la permutation identité, alors il est possible de calculer le nombre de points d'arrêt entre une permutation et l'identité de permutation dans O (n). De même, dans le cas d’intervalles totaux, un algorithme naïf peut calculer le nombre d’intervalles totaux au carré du temps. Ce résultat de complexité reste valable même dans le cas de la version non signée du sous-problème 2-SBR. En revanche, la complexité des deux stratégies dans le cas de la distance des intervalles conservés n'est pas encore définie. Christie et al.) ont montré la NP-complétude de la version signée (resp. non signée) du problème. En revanche, la complexité du problème du calcul de la distance de rupture dans le cas de l'utilisation d'accouplements est inconnue. C HAPITRE 9 Soit GetH deux génomes équilibrés tels qu'il existe une seule famille de gènes dupliqués et qu'aucun segment de gène dupliqué consécutif ne contienne plus de gènes L. Il existe une union qui minimise la distance de point d'arrêt entre les gènes dans GetH , de telle sorte que chaque segment de gène dupliqué dans G est parfaitement lié à son homologue dans H. Soit GetH deux génomes équilibrés de telle sorte qu'il y ait une seule famille de gènes dupliqués et aucun segment de gène dupliqué, les doublons consécutifs ne contiennent pas plus de Lgènes. Données : prenez deux génomes équilibrés en H, de telle sorte qu'il existe une seule famille de gènes dupliqués et qu'aucun segment de gène dupliqué consécutif ne contienne plus de gènes. C HAPITRE 10 Dans cette section, nous abordons le problème du calcul de la distance des intervalles conservés en présence de gènes dupliqués à l'aide d'une stratégie d'échantillonnage. Dans cette section, nous abordons le problème du calcul de la distance des intervalles conservés en présence de gènes dupliqués à l'aide de la liaison. D'après le lemme 10.3, nous concluons donc qu'il existe au plus p(p-1)2 intervalles conservés pour chaque segment de gènes dupliqués de longueur p. D’après le lemme 10.4, nous savons que Dic(G, H) = k, et que quel que soit le lien entre les gènes GetH, chaque intervalle conservé I a la forme [α, β] ou I = [p, q ], tel que S [p, q] est un segment constitué uniquement de gènes dupliqués. Enfin, nous avons prouvé que le problème reste NP-complet dans le cas où le graphe peut être imbriqué sur deux pages. Un autre axe de recherche que nous avons suivi concerne le calcul des distances intergénomiques en présence de gènes dupliqués. Nous avons pris en compte quelque chose qui a été peu étudié jusqu'à présent, la présence de gènes dupliqués dans le calcul de deux distances évolutives : la distance du point de rupture et la distance des intervalles conservés. Nous avons également prouvé que l’utilisation de l’instanciation dans le cas de la distance entre intervalles conservés induit la NP-complétude du problème. In Proceedings of the 13th Annual European Symposium on Algorithms (ESA'05), To appear in Lecture Notes in Computer Science. In Proceedings of 4th Workshop on Algorithms in Bioinformatics (WABI’04), Lecture Notes in Computer Science, pages 38–49. In Proceedings of the 6th Annual European Symposium on Algorithms (ESA'98), Lecture Notes in Computer Science, pages 91–102. In Proceedings of the 15th Annual Symposium on Combinatorial Pattern Matching (CPM’04), volume 3109 Lecture Notes in Computer Science, pages 1–13. Liste des tableaux Concepts généraux Par souci de lisibilité, les longueurs des régions mono-chaînes n’ont pas été respectées. a) Un graphe cubique planaire connexe G pa. c) Exemple du problème L-EDIT(NESTED, NESTED) issu de la construction UA. 62 4.6 Deux nouvelles opérations d'édition entre deux arbres représentant des structures secondaires. a) Une opération d'appairage/dissociation correspondant à la coupure d'arc. 73 6.1 Illustration de la construction d'une collection de trapèzes à partir d'une collection de i. a) une collectionD de 2 intervalles et (b) la collection correspondante de trapèzes. 133 10.3 Un segment de gènes dupliqués SdeG couplé à deux segments disjoints TetT0deH.134 10.4 Occurrence du problème MCIM associé à l'apparition du problème MINIMUM BIN PA. À l’intérieur de chacune de leurs cellules, les eucaryotes possèdent un noyau : un petit sac entouré d’une membrane semi-perméable qui renferme les chromosomes. Les eubactéries et les archées n'ont pas de véritable noyau, mais une structure beaucoup plus simple, non entourée de membrane. Feuille bêta Structure d'une molécule biologique formée par le repliement en accordéon d'une série d'acides aminés sur elle-même. Hélice alpha Structure d'une molécule biologique formée par le repliement en tire-bouchon d'une séquence d'acides aminés sur elle-même. Théorie de la complexité L'étude formelle de l'efficacité des algorithmes ainsi que de la difficulté inhérente des problèmes. Étape de traduction de la synthèse protéique (fabrication) durant laquelle le brin d'ARN messager obtenu lors de la transcription est converti en une chaîne d'acides aminés qui produira une protéine. Transcription Processus de production d'un ARNm à partir de la séquence codante d'un gène qui sert de matrice. Transformation polynomiale Il existe une transformation polynomiale d'un problème de décision P1 en un problème de décision P2 si, étant donné une instance I1 de P1, il est possible de construire une instance I2 de P2 en temps polynomial (par rapport à la taille de I1) telle que la réponse à I1 est « oui » si et seulement si la réponse à I2 est également « oui ». Combinatoire et Bio-informatique: Comparaison de structures d’ARN et calcul de distancesConclusion et perspectives
C HAPITRE 7

C HAPITRE 8
Distances et permutations
Distances et séquences d’entiers signés

Couplage et distance de breakpoints en présence

La distance d’intervalles conservés en présence de

Conclusion générale
Bibliographie
Comparaison de structures de molécules d’ARN
Glossaire

![Figure 1.12 – Structure secondaire de l’ARN ribosomal 16s d’Escherichia Coli [60] (vue partielle).](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465062.70396/28.918.213.646.282.692/figure-structure-secondaire-arn-ribosomal-escherichia-coli-partielle.webp)
![Figure 1.13 – Structure tertiaire d’une molécule d’ARN: un pseudo-noeud de l’ARN ribosomal 16s d’Escherichia Coli [60] (vue partielle).](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465062.70396/29.918.256.604.111.319/figure-structure-tertiaire-molécule-pseudo-ribosomal-escherichia-partielle.webp)