Mme. COSSY Janine Professeur, Ecole Supérieure de Physique et Chimie Industrielle de la ville de Paris. Nicolaou, la pratique de la synthèse totale demande de l'ingéniosité, un certain sens artistique, une bonne pratique de laboratoire, de la persévérance et une certaine dose de caractère.
1. ) Les'Amphidinols'
1.1. ) Introduction)
Parmi eux, l'amphidinolide A I.4 possède un cycle à 20 chaînons et une excellente activité contre la prolifération des cellules cancéreuses ; 3 L'amphidinolide N I.5, un macrocycle à 24 chaînons, présente une toxicité élevée contre les cellules de leucémie lymphoïde et également une activité dominante contre la prolifération des cellules cancéreuses. Cependant, les lutéophanols ont des propriétés antibactériennes, tandis que les AM ont des propriétés antifongiques et hémolytiques.
1.2. ) Origine)des)Amphidinols)
1.3. ) Extraction)des)amphidinols)
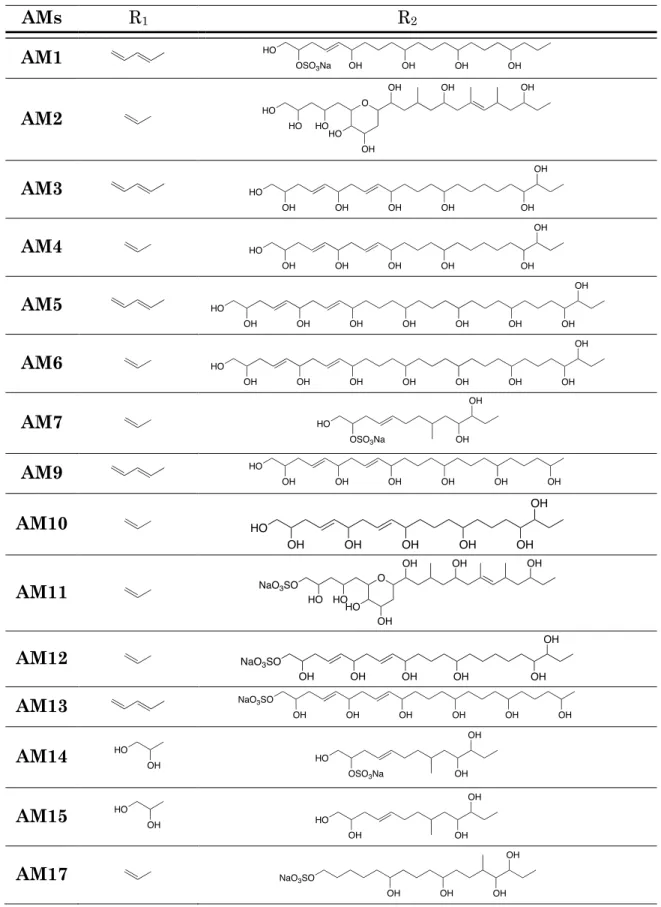
1.4. ) Structures)des)amphidinols)
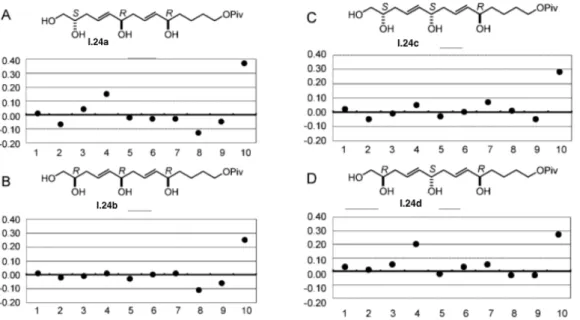
Ainsi, l'analyse conformationnelle basée sur les constantes de couplage a permis d'élucider la configuration relative des centres stéréogéniques des séquences 1,2 et 1,3 des portions C20-C27, C44-C45 et C50-C51 d'AM3 (Figure 6 ). Enfin, la configuration absolue du centre C2 a été attribuée par décomposition chimique de AM3 sous l'action de OsO4/NaIO4, donnant I.17a.
1.5. ) Biosynthèse)des)amphidinols)
Les insaturations M=m de AM2 et AM4 en C30-C31 et C52-C53 pourraient provenir d'une déshydratation impliquant des hydroxyles porteurs d'une méthine et de ses méthylènes adjacents. Les motifs m-m présents dans AM2 et AM4 proviendraient d'une réaction de type Favorskii,31 qui élimine un c-carbonyle d'une séquence c-m (Schéma 21).
1.6. ) Bioactivités)des)amphidinols)
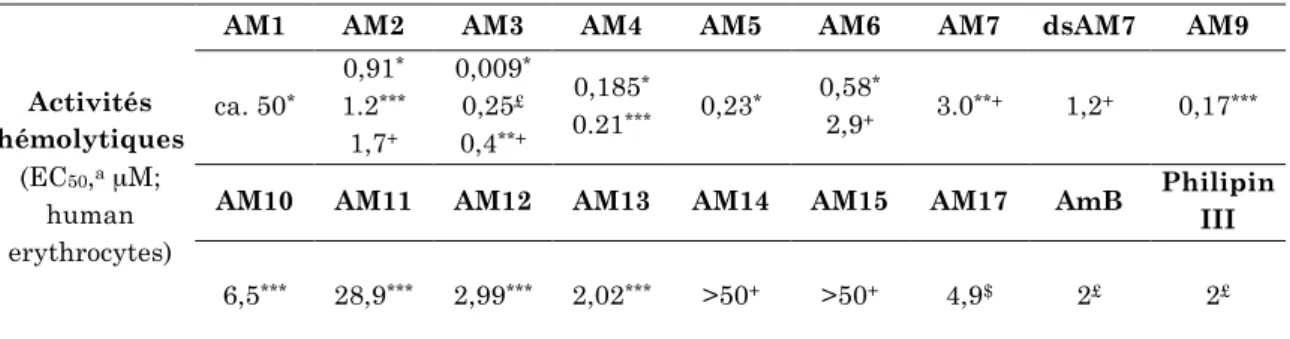
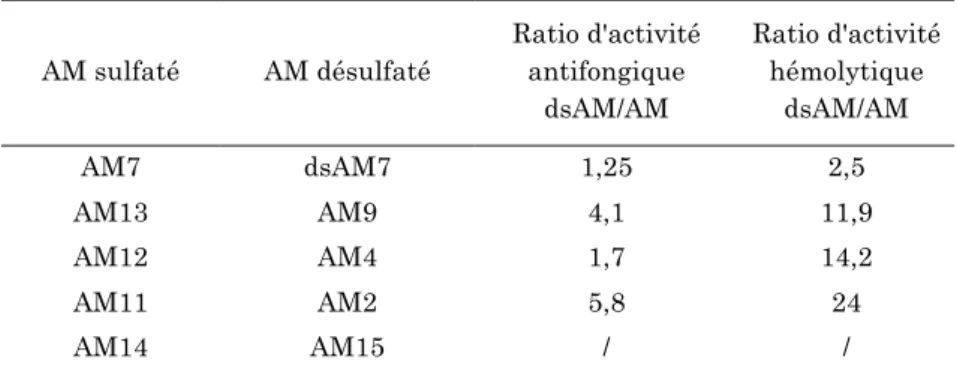
AM, en particulier AM3, ont des activités antifongiques particulièrement fortes, mais sont toujours dépassées par AmB et Filipin III. En revanche, l'AM3 révèle une activité hémolytique bien supérieure à celle de l'AmB et de la filipine III.
1.7. ) Conclusion)
2. ) L’amphidinol'3'
2.1. ) Précédents)dans)la)littérature)
2.2. ) Travaux)de)L.A.)Paquette)et)al.)
Un clivage oxydatif de l'alcène I.58 suivi d'une oléfination Julia-Kocienski58, d'une dihydroxylation asymétrique de Sharpless57 et d'une oxydation de l'alcool primaire préalablement déprotégé donne l'aldéhyde I.61. Le composé I.71 est obtenu par couplage de Wittig entre l'aldéhyde dérivé de I.63 et le synthon I.67 suivi d'une hydrogénation de la double liaison résultante.
2.3. ) Travaux)de)S.D.)Rychnovsky)et)al.)
La synthèse du corps central part de l'acide D-(-)-tartrique, qui est converti en aldéhyde I.75 en 5 étapes. L'allylation de l'hémicétal suivie d'une ozonolyse donne l'aldéhyde I.80, qui, avec le sulfoxyde I.81 dans des conditions de Knoevenagel 68, conduit au motif tétrahydropyrane commun I.82 du corps central AM3 en 16 étapes avec un rendement global de 9,9 % (Schéma 37 ). Rychnovsky a développé une synthèse convergente remarquable du fragment C31-C52 comprenant les motifs THP d'AM3.
2.4. ) Travaux)de)W.R.)Roush)et)al.)
Les déconnexions des différents fragments sont les principaux problèmes de la synthèse totale d'AM3. Plusieurs stratégies ont été envisagées lors de notre approche de la synthèse du fragment polyhydroxyle de l'AM3. Dans le cas de l'alcool III.9, la synthèse du deuxième synthon de métathèse croisée part de l'octan-1,7-diène, qui sera monooxydé et la fonction hydroxyle résultante protégée.
Ainsi, dans le cas de III.80 3JHA-HB = 15,2 Hz démontre la configuration E de la double liaison de l'isomère majoritaire (Figure 3). Parallèlement à la synthèse de l'aldéhyde III.7, la synthèse du dérivé 1,3-dithian III.11 a été développée. La première étape de la synthèse de l'époxyde est l'ouverture de la β-propiolactone III.89 à partir de l'anion lithium du méthyl-para-tolylsulfoxyde II.3.
La synthèse débute donc par la réduction de l'acide L-malique III.15, au même titre que la synthèse du dithiane III.117.
2.5. ) Travaux)de)J.)Cossy)/)F.)Colobert)/)E.)Markó)
2.6. ) Travaux)de)M.T.)Crimmins)et)al.)
Crimmins a réalisé uniquement la synthèse du corps central de l'AM3 et ne fournit pas d'information sur le type de déconnexions entre les chaînes polyhydroxyle/polyène et le corps central. Crimmins propose une synthèse convergente du corps bistétrahydropyranique de l'AM3, qui diffère des synthèses précédentes. Une réduction diastéréosélective avec (R)-CBS77 permet un contrôle configurationnel parfait de la fonction hydroxyle en C39, et un clivage oxydatif Johnson-Lemieux60 de la double liaison terminale complète la synthèse de l'aldéhyde I.180 (Schéma 72).
2.7. ) Travaux)de)M.)Murata)et)al.)
La synthèse du motif tétrahydropyrane commence par la métathèse croisée entre l'iodure de vinyle I.20b décrite précédemment21 et le composé I.182, suivie d'une dihydroxylation asymétrique Sharpless.57 L'homologation est réalisée par un couplage Migita-Kosugi-Stille entre l'iodure de vinyle I.183 et vinylstannane I.184, tandis que l'action du D-(-)-DET permet l'époxydation stéréosélective Sharpless56 de la double liaison α-hydroxylée. Le cycle THP est formé dans un environnement acide, tandis que les deux derniers centres sont introduits par dihydroxylation asymétrique Sharpless, donnant accès au motif THP commun de l'AM3 I.186 (Schéma 76). Cette synthèse a pour point de départ l'iodure de vinyle déjà utilisé lors de la synthèse précédente, et les centres asymétriques sont introduits soit par dihydroxylation asymétrique Sharpless, soit par époxydation asymétrique Sharpless, en 10 étapes avec un rendement total de 29 %.
3. ) Conclusion'
4. ) Synthèse'de'l’avancement'des'différents'groupes'
5. ) Bibliographie'
1. ) Introduction'
2. ) Rétrosynthèse'
L'ajout du dérivé lithium d'un bromure de vinyle à l'aldéhyde II.25 introduirait le centre stéréogénique C24 selon une approximation de Felkin-Ahn. L'aldéhyde II.25 proviendrait de l'acide penténoïque II.16, et les centres asymétriques C25 et C27 résulteraient respectivement d'une réduction. L'utilisation du corps central II.26 rend la synthèse du fragment C17-C30 très convergente, les aldéhydes II.15 et II.25 étant hautement fonctionnalisés.
3. ) Généralités'sur'les'sulfoxydes'
3.1. ) Les)sulfoxydes)
111 L’accès aux sulfoxydes énantiopurs est également un véritable point fort des sulfoxydes et est largement décrit dans la littérature. L'une des méthodes les plus anciennes de synthèse de sulfoxydes chiraux non racémiques a été développée par K.K.
3.2. ) Synthèse)des)sulfoxydes)
L'anion lithié (+)-(R)-méthyl-para-tolylsulfoxyde II.3 est obtenu par action du LDA, fraîchement préparé, puis ajouté à l'ester pour donner le β-cétosulfoxyde II.4. L'action de l'halogénure de zinc sur le (R)-β-cétosulfoxyde II.4 entraîne la formation de deux chélates A' et B' ; Le chélate B' est désavantagé en raison de la position pseudo-axiale du para-solvant, qui empêche l'approche de DIBAL-H. Schéma 87 : États de transition de réduction du β-cétosulfoxyde par DIBAL-H en présence de ZnX2.
4. ) Travail'effectué'
La prochaine étape de la synthèse est la réduction de la fonction sulfoxyde du III.94 en thioéther. Les groupes protecteurs de III.117 sont directement impliqués dans l'échec de la réaction de couplage. Tests de couplage du dithiane III.137 avec l'aldéhyde II.7 et test de protection avec le groupe MTM.
La formation de l'intermédiaire III.148, par élimination, est irréversible et peut subir une attaque nucléophile par la fonction alcool (Schéma 207).241. Cependant, la thioacétalisation de l'aldéhyde III.154 en dithiane correspondant n'a pas donné les résultats escomptés.
4.1. ) Synthèse)de)l’aldéhyde)15)
4.2. ) Synthèse)de)l’aldéhyde)25)
4.3. ) Introduction)du)corps)central):)l’allylsilane)26)
Ainsi, il a été possible d'obtenir l'alcool III.9 à partir de l'acide -(+)-camphorsulfonique via Oppolzer sultam en 7 étapes avec un rendement total de 37 %. L'anion sulfoxyde lithié II.3 est un nucléophile dur et ne devrait conduire qu'à l'ouverture α de la β-propiolactone III.89. Synthèse de l'époxyde à partir de l'acide L-malique La synthèse de l'époxyde III.105 portant un éther paraméthoxybenzylique a été décrite par C.L.
Cette synthèse constitue un accès rapide et efficace à l'époxyde III.105 à partir de l'acide L-malique III.15 en 6 étapes. Lorsque le groupement TBS est présent dans la fonction β hydroxyle du dithiane III.135, aucun produit de couplage n'est observé.
4.4. ) Vers)le)fragment)C172C30)porteur)de)la)fonction)cétone)terminale)
5. ) Problémes'soulevés'
Cet alcool α-méthylé pourrait être obtenu énantiomériquement pur via Oppolzer sultam, synthétisé à partir de l'acide (+)-camphorsulfonique III.10. Une simple évaporation de l'excès de chlorure de thionyle suivie d'une recristallisation donne du chlorure de sulfonyle III.65 avec un rendement de 97 % (graphique 159). Le 1,2-Epoxybutane est ouvert par le 2-lithio-1,3-dithiane III.35, après quoi la fonction hydroxyle est protégée par un MOM (Schéma 197).
Le modèle du dithiane III.130 est similaire au III.117 avec la fonction dithiane β hydroxyle. La fonction hydroxyle primaire restante est ensuite oxydée en aldéhyde III.151 par oxydation de Swern (schéma 210).
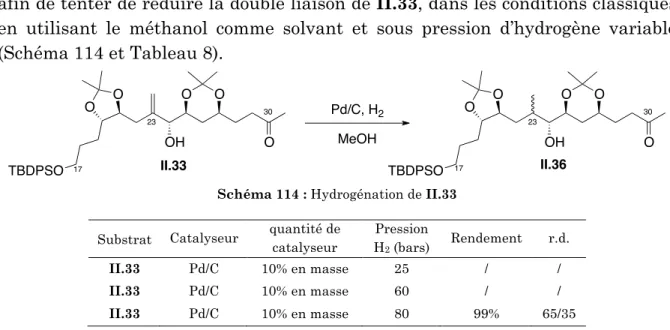
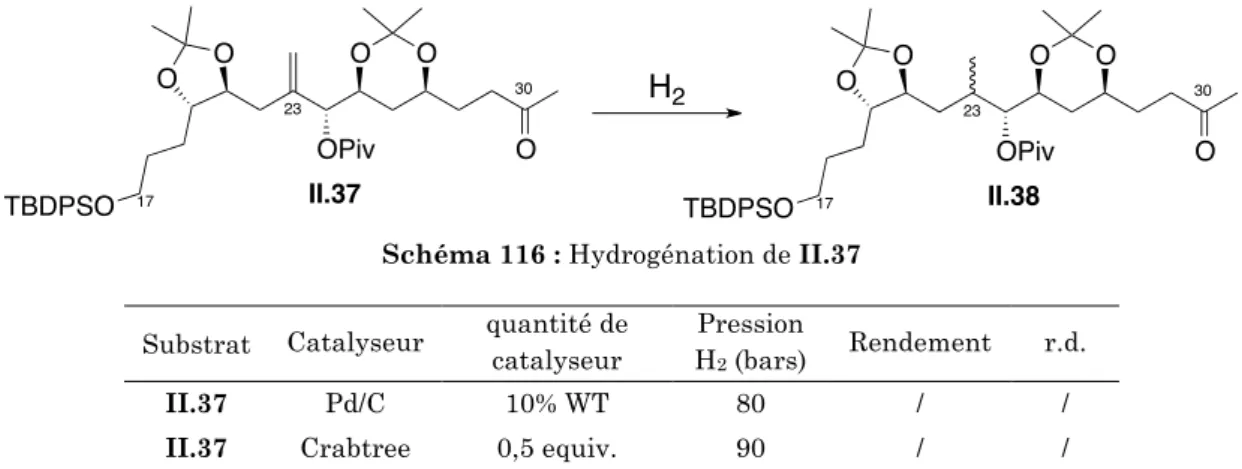
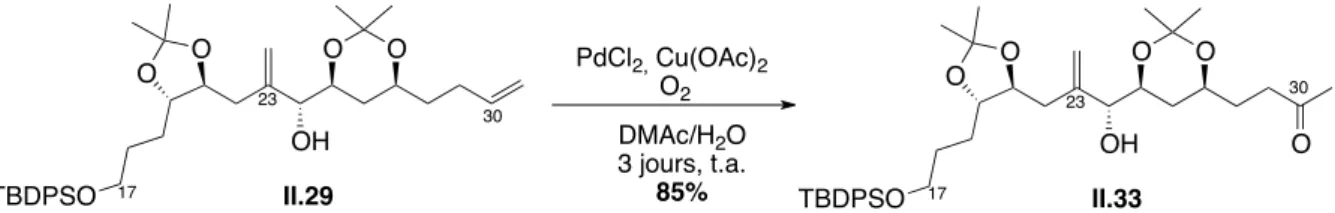
5.1. ) Réduction)de)l’insaturation)C162C17)
5.2. ) Oléfination)C302C31)
6. ) Conclusion'de'la'synthèse'du'fragment'C17OC30'
7. ) Schéma'récapitulatif'
8. ) Bibliographie'
1. ) Introduction'
2. ) Rétrosynthèse'
La fonction alcool est ensuite protégée par un ester de pivaloyle III.8 dans des conditions standards (Schéma 165).212. La fonction alcool de III.80 est ensuite oxydée par IBX III.82 qui a été préalablement synthétisé avec d'excellents rendements (Schéma 172). La synthèse de l'époxyde III.14 portant le centre stéréogénique C27 sera réalisée à l'aide d'un sulfoxyde comme inducteur de chiralité ou d'acide L-malique.
L'époxy III.105 est ensuite obtenu à partir de l'acide L-malique en six étapes par un jeu de protection/déprotection et une fermeture de cycle par élimination d'un sulfonate. Ainsi, chacun des deux groupes de protection du III.117 est remplacé individuellement par un autre groupe de protection.
3. ) Généralités'sur'les'1,3Odithianes'
3.1. ) Carbanions)adjacents)aux)atomes)de)soufre)
La première mention de la stabilisation des charges négatives dans α des sulfures a été décrite par Gilman et Webb148 ; la déprotonation avec une base forte suivie d'une carboxylation a lieu en ortho dans le cas de l'anisole, mais sur méthyle dans le cas du méthyl-phényl- sulfure (Annexe 132). Avant ces travaux, peu de preuves avaient été trouvées sur l'effet des orbitales d sur l'acidité des protons α dans les sulfures ; seules la polarisabilité et la longueur de la liaison CS ont fourni quelques réponses. Streitweiser a montré par calcul que l'acidité des dérivés du 1,3-dithiane 2-substitué ne dépendait que peu de la nature du substituant151, tandis qu'E.J.
3.2. ) Introduction)des)1,32dithianes)
3.3. ) Création)de)la)liaison)C2C)à)partir)du)1,32dithiane)
3.4. ) Hydrolyse)du)1,32dithiane)
3.5. ) Transformation)en)autres)fonctions)
En général, l'ouverture des époxydes avec le 2-lithium-1,3-dithiane a lieu entre -20 °C et température ambiante. Dans le cas des cétones α,β-insaturées, le 2-lithio-1,3-dithiane peut être ajouté en position 2 ou 4, selon les conditions de réaction. La plupart des méthodes d’hydrolyse du 1,3-dithiane coordonnée avec des métaux suivent un mécanisme similaire.
4. ) Travail'effectué'
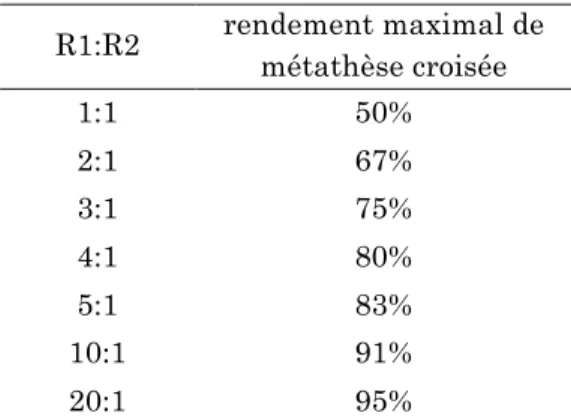
L'oléfine ajoutée en excès lors de la métathèse croisée était III.8, le composé le moins développé et le plus facilement accessible. La fonction hydroxyle primaire de III.110 est ensuite activée sélectivement par le chlorure de 2,4,6-triisopropylbenzènesulfonyle III.111. Lorsque le groupe TBS est remplacé par un groupe MOM III.137, la réaction de couplage s'effectue avec un rendement de 58 %.
4.1. ) Synthèse)de)l’alcool)9)
4.2. ) Métathèse)croisée)
La formation de III.117 montre clairement la réalisation du réarrangement de Brook, la charge négative doit probablement être déplacée au pied de la fonction dithiane. Ces mêmes conditions sont ensuite appliquées à un substrat plus complexe, pour imiter au mieux le dithiane III.117. Les conditions de couplage développées entre les ditithiens modèles et l'aldéhyde modèle sont ensuite appliquées au substrat III.117 (Schéma 199).
4.3. ) Synthèse)du)dithiane)11)et)couplage)avec)l’aldéhyde)83)
4.4. ) Hydrolyse)du)dithiane)164)
Le rendement est relativement faible et est dû à la captation simultanée du produit d'oxydation de l'alcool III.142, réaction qui ne peut être évitée.
4.5. ) Accès)au)fragment)C142C29)
4.6. ) Perspectives)
5. ) Conclusion'
6. ) Schéma'récapitulatif'
7. ) Bibliographie'