Je tiens tout particulièrement à remercier le Docteur Morwenna Pearson-Long pour sa très forte implication dans mes travaux de thèse à travers son encadrement dynamique et bénévole, ainsi que son aide précieuse dans la rédaction et la correction de la partie expérimentale de ce document. II - Développement d'une méthode de formation de complexes chiraux du titane 93 II.1 - Stratégies de formation de complexes entre un diol et Ti(Oi-Pr)4.
Introduction
Dans le cadre de cette thèse, nous allons maintenant décrire des méthodes de cyclopropanation de dérivés d'acide carboxylique au moyen de complexes dérivés de XTi(Oi-Pr)3. Ainsi, après un rappel de la structure, des méthodes de formation et de la réactivité des complexes d'oléfines de titane, leurs utilisations pour la synthèse des cyclopropanols ainsi que des cyclopropylamines tertiaires et primaires à partir des dérivés d'acide carboxylique correspondants seront présentées.
I - Présentation des complexes oléfiniques de titane
Dans le cas de réactions de cyclopropanation de dérivés acides, les complexes oléfiniques du titane utilisés contiennent des groupements alcoxy, qui ne stabilisent pas suffisamment le complexe pour permettre son isolement et son analyse. La formation de I.1 permet de mettre en évidence le caractère 1,2-dicarbanionique du complexe oléfinique du titane A, confirmant ainsi sa représentation par un métallacycle.

II - Synthèse de cyclopropanols
L'insertion de la fonction ester dans le complexe A est sous contrôle cinétique, Wu et al. Pour cette synthèse, le complexe I.9 donne les meilleurs résultats lors de la réaction d'échange de ligand.
III - Synthèse de cyclopropylamines tertiaires
L'élimination de cette amine d'une manière similaire à l'élimination de l'alcoxyde dans le mécanisme de cyclopropanation de l'ester (voir diagramme I-10) conduirait à la formation d'un amide instable. De la même manière que dans le cas des esters, la cyclopropanation des amides peut être précédée d'un échange de ligand entre le complexe oléfinique du titane issu d'un réactif de Grignard et un alcène.
IV - Synthèse de cyclopropylamines primaires
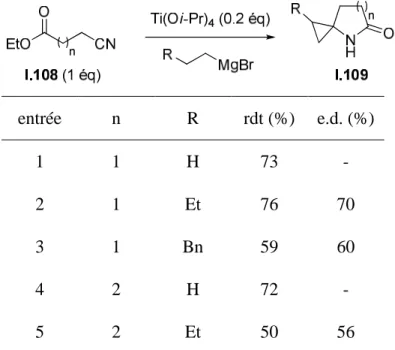
En interagissant avec l'atome d'azote de l'intermédiaire B, l'acide de Lewis (typiquement BF3.OEt2) augmente l'électrophilie de la fonction imine métallisée. Cependant, Szymoniak et Bertus ont noté que les substrats contenant un atome d'oxygène en α de la fonction nitrile étaient capables de donner une cyclopropylamine avec de bons rendements sans ajouter d'acide de Lewis au milieu réactionnel (Schéma I-57).54. La tolérance fonctionnelle de la réaction de cyclopropanation du nitrile permet un accès facile à plusieurs types de cyclopropylamines fonctionnalisées (Figure I-4).
L'ajout d'un nitrile après élévation de la température à -20°C conduit alors à la formation de cyclopenténylamines I.121 (schéma I-70).63.
V - Conclusion et objectifs de la thèse
BIBLIOGRAPHIE
Laroche, C., Développements dans la réaction de conversion des nitriles en cyclopropylamines ; thèse de l'Université de Reims, 2006.
Etude de la cyclopropanation asymétrique d’un cyanoester par une méthode de formation rapide de
Nous discuterons ensuite du développement d'une méthode d'évaluation (criblage) de ligands chiraux (diols) sur le titane, après quoi les résultats de son application à la cyclopropanation asymétrique de cyanoesters seront présentés.
I – Synthèse asymétrique de cyclopropanes
Cependant, le contrôle de la diastéréosélectivité reste un problème important dans ce type de réaction. L'oxytanecyclopentane B résulterait de l'introduction de la fonction ester au niveau de la liaison Ti-C la plus substituée du complexe A. Cependant, il a été démontré, quelques années plus tard, que l'introduction de la fonction ester était moindre au niveau de la liaison Ti-C la moins substituée. liaison de A (voir paragraphe II.2.a de la partie I).
36 Laroche, C., Développements dans la réaction de transformation des nitriles en cyclopropylamines ; thèse de l'Université de Reims, 2006.
II - Développement d'une méthode de formation de complexes chiraux de titane
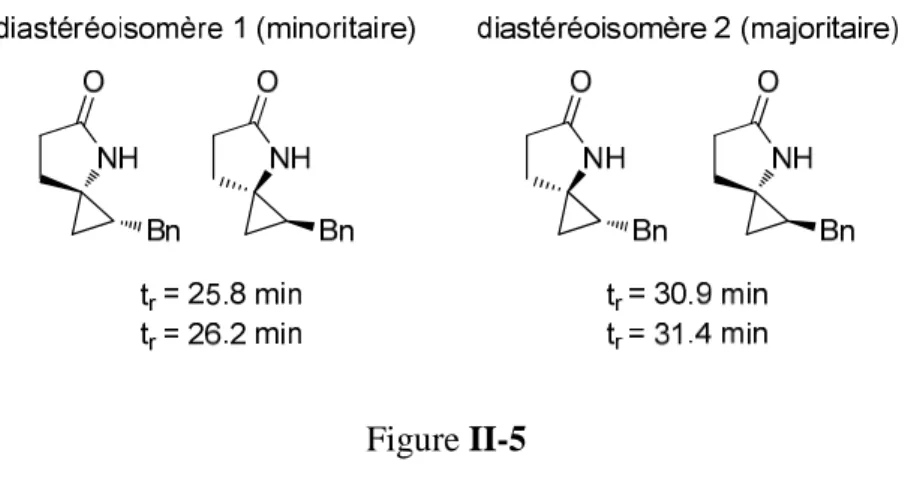
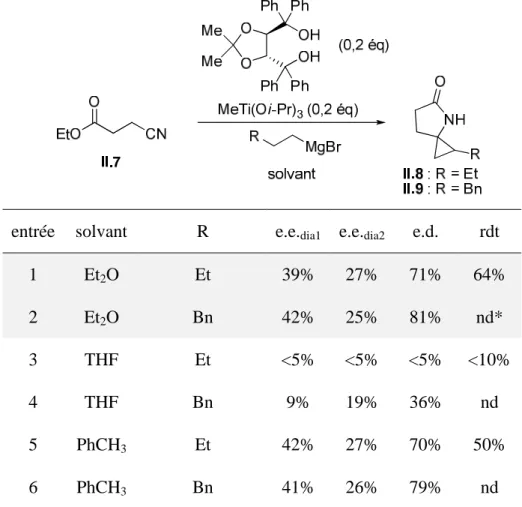
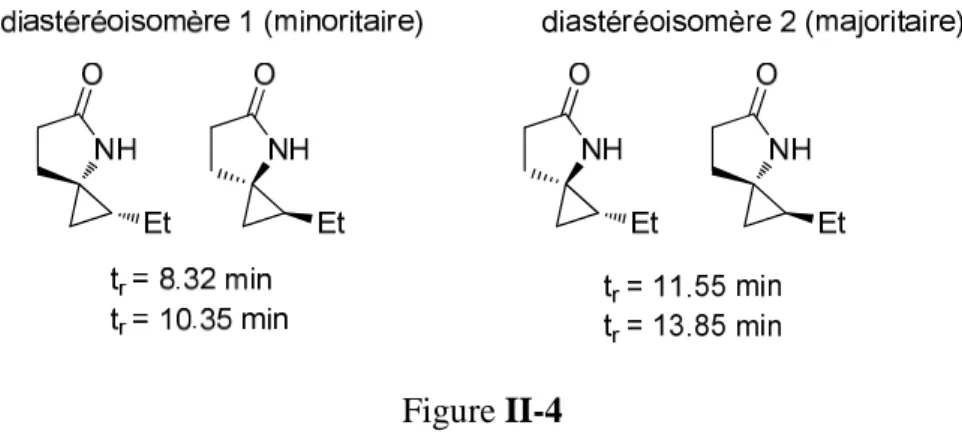
Dans le cas du spirolactame II.8 (substitué par un groupe R = Et), les excès énantiomériques et diastéréoisomères ont été déterminés par HPLC chirale sur un appareil Waters MILLIPORE® Modèle 510. Nous avons ensuite réalisé la cyclopropanation du cyanoester II.7 dans l'éther, successivement utilisé comme catalyseur. L'utilisation du bisTADDOLate de titane comme catalyseur de la réaction de cyclopropanation du cyanoester II.7 a conduit à des excès énantiomériques modérés pour les deux diastéréoisomères des spirolactames II.8 (point 3) et II.9 (point 4), l'excès du diastéréoisomère minoritaire étant supérieur à celui de la majorité.
De manière surprenante, le rendement en II.8 est bien inférieur avec le bisTADDOLAT de titane comme catalyseur qu'avec le Ti(Oi-Pr)4.
III – Evaluation de diols chiraux pour la cyclopropanation asymétrique du 2-cyanopropanoate d'éthyle
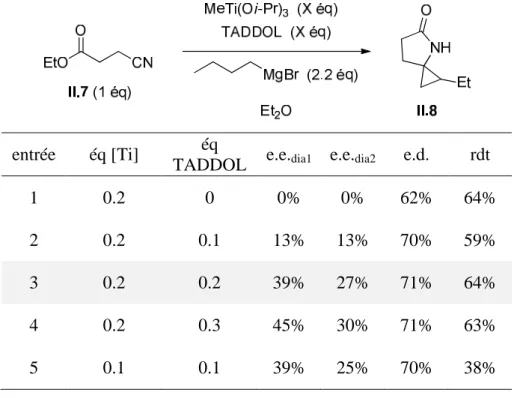
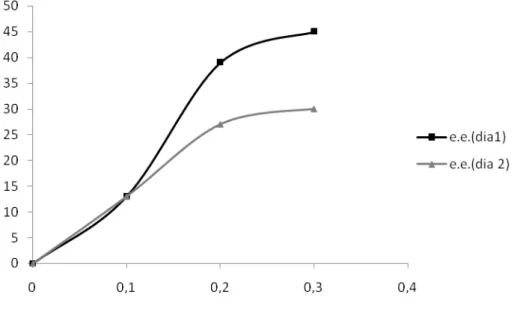
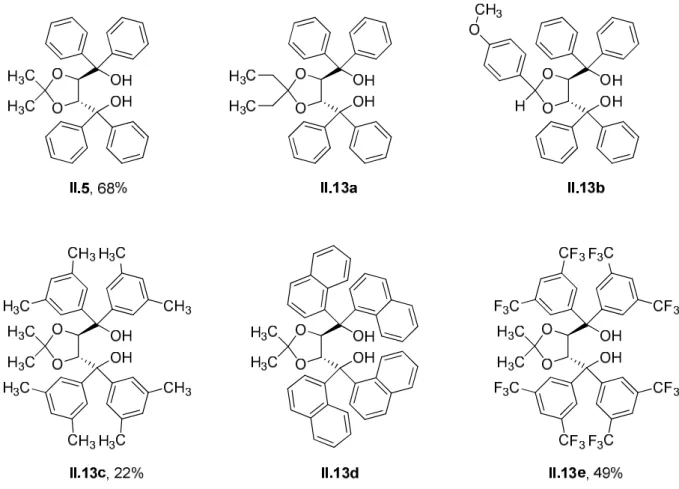
Les dérivés II.13a-b ont les mêmes substituants que le TADDOL original dans α des groupes hydroxyle, et des substituants différents sur la partie acétonide. À l'inverse, les dérivés II.13c-e ont les mêmes substituants sur le fragment acétonide et différents substituants α sur les hydroxyles. L'utilisation d'un ligand contenant deux substituants éthyle à la place des méthyles du TADDOL original (entrée 1) a conduit pour la synthèse du spirolactame II.8 à une légère réduction de l'excès énantiomérique du diastéréoisomère minoritaire (dia1) dans l'éther (entrée 2), et à une diminution plus importante (10 %) du toluène (entrée 3).
En revanche, pour la synthèse des spirolactames II.8 et II.9 dans l'éther, le remplacement des méthyles du TADDOL d'origine par un groupement aromatique et un hydrogène a entraîné une réduction significative des excès énantiomériques des deux diastéréoisomères.
IV - Conclusion
Partie expérimentale
Conditions générales d’analyses
Le solvant est évaporé sous pression réduite et le résidu est purifié par chromatographie sur gel de silice (cyclohexane-AcOEt 85:15) pour obtenir le cyanoester II.7 (3,66 g, 38 %) sous forme d'un liquide incolore. Le résidu contient du spirolactame II.8 sous forme de deux diastéréoisomères séparables par chromatographie sur gel de silice (cyclohexane-AcOEt 25:75 puis AcOEt-i-PrOH 95:5). Le résidu contient du spirolactame II.8 sous forme de deux diastéréoisomères séparables par chromatographie sur gel de silice (cyclohexane-AcOEt 25:75 puis AcOEt-i-PrOH 95:5).
Le solvant a été évaporé sous pression réduite et le résidu a été purifié par chromatographie sur gel de silice (CH 2 Cl 2 pur) pour donner TADDOL II.5 (7,30 g, 68 %) sous la forme d'un solide blanc.
Etude de la formation de 1,4-dicétones via l’utilisation d’un complexe oléfinique de titane
I - Objectif initial : accès aux dérivés de l’ACC
Nous avons proposé de tester notre stratégie avec un substrat modèle, le cyanométhylbenzoate III.1a (R = Ph). En 1913, Aloy et Rambaud rapportèrent que le benzoate de cyanométhyle pouvait être synthétisé avec un rendement de 45 % en ajoutant du chlorure d'acide à une solution aqueuse de KCN et de formaldéhyde (schéma III-2).1 La réaction fut ensuite étendue à un acide α,β-insaturé. . chlorures.2. Une version de cette méthode publiée en 19763 consiste en la réaction du chlorure d'acide en milieu anhydre avec le glycolonitrile préalablement formé par transhydrocyanation entre la cyanhydrine de la 2-butanone et le formaldéhyde.4, 5 Le cyanométhylbenzoate est synthétisé par cette voie avec une efficacité de 66% (schéma III-3).
Mowry précise dans sa publication2 que la méthode qu'il utilise donne de faibles rendements avec des chlorures d'acides saturés et qu'il est préférable d'obtenir des esters cyanométhyliques saturés en faisant réagir le carboxylate de sodium de l'acide correspondant avec le chloroacétonitrile en présence d'une amine tertiaire.6.
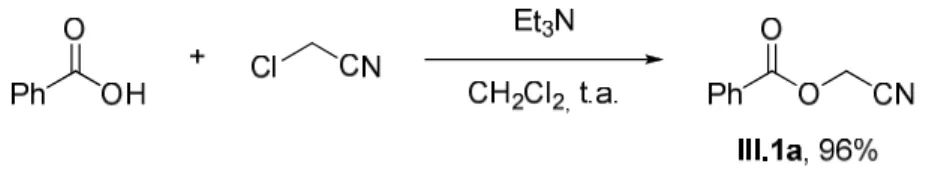
II - Méthodes de synthèse des 1,4-dicétones
Ceux-ci peuvent alors réagir avec les chlorures d'acyle en présence d'une quantité catalytique de PdCl2(PPh3)2 (Diagramme III-8).10. Récemment, des 1,4-dicétones ont été obtenues à partir de dialkylzinc, de monoxyde de carbone et d'une énone en présence d'une quantité catalytique de Pd2(dba)3 (Schéma III-13).15 Ce « carbonylant d'addition conjugué » peut même être transporté dehors. avec des aldéhydes insaturés (R' = H). Il a également été récemment démontré que les énolates de zinc pouvaient être couplés efficacement avec des α-chlorocétones en présence d'une quantité catalytique de PdCl2(dppf)2.17. Cependant, cette méthode semble limitée aux composés ayant un groupe aryle en position 2 et en la 3 positions.
Les nitrooléfines ont également été utilisées comme équivalent de cation α-carbonyle.18 Une réaction de Michael entre des éthers d'énol silylés et des nitrooléfines en présence d'acide de Lewis (TiCl4 ou SnCl4) conduit à un nitronate, qui subit ensuite une réaction de Nef pour conduire au correspondant. 1,4-dicétones (schéma III-15).
III - Optimisation de la réaction : variation des conditions opératoires
Nous nous sommes fixés pour objectif de modifier la sélectivité de la réaction vers la formation d'un minimum de sous-produits, afin d'augmenter la proportion de 1,4-dicétone dans le mélange réactionnel brut. La proportion d'aminocyclopropane III.3a formée augmente, entraînant une diminution de la proportion de 1,4-dicétone dans le mélange. Lorsqu'on utilise du méthyltriisopropoxytitane à la place du tétraisopropoxytitane, on observe une légère augmentation de la proportion de 1,4-dicétone par rapport à celle de l'aminocyclopropane.
Le faible effet de la concentration du substrat cyanoester sur la proportion et le rendement isolé de 1,4-dicétone est cohérent avec un mécanisme de réaction intramoléculaire.
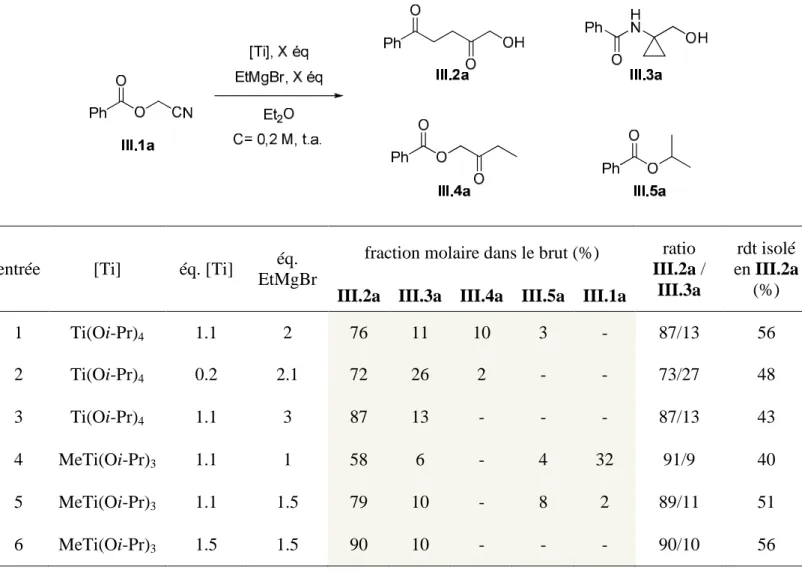
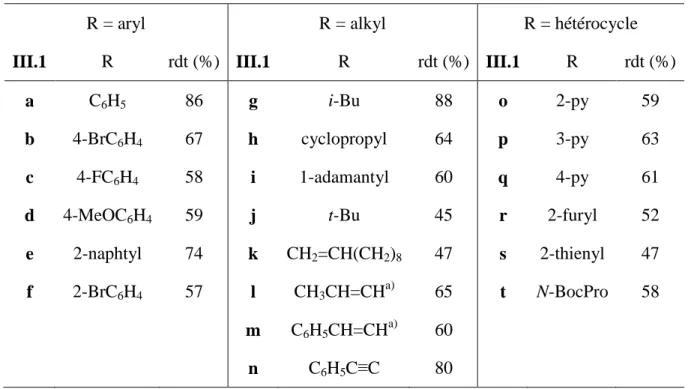
IV - Exploration de la réaction : variation des substrats
Lors de la réaction impliquant les cyanoesters III.1, qui incluent un groupe thiophène (entrée 8), le cyanoester est présent en quantité importante dans le pétrole brut et la proportion de 1,4-dicétone est inférieure au niveau de référence. Par exemple, le cyanoester III.1t issu de la L-proline protégée par un groupement Boc a été testé comme substrat de la réaction dans l'éther (entrée 9) puis dans le THF (entrée 10). Dans le cas d'un substituant méthyle (entrée 2), le rendement en 1,4-dicétone III.2v obtenu est comparable à celui observé sans substituant.
Le cyanoester III.1v (R1=Me) a ainsi été soumis aux conditions de formation de 1,4-dicétones avec le bromure de butylmagnésium (R2=Et) comme réactif de Grignard.
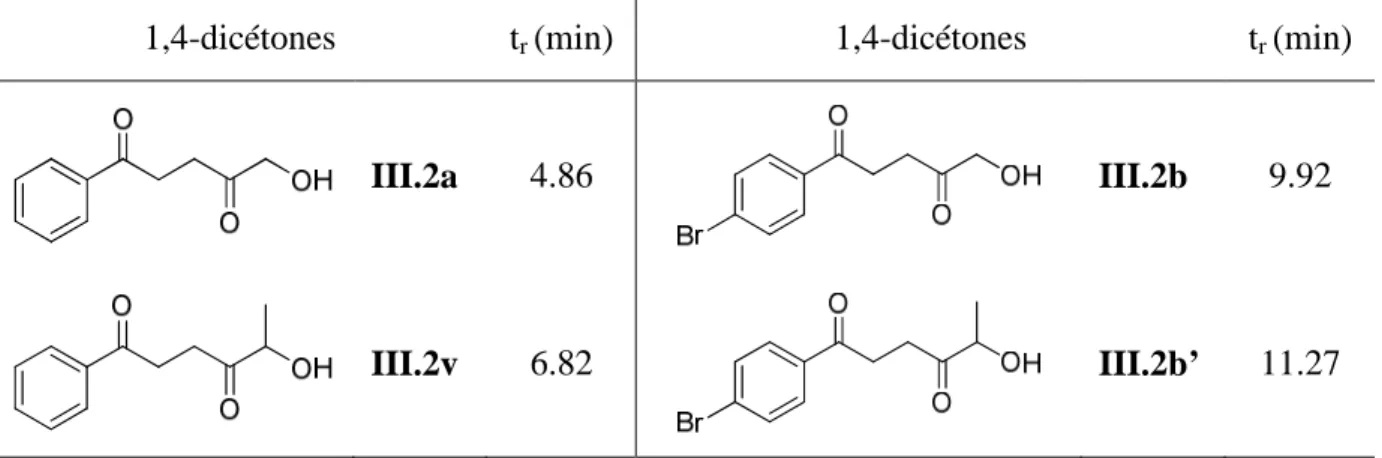
V - Elaboration d’une proposition de mécanisme réactionnel
Ainsi, dans le cas d'un mécanisme intermoléculaire, le mélange obtenu après la réaction sera constitué de quatre 1,4-dicétones III.2b, III.2v, III.2b' et III.2a. Ainsi, la présence ou l'absence des 1,4-dicétones III.2b' et III.2a permettront d'affirmer que le mécanisme est respectivement intermoléculaire ou intramoléculaire. Le chromatogramme de la réaction ne présente que deux pics dont les temps de rétention permettent d'identifier les composés correspondants : ce sont les 1,4-dicétones III.2v et III.2b.
Ainsi, les 1,4-dicétones III.2a et III.2b' ne se sont pas formées au cours de la réaction, ce qui permet d'affirmer que le mécanisme de formation des 1,4-dicétones est strictement intramoléculaire.
VI - Cyclisation des 1,4-dicétones
En revanche, aucune réaction n'a été observée lors du chauffage de la 1,4-dicétone III.2a en présence de TsOH sous reflux de THF. Enfin, nous avons testé la cyclisation de la 1,4-dicétone III.2a en thiophène via le réactif de Lawesson dans le toluène. En général, la 1,4-dicétone III.2a ne semble pas être un substrat approprié pour la préparation d'hétérocycles insaturés à cinq chaînons.
Il nous a semblé intéressant de tester cette réaction de cyclisation sur la 1,4-dicétone III.2x, qui permettrait d'obtenir une cyclopenténone substituée (Schéma III-41).
VII - Conclusions et perspectives
En revanche, les cyclopenténones III.11 et III.12 ont été formées avec de bons rendements par aldolisation intramoléculaire des 1,4-dicétones III.2a et III.2x. Il est intéressant de noter que la cyclopenténone III.11 pourrait également être synthétisée en une seule étape à partir du cyanoester III.1a via la réaction d'hydrolyse basique de la formation de III.2a (Schéma III-45). Purification par chromatographie sur gel de silice (cyclohexane – AcOEt 90 :10). 2S)-pyrrolidine-1,2-dicarboxylate de 2-cyanométhyle et de 1-tert.butyl III.1t.
Le cyanoester brut est purifié par chromatographie sur gel de silice (cyclohexane 100% puis cyclohexane-AcOEt 60:40) pour donner III.1j sous forme d'huile jaune clair (6,30 g, 45%).