UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE CENTRO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
RASTREAMENTO POPULACIONAL PARA DOENÇA DE GAUCHER EM TABULEIRO DO NORTE - CEARÁ - BRASIL
RIGOBERTO GADELHA CHAVES
RIGOBERTO GADELHA CHAVES
RASTREAMENTO POPULACIONAL PARA DOENÇA DE GAUCHER EM TABULEIRO DO NORTE – CEARÁ – BRASIL
Dissertação apresentada ao Programa de Pós-Graduação em Ciências da Saúde, da Universidade Federal do Rio Grande do Norte, como requisito parcial para obtenção do título de Mestre em Ciências da Saúde, aprovada em 31 de maio de 2011.
Orientador: Prof. Dr. Geraldo Barroso Cavalcanti Júnior
UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE CENTRO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
Coordenadora: Profa. Dra. Técia Maria de Oliveira Maranhão
RIGOBERTO GADELHA CHAVES
RASTREAMENTO POPULACIONAL PARA DOENÇA DE GAUCHER EM TABULEIRO DO NORTE – CEARÁ – BRASIL
,
Banca Examinadora
Prof. Dr. Geraldo Barroso Cavalcanti Júnior Orientador
Profa. Dra. Ana Maria Martins UNIFEP
AGRADECIMENTOS
Ao Senhor Deus, por ter concedido saúde e motivação a mim para a conclusão do Curso de Mestrado.
Aos meus pais, Sr. Alcides Monteiro Chaves e Sra. Lucinda Gadelha Chaves, que me ensinaram os verdadeiros valores da vida.
A minha esposa, Edineide e aos meus filhos, Marília e Mateus, que souberam sublimar a minha ausência, durante todo o tempo dedicado ao projeto e estimularam a superar as dificuldades advindas de um trabalho envolvendo uma comunidade.
Aos gestores e demais autoridades municipais, em nome de João Márcio da Silva, Secretário de Saúde de Tabuleiro do Norte, pelo apoio às atividades da pesquisa.
À Genzyme do Brasil, por colaborar no suporte financeiro e de logística, para a realização do estudo.
À Associação Cearense de Profissionais Atuantes em Doenças Genéticas, Pacientes, Familiares e Voluntários (ACDG), pelo apoio e viabilização de recursos financeiros para realização do estudo.
Ao Centro de Ciências da Saúde da Universidade Federal do Rio Grande do Norte, na pessoa da Profa. Dra. Técia Maria Oliveira Maranhão, Coordenadora do Curso de Pós-graduação, pelo generoso acolhimento dispensado a mim durante a realização do mestrado.
Aos professores do mestrado, pelos importantes ensinamentos transmitidos, fazendo com que eu veja o mundo das ciências com mais clareza e espírito crítico.
Ao orientador, Prof. Dr. Geraldo Barroso Cavalcanti Jr., pelo zelo profissional na minha formação de mestre, valorizando sempre os princípios da ética em pesquisa.
À Dra. Erlane Marques Ribeiro, por sua colaboração na fase inicial do projeto e pelo incentivo à realização deste estudo.
À Dra. Elisa Sobreira e ao Dr. Tarek Ebrahim, membros da Genzyme do Brasil e Genzyme Corporation, pela contribuição para meu crescimento intelectual, facilitando o acesso a artigos e a eventos científicos.
Ao Dr. Antônio Filgueiras, por trilhar para mim os caminhos da Universidade Federal do Rio Grande do Norte.
Aos colegas de trabalho da Secretaria de Saúde de Tabuleiro do Norte, aos membros da Equipe da Unidade Básica de Saúde Alcides Monteiro Chaves, pela valiosa colaboração nas diversas atividades da pesquisa.
Aos colegas colaboradores da pesquisa, co-autores do Artigo Científico, por dividirem comigo a responsabilidade deste trabalho.
“Ser feliz é deixar de ser vítima dos problemas e se tornar um autor da própria história. É atravessar desertos fora de si, mas ser capaz de encontrar um oásis no recôndito da sua alma.”
LISTA DE ABREVIATURAS
CE: Ceará
CREIM: Centro de Referência dos Erros Inatos do Metabolismo
DDL: Doença de Depósito Lisossômico
DG: Doença de Gaucher
EIM: Erro Inato do Metabolismo FDA: Food and Drug Administration
GBA: β-glucocerebrosidase GELs: Glicoesfingolipídios
GlcCer: Glicosilceramida
HCPA Hospital das Clínicas de Porto Alegre ICGG: International Collaborative Gaucher Group
IL-1α : Interleucina-1 alfa
IL-6: Interleucina 6
PCR: Reação em Cadeia da Polimerase
PSGBA: Pseudogene da β-glucocerebrosidase
SSPF: Sangue seco em papel de filtro SUS: Sistema Único de Saúde
TCLE Termo de Consentimento Livre e Esclarecido TNF-α: Fator de necrose tumoral alfa
TRE: Terapia de reposição enzimática
LISTA DE TABELAS
Tabela 1 Características clínicas dos tipos da doença de Gaucher... 19
Tabela 2 Comparação entre os indivíduos do “grupo de risco para DG” e “normais” de acordo com a atividade da GBA em sangue seco em
papel de filtro... 42 Tabela 3 Distribuição dos indivíduos conforme resultados das atividades
enzimáticas da GBA e quitotriosidase em sangue seco em papel de filtro... 42 Tabela 4 Critérios de classificação dos voluntários de acordo com o nível da
atividade de β-glucocerebrosidase em leucócitos periféricos... 43 Tabela 5 Distribuição de frequência dos indivíduos de acordo com atividade
de GBA em leucócitos periféricos... 44 Tabela 6 Análise enzimática e molecular dos indivíduos com atividade de
LISTA DE FIGURAS
Figura 1 A Célula de Gaucher... 07
Figura 2 Fatos históricos da doença de Gaucher... 08
Figura 3 Representação da estrutura química da D-glucosyl-ceramida, o substrato estocado na doença de Gaucher... 11
Figura 4 O Raio-X da refinada estrutura da β-glucocerebrosidase humana... 12
Figura 5 Reação catalisada pela β-glucocerebrosidase humana... 12
Figura 6 Localização geográfica e principais dados de Tabuleiro do Norte -CE... 17 Figura 7 Prevalência da doença de Gaucher no Brasil... 18
Figura 8 Paciente da doença de Gaucher com acentuada
hepatoesplenomegalia e comprometimento ósseo observado na imagem de Ressonância Magnética dos fêmures... 20 Figura 9 Desenho do Estudo... 32
LISTA DE GRÁFICOS
Gráfico 1 Correlação entre sexo e idade dos participantes... 39
Gráfico 2 Distribuição dos participantes por faixas etárias... 39 Gráfico 3 Relação entre a idade dos participantes e a dosagem da
β -glicosidase ácida em sangue seco em papel de filtro em Tabuleiro do Norte... 40 Gráfico 4 Relação entre as atividades da β-glicosidase ácida e
LISTA DE APÊNDICES
APÊNDICE A Mapa conceitual do Estudo Populacional da Doença de Gaucher em Tabuleiro do Norte–CE: Eixos Estratégicos e as Principais Ações... 64 APÊNDICE B Atividades da β-glicosidase ácida e quitotriosidase em amostras
LISTA DE ANEXOS
ANEXO A Artigo Científico a ser publicado: Successful screening for Gaucher disease in a high-prevalence population in Tabuleiro do Norte (northeastern Brazil): a cross-sectional study………….. 66 ANEXO B Certificado da apresentação no XII Simpósio Latino-Americano
de Doenças de Depósito Lisossômico... 73 ANEXO C Certificado da apresentação no XX Congresso Brasileiro de
Genética Médica... 74 ANEXO D Poster exposto no XIII Simpósio Latino-Americano de Doenças
de depósito Lisossômico... 75 ANEXO E Certificado da premiação no XIII Simpósio Latino-Americano de
Doenças de Depósito Lisossômico ... 76 ANEXO F Poster apresentado no IX Congresso Brasileiro de Medicina de
Família e Comunidade... 77 ANEXO G Certificado da participação como debatedor no X Congresso
Brasileiro de Medicina de Família e Comunidade... 78 ANEXO H Convite para apresentação no Third NIH Workshop on Gaucher
Disease and Parkinsonism……… 79
ANEXO I Reportagem na Revista ISTOÉ... 80
SUMÁRIO
1 INTRODUÇÃO ... 1
2 REVISÃO DA LITERATURA ... 4
2.1 ERROS INATOS DO METABOLISMO ... 4
2.2 AS DOENÇAS DE DEPÓSITO LISOSSÔMICO ... 4
2.3 A DOENÇA DE GAUCHER ... 6
2.3.1 Fatos históricos da doença de Gaucher ... 7
2.3.2 Bases celulares e bioquímicas da doença de Gaucher ... 9
2.3.3 Aspectos genéticos das mutações da doença de Gaucher ... 14
2.3.4 Aspectos epidemiológicos da doença de Gaucher ... 16
2.3.5 Aspectos clínicos da doença de Gaucher ... 18
2.3.6 Diagnóstico da doença de Gaucher ... 20
2.3.7 Diagnóstico de heterozigotos para a doença de Gaucher ... 22
2.3.8 Avaliação familiar na doença de Gaucher ... 23
2.3.9 Rastreamento populacional para a doença de Gaucher ... 24
2.3.10 Tratamento da doença de Gaucher ... 26
3 OBJETIVO GERAL ... 29
3.1 OBJETIVOS ESPECÍFICOS ... 29
4 METODOLOGIA ... 30
4.1 DESENHO DO ESTUDO ... 31
4.2 EXAMES LABORATORIAIS ... 33
4.2.1 Avaliação das atividades enzimáticas em sangue seco em papel filtro ... 33
4.2.3 Análise molecular das mutações ... 35
4.2.4 Análise dos Dados ... 36
5 RESULTADOS ... 38
6 DISCUSSÃO DOS RESULTADOS ... 47
7 CONCLUSÕES ... 50
8 CONSIDERAÇÕES FINAIS... 52
REFERÊNCIAS ... 54
APÊNDICES ... 62
RESUMO
A doença de Gaucher (DG) é uma patologia de depósito de gordura nos lisossomos, de herança autossômica recessiva, caracterizada pelo acúmulo do substrato glicosilceramida, principalmente nas células do sistema reticuloendotelial, em razão da deficiência da enzima β-glicosidase ácida (GBA). O diagnóstico, comumente, é feito pela dosagem da atividade da GBA em leucócitos periféricos. Tabuleiro do Norte (TN), Ceará, Brasil, é um município com cerca de 28.000 habitantes com a prevalência da DG de 1:4.000 habitantes, possivelmente a mais elevada do Brasil. O objetivo da dissertação é avaliar o rastreamento para DG realizado em TN com base na análise das atividades enzimáticas da GBA e da quitotriosidase em amostras Sangue Seco em Papel de Filtro (SSPF). Entre 01 de junho de 2007 a 31 de maio de 2008, 740 indivíduos residentes e descendentes de famílias de TN participaram do rastreamento para DG a partir de amostras de SSPF. Indivíduos com atividade GBA<2,19 nmol/h/mL foram selecionados para análise da atividade da GBA e da quitotriosidase em leucócitos periféricos e no plasma, respectivamente. Os indivíduos com maiores riscos de DG (atividade de GBA em leucócitos periféricos <5,6 nmol/h/mg de proteína) foram referenciados para análise molecular para confirmação diagnóstica. A triagem com amostras de SSPF identificou 135 indivíduos (18,2%) com atividade da GBA<2,19 nmol/h/mL, dos quais 131 permaneceram no estudo. Em dez destes (7,6%), a atividade da GBA em leucócitos variou de 2,6-5,5 nmol/ h/mg de proteína, considerados suspeitos da DG. A análise molecular subsequente revelou, entretanto, que se tratava de seis indivíduos heterozigotos para a mutação G377S e, em quatro deles, não foram identificadas mutações da DG. A análise enzimática de amostras de SSPF mostrou ser uma estratégia eficaz de triagem da DG em populações com alto risco, mas a medida da atividade da GBA em leucócitos deve ser realizada para confirmação diagnóstica. O diagnóstico de DG em indivíduos assintomáticos não deve ser firmado baseando-se apenas na análise da atividade da GBA em leucócitos, sendo necessária, também, a confirmação diagnóstica pela análise molecular.
ABSTRACT
Background. Gaucher Disease (GD) is a hereditary lysosomal storage disorder characterized by the accumulation of glucosylceramide, mainly in the cells of the reticuloendothelial system, due to a deficiency of the enzyme acid β-glucosidase (GBA). Diagnosis is usually based on measurement of GBA activity in peripheral leukocytes. The purpose of this study was to evaluate the ability of screening for GBA and chitotriosidase activity using Dried Blood Spots on Filter Paper (DBS-FP) to identify individuals at high risk for GD in high-risk populations such as that of Tabuleiro do Norte, a small town in Northeastern Brazil. Methods. Between June 1, 2007 and May 31, 2008, 740 consented residents and descendants of traditional families from Tabuleiro do Norte were submitted to screening with DBS-FP. Subjects with GBA activity <2.19 nmol/h/mL were referred to analysis of GBA and chitotriosidase activity in peripheral leukocytes and in plasma, respectively. Subjects at highest risk for GD (GBA activity in peripheral leukocytes <5.6 nmol/h/mg protein) were submitted to molecular analysis to confirm diagnosis. Results. Screening with DBS-FP identified 135 subjects (18.2%) with GBA activity <2.19 nmol/h/mL, 131 of whom remained in the study. In 10 of these (7.6%), GBA activity in leukocytes was 2.6–5.5 nmol/h/mg protein. Subsequent molecular analysis confirmed 6 cases of heterozygosity and 4 normals for GD. Conclusion. DBS-FP assay was shown to be an effective initial GD screening strategy for high-prevalence populations in developing regions. Diagnosis could not be established from GBA activity in leukocytes alone, but required confirmation with molecular analysis.
1 INTRODUÇÃO
A doença de Gaucher (DG) é uma enfermidade de depósito lisossômico causada pela deficiência de atividade da enzima glicosilceramidase (β -glucocerebrosidase; GBA; β-glicosidase ácida; EC.3.2.1.45) (BRADY; KANFER, et
al.,1966) que leva ao acúmulo do substrato glicosilceramida (GlcCer), particularmente, nas células do sistema reticuloendotelial. (AMARAL; MARCAO, et
al., 2000; CHARROW; ANDERSSON, et al., 2000; BEUTLER; GRABOWSKI, 2001). A DG é uma doença genética, herdada de forma autossômica recessiva, e suas principais manifestações clínicas são hepatoesplenomegalia, anemia, plaquetopenia
(com tendência a sangramentos espontâneos), e dores ósseas (BARRANGER; O'ROURKE, 2001).
A DG é pan-étnica, com incidência estimada de 1:40.000 nos Estados Unidos da América, sendo registrada uma maior prevalência (1:400 a 1:800) entre os judeus Ashkenazi (GRABOWSKI, 2004).
O International Collaborative Gaucher Group (ICGG), Gaucher Registry, é o maior cadastro de pacientes com DG do mundo e conta com dados coletados por
médicos em 56 países (2011)1. Em novembro de 2010, havia 5.915 pacientes com DG cadastrados no ICGG, dos quais 561 (9,4%) são brasileiros (ICGG GAUCHER REGISTRY, 2011). Dentre os pacientes brasileiros com DG, 17(3,62%) são do
Ceará e sete doentes (35% dos casos da DG do Ceará) são provenientes de
Tabuleiro do Norte, um município com cerca de 28.000 habitantes, situado no leste
do Estado do Ceará, a 209 km de Fortaleza.
Especula-se que a elevada prevalência de DG em Tabuleiro do Norte,
estimada em 1:4 000 habitantes, provavelmente a maior do Brasil, poderia decorrer da baixa taxa de migração, do elevado nível de endogamia ao longo de muitas gerações e, ainda, da possível influência da origem judaica de algumas famílias
locais que vieram de Portugal, para a Região Nordeste do Brasil, por ocasião das guerras contra os holandeses, na segunda metade do século XVII (VIEIRA, 2000). A
elevada prevalência da doença no Município fomentou a realização do Estudo Populacional da Doença de Gaucher em Tabuleiro do Norte que se constituiu num grande desafio, por envolver uma patologia rara e uma comunidade de limitados
recursos financeiros e de infraestrutura.
O desenho inicial desse estudo populacional foi fundamentado nas estratégias
de Educação em Saúde, no estudo da genealogia das famílias envolvidas e no rastreamento populacional para DG, com suporte na análise das atividades enzimáticas da GBA e quitotriosidase, em amostras de sangue seco em papel de
filtro (SSPF), seguida de exames confirmatórios das atividades de GBA, em leucócitos e quitotriosidase no plasma (APÊNDICE A).
Para esclarecer dúvidas diagnósticas foi acrescentado o rastreamento das mutações da DG mais freqüentes no Brasil (N370S, L444P, G377S e 55del) entre os
2 REVISÃO DA LITERATURA
A doença de Gaucher (DG) é hereditária, causada por um erro inato do metabolismo dos glicoesfingolipídios (GELs), resultando no acúmulo de um material degradado incompletamente, denominado de glicocerebrosídio, dentro dos
lisossomos de células da linhagem monócito-macrófago (BEUTLER, 1999). Por isso, a DG se configura como uma enfermidade multissistêmica e um exemplo típico de
Doença de Depósito Lisossômico (DDL) resultante de uma deficiência inata da enzima glicosilceramidase (β-glucocerebrosidase; β-glicosidase ácida; GBA; EC 3.2.1.45) (BRADY; KANFER, et al., 1966; WEINREB; BRADY, et al., 1968).
2.1 ERROS INATOS DO METABOLISMO
Os Erros Inatos do Metabolismo (EIM) causam doenças metabólicas hereditárias que resultam da falta da atividade de uma ou de mais enzimas
específicas ou de defeitos no transporte de proteínas. Em geral, esse bloqueio provoca aumento do precursor da fase de comprometimento metabólico e
diminuição de intermediários subsequentes, ou ativação de rotas metabólicas alternativas, levando à produção de substâncias que não são encontradas de forma rotineira no organismo. As consequências podem ser, portanto, o acúmulo de
substâncias que estariam presentes em pequenas quantidades, deficiência de produtos intermediários críticos, deficiência de produtos finais específicos ou ainda o
número de doenças resultantes desses distúrbios metabólicos. Nos dias atuais
várias centenas de transtornos metabólicos hereditários são conhecidos, muitos dos quais correspondem a doenças graves que evoluem com frequência para morte ou
podem causar seqüelas importantes, como a deficiência mental dos indivíduos acometidos (OLIVEIRA; SANTOS, et al., 2001).
Cerca de 700 doenças relacionadas aos EIM foram descritos. A incidência é
comumente estimada em 1:2.500 a 1:5.000 nascidos vivos. Acredita-se, entretanto, que, pela enorme heterogeneidade das manifestações clínicas e pelas dificuldades
de diagnosticar os casos, o quadro real é subestimado e ainda faltam estudos epidemiológicos mais amplos sobre a incidência geral das doenças dos EIM (PAMPOLS, 2010).
A detecção precoce dos EIM tem a vantagem adicional de permitir o aconselhamento genético para os pais, com a opção do diagnóstico pré-natal em
gestações subsequentes. Na ausência de uma história familiar, a única maneira prática de identificar os indivíduos afetados assintomáticos é por meio de um programa de triagem neonatal (MEIKLE, et al., 2004).
2.2 DOENÇAS DE DEPÓSITO LISOSSÔMICO
As Doenças de Depósito Lisossômico (DDL) representam um grupo de cerca
de 50 patologias genéticas que apresentam deficiências de proteínas lisossômicas e não lisossômicas, causadas por mutações em genes codificadores de enzimas ou cofatores, resultando no acúmulo de substratos não degradados dentro dos
natureza do substrato é utilizada para agrupar as DDL em grandes categorias,
incluindo mucopolissacaridoses, lipidoses, glicogenoses e oligossacaridoses (MEIKLE, et al., 2004).
As DDL são doenças raras, com valores de prevalências isoladas variando de 1:50.000 nascimentos até 1:4.000.000 nascimentos, mas uma incidência combinada de aproximadamente um em 1.500 – 7.000 nascidos vivos (STARETZ-CHACHAM;
LANG, et al., 2009).
Ao longo da história, as DDL não foram consideradas doenças do
recém-nascido e os médicos, em geral, são ensinados que recém-recém-nascidos com DDL parecem normais ao nascimento e que os sintomas se desenvolvem de forma progressiva ao longo dos primeiros meses de vida ou mesmo depois de muitos
anos. Uma parcela desses pacientes, no entanto, pode ser de oligossintomáticos logo nos primeiros dias de vida ou, até mesmo, antes do nascimento, ou podem ter
sintomas transitórios no período neonatal (STARETZ-CHACHAM; LANG, et al., 2009), pois o depósito do substrato nas DDL se inicia na vida fetal e, dependendo da sua velocidade, torna-se evidente nos primeiros anos ou, até mesmo, somente após
a quarta ou quinta década de vida.
As DDL são patologias com manifestações clínicas progressivas, graves e
muitas são incuráveis. Para a maioria das DDL, o diagnóstico permite apenas medidas preventivas, tais como: o aconselhamento genético, detecção de
heterozigotos e o diagnóstico pré-natal. Embora as DDL estejam entre as primeiras doenças genéticas para as quais os defeitos bioquímicos primários foram elucidados, ainda há muito a aprender sobre os mecanismos patogenéticos
2.3 DOENÇA DE GAUCHER
A doença de Gaucher é a DDL mais frequentemente encontrada entre
humanos (BEUTLER; GRABOWSKI, 2001) e resulta da deficiência da β-glucocerebrosidase (GBA; Β-GLICOSIDASE ÁCIDA; EC. 3.2.1.45) (BRADY; KANFER, et al., 1966). Essa doença de herança autossômica recessiva, embora
rara, pode resultar também de mutações nos genes determinantes na produção de pró-saposinas ou ativadores, causando graves fenótipos da DG (BEUTLER;
GRABOWSKI, 2001).
Até o ano de 2001, cerca de 200 diferentes mutações no gene codificador da GBA lisossômica já haviam sido descritas (BEUTLER; GRABOWSKI, 2001) e, como
resultado, o GlcCer é degradado de forma mais lenta do que em células normais, acumulando-se dentro das células, primeiros nos fagócitos mononucleares. Esses
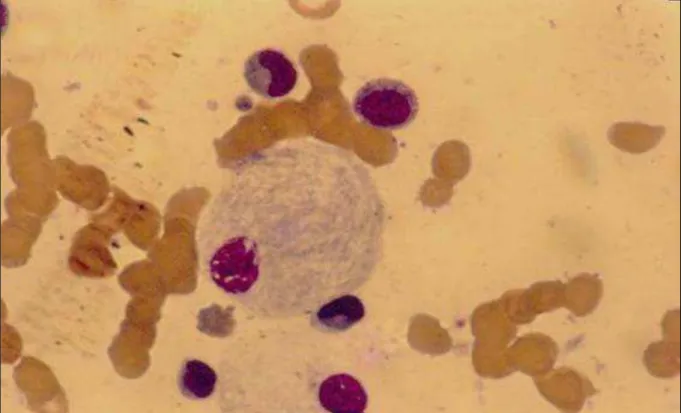
Figura 1. A Célula de Gaucher
Fonte: Foto cedida pelo Prof. Geraldo B. Cavalcanti, ano 2010.
2.3.1 Fatos Históricos da doença de Gaucher
A história da DG começa em 1882 quando o médico francês Philippe C. E. Gaucher descreveu, em detalhes, as características clínicas apresentadas por uma
paciente de 32 anos de idade que tinha o baço aumentado de volume e uma célula grande e incomum (Figura 2). A denominação de doença de Gaucher em sua homenagem, porém, foi dada pela primeira vez somente em 1905 por N. E. Brill
(AERTS, VAN BREEMEN et al., 2005).
No início do século XX, foi sugerido que a DG era familiar, contudo, o
lipídios como material estocado foi reconhecido somente em 1916 (BEUTLER;
GRABOWSKI, 2001).
Em 1934, H. Aghion, na França, identificou o material estocado como um glicocerebrosídio (AERTS; BREEMEN, et al., 2005), mas somente nas décadas de
1950 e 1960, Brady e seus colegas demonstraram que o acúmulo do GlcCer era causado pela atividade deficiente da enzima GBA. Uma vez que o defeito enzimático
havia sido descoberto, o conceito de Terapia de Reposição Enzimática (TRE) foi desenvolvido na qual a enzima deficiente pode ser suplementada por uma enzima ativa (FUTERMAN; MEER, 2004). Depois, Weinreb, et al., (1968) localizaram o
defeito enzimático dentro dos lisossomos e a DG passou a integrar o Grupo das DDL (BEUTLER; GRABOWSKI, 2001).
Figura 2. Fatos históricos da doença de Gaucher.
Em meados da década de 1980, os estudos de Beutler e Ginns permitiram a
localização e o sequenciamneto do gene produtor da GBA no cromossomo 1 (GINNS; CHOUDARY, et al., 1985). Apesar do grande progresso na compreensão
das bases genéticas, moleculares e bioquímicas da doença, quase nada se sabe sobre como o acúmulo de GlcCer nos lisossomos causa a doença em nível celular (FUTERMAN; SUSSMAN, et al., 2004).
Os estudos de Barton e Brady nos Estados Unidos no início dos anos 90 deram origem à TRE para a DG. A GBA foi purificada de tecidos placentários para realização da primeira TRE para tratamento da DG Tipo I, aprovada pelo U.S. Food
and Drug Administration (FDA) em 1991. A seguir, a GBA passou a ser obtida de
células de ovários de hamster chinês, sendo modificada por tecnologia
recombinante, originando um novo medicamento, Imiglucerase, que foi aprovado pelo FDA para TRE em 1994, em substituição ao medicamento que estava sendo
utilizado (Alglucerase). A eficácia dessa terapia tem sido confirmada por vários estudos ao longo do tempo. Em 2006, mais de 4.300 pacientes já estavam realizando a TRE, com eficácia e segurança, permitindo avanços no tratamento da
DG Tipo I (BRADY, 2006).
2.3.2Bases Celulares e Bioquímicas da doença de Gaucher
Os lisossomos são organelas celulares, contendo enzimas líticas, a maioria delas solúveis, que atuam em pH ácido para degradação de substâncias localizadas em seu interior (FUTERMAN; MEER, 2004). A descoberta das funções dos
respeito do papel dos lisossomos nas doenças, em especial, nas doenças que
resultam da "falta do correto funcionamento das enzimas lisossômicas (FUTERMAN; MEER, 2004). Essa descoberta foi fundamental para aprofundamento no estudo da
DG, pois essa patologia se encontra localizada nos lisossomos (AMARAL; PINTO, et al., 1996).
As enzimas lisossômicas, cerca de 60 hidrolases ácidas e uma dezena de
proteínas acessórias, permitem a degradação sequencial de quase todas as macromoléculas, incluindo os lipídios, glicosaminoglicanos, oligossacarídeos,
proteínas e ácidos nucléicos (SANDHOFF; KOLTER, 2003). Essas proteínas são direcionadas aos lisossomos para serem degradadas, principalmente, pela via dos receptores da manose-6-fosfatase. Transportadores específicos na membrana
lisossômica regulam a exportação dos produtos do catabolismo intralisossômico para o citoplasma, onde podem ser reutilizados (AERTS; HOLLAK, et al., 2003).
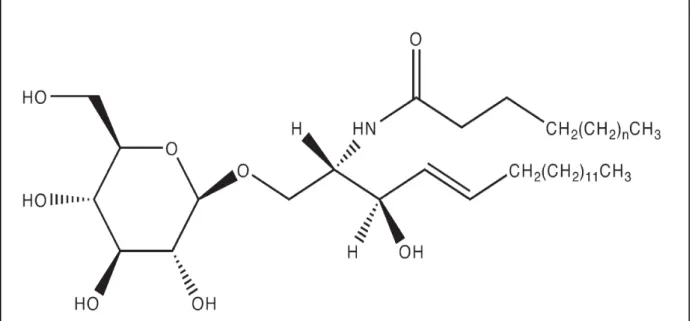
O substrato GlcCer acumulado na DG é um glicoesfingolipídio e, por isso, a DG é classificada como uma esfingolipidose, como mostra a Figura 3. Os GELs são componentes essenciais das membranas das células eucarióticas, sintetizados no
retículo endoplasmático e no Complexo de Golgi. Os GELs estão localizados, principalmente, na camada externa das membranas plasmáticas e são direcionados
para o interior dos lisossomos onde são degradados (SILLENCE; PLATT, 2004). As macromoléculas, exógenas e endógenas, incluindo o GlcCer, são entregues aos
Figura 3 - Representação da estrutura química da D-glucosyl-ceramida, o substrato estocado na doença de Gaucher. N= 10-18
Fonte: Sawkar; D'haeze, et al., 2006.
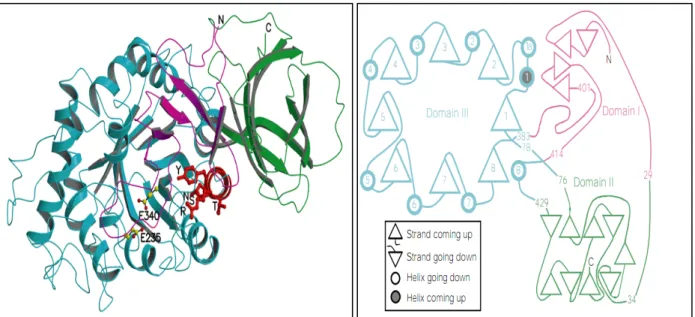
A GBA, mostrada na Figura 4, é uma proteína de membrana periférica que realiza a reação da hidrólise da ligação da β-glicosil do GlcCer nos lisossomos, conforme apresentada na Figura 5 (SAWKAR; D'HAEZE, et al., 2006). Para isso, a
GBA requer a ação coordenada da saposina C (cofator) e de lipídios carregados de íons negativos para que ocorra a atividade máxima da enzima (DVIR; et al., 2003). De modo surpreendente, ainda não está completamente esclarecido o mecanismo
pelo qual a GBA é direcionada de seu local de síntese no retículo endoplasmático para os lisossomos (RIJNBOUTT; AERTS, et al., 1991). Sabe-se, contudo, que as
saposinas são proteínas não enzimáticas que possuem pequena cadeia de oligossacarídeos que estimulam a clivagem enzimática de moléculas de armazenamento (esfingolipídios) e a saposina C é a responsável pela ativação de
Figura 4- O Raio-X da refinada estrutura da β-glucocerebrosidase humana. Fonte: Adaptado de Dvir; Harel, et al. (2003).
Figura 5– Reação catalisada pela β-glucocerebrosidase humana. Fonte: Adaptado de Dvir; Harel, et al., 2003.
A maioria das mutações conhecidas da GBA diminui, de forma parcial ou total,
a atividade catalítica ou reduz a estabilidade da enzima (GRACE; NEWMAN, et al., 1994). Embora a atividade da GBA esteja diminuída de modo acentuado nos pacientes com DG, estima-se que os níveis residuais de atividade enzimática da
AERTS, et al., 1990; MEIVAR-LEVY; HOROWITZ, et al., 1994; AMARAL; PINTO, et
al., 1996; STEWART; JONES, 1999; RUDENSKY; PAZ, et al., 2003).
Embora tenha sido dito que a gravidade da DG depende do nível de atividade
residual da GBA (BEUTLER; GRABOWSKI, 2001), isso é difícil de ser provado para a maioria das mutações (MEIVAR-LEVY; HOROWITZ, et al., 1994), pois outros fatores também podem estar relacionados às manifestações clínicas da DG (COX,
2001). Acredita-se que a célula de Gaucher possa estimular a liberação de citocinas que podem contribuir para desenvolver a doença, tais como: interleucinas-1α (IL-1α),
interleucina-6 (IL-6) (ALLEN; MYER, et al., 1997), e o fator de necrose tumoral alfa (TNF-α) (MICHELAKAKIS; SPANOU, et al., 1996).
Os macrófagos, repletos de inclusões do substrato GlcCer (Células de
Gaucher), podem ser encontrados na medula óssea, fígado e baço (MACARIOLA; STAAT, et al., 2001; MARTINS; LOBO, et al., 2003), e estão também implicados em
muitos fenômenos sistêmicos que acompanham a doença, incluindo distúrbios do metabolismo energético, diversas anormalidades de proteínas plasmáticas e outros constituintes do sangue, tais como: imunoglobulinas, fatores de coagulação,
lipoproteínas, ferritina, transcobalamina II, enzima conversora de angiotensina, enzimas lisossômicas e quitotriosidase. Está claro que ocorre uma resposta
inflamatória em indivíduos afetados e que o fenótipo clínico decorre de um efeito oriundo do armazenamento de macrófagos, além da presença física das células de
Algumas substâncias se elevam acentuadamente nos pacientes com DG e
podem ser utilizadas como marcadores da atividade da doença. Um exemplo delas é a quitotriosidase, uma cistinase humana, que se mostra enormemente elevada no
plasma de pacientes sintomáticos da DG. A enzima é sintetizada nos macrófagos doentes e sua atividade elevada se correlaciona com o estoque de GlcCer nos tecidos, servindo de parâmetro de gravidade da doença. Em pacientes
assintomáticos da DG, entretanto, a quitotriosidase pode estar levemente alterada (HOLLAK; WEELY, et al., 1994).
Um interessante aspecto que pode representar uma armadilha no uso da quitotriosidase como marcador para DG é a completa ausência da atividade dessa enzima em 3 a 5% da população geral. Isso resulta da herança de um alelo nulo do
gene da quitotriosidase humana que decorre de uma duplicação intragênica encontrada com elevada frequência na população. A presença de um alelo dessa
mutação (heterozigoto) poderia reduzir o potencial aumento da quitotriosidase como consequência da atividade da DG (HOLLAK; WEELY, et al.,1994).
2.3.3 Aspectos Genéticos das Mutações da doença de Gaucher
A DG possui mecanismo de herança autossômica recessiva, causada por alteração no gene codificador da enzima GBA que se encontra localizado no
cromossomo 1 (região q21). O gene abrange aproximadamente 7kb de DNA do genoma (ROZENBERG, et al., 2006), dividido em 11 éxons que codificam uma proteína de 497 aminoácidos (HRUSKA; LAMARCA, et al., 2008). Existe, também,
localiza a 16 kb do gene funcional, abrangendo apenas 5,7 kb de DNA, portanto
menor, em decorrência de algumas perdas nucleotídicas em relação ao gene funcional (HOROWITZ; WILDER, et al., 1989). Esse PSGBA apresenta acentuada
homologia com o gene funcional, com 96% de identidade entre seus nucleotídeos (BEUTLER; GRABOWSKI, 2001), o que complica a análise da mutação na região (DEPAOLO, et al., 2009).
As mutações N370S, L444P, IVS2+1 e 84GG representam 90% dos alelos mutantes na população de judeus Ashkenazi, apesar de constituírem menos de 75%
dos alelos nas populações não Ashkenazi (EMRE; GURAKAN, et al., 2008). No Brasil, as mutações N370S, L444P e G377S são as mais frequentes e representam 47%, 27%, e 2,2% de todas as mutações, respectivamente (ROZENBERG, et al.,
2006).
As mutações são classificadas em brandas, graves e nulas, e a combinação
entre elas determina a gravidade da doença. Por exemplo, os pacientes homozigotos com mutação N370S desenvolvem as formas não neuropáticas e os homozigotos para a mutação L444P, frequentemente, desenvolvem a forma
neuropática da doença (GRABOWSKI, 2008). Apesar disso, a correlação genótipo-fenótipo é limitada e incompleta, pois há muitas variações entre pacientes com a
mesma forma clínica e entre os que apresentam o mesmo genótipo (HRUSKA; LAMARCA, et al., 2008; SIDRANSKY, 2004). Genes modificadores, proteínas
apenas na genotipagem para predizer o prognóstico ou afirmar a necessidade de
terapia (KOPRIVICA, et al., 2000).
2.3.4 Aspectos Epidemiológicos da doença de Gaucher
A DG é pan-étnica, com incidência estimada de 1:40.000 nos Estados Unidos,
sendo registrada a maior prevalência entre os judeus Ashkenazi (1:400 a 1:800) (GRABOWSKI, 2004). Em 2007, havia 4936 pacientes cadastrados no ICGG. Os
Estados Unidos da América (1780 pacientes, 36%), Israel (683 pacientes, 13,8%), Brasil (496 pacientes, 10%) e Reino Unido (235 pacientes, 4,7%) são os destaques na epidemiologia da DG (ICGG GAUCHER REGISTRY, 2007).
Em novembro de 2010, o número de pacientes cadastrados no ICGG já era 5915 pacientes, dos quais 561 estavam no Brasil (ICGG GAUCHER REGISTRY,
2011). Dos pacientes brasileiros, vinte deles (3,5%) estão no Ceará2, um estado com cerca de 8,2 milhões de habitantes. Desses, sete (35% pacientes) provêm de Tabuleiro do Norte, um pequeno município com cerca de 28.000 habitantes, no
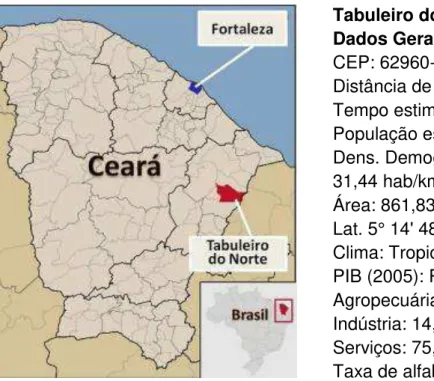
Estado do Ceará, na Região Nordeste do Brasil (Figura 6).
A Associação Nacional de Pacientes com doença de Gaucher informou que,
em maio de 2011, havia 614 pacientes com DG em todo Brasil, com predomínio de pacientes na Região Sudeste (57% dos pacientes). São Paulo (178 pacientes),
Minas Gerais (84 pacientes) e Rio de Janeiro (76 pacientes) são os estados
brasileiros com maior número de pacientes com a DG3, conforme Figura 7.
Tabuleiro do Norte-CE Dados Gerais
CEP: 62960-000
Distância de Fortaleza: 209 km
Tempo estimado de viagem: 2 h 59min População estimada (2007): 28.291 hab Dens. Demográfica (2000):
31,44 hab/km² Área: 861,838 km²
Lat. 5° 14' 48'' Longitude: 39° 07' 50'' Clima: Tropical quente semiárido PIB (2005): R$ 81.832.000 Agropecuária: 10,12 % Indústria: 14,03 % Serviços: 75,03 %\
Taxa de alfabetização (2000): 72,2 %
Figura 6. Localização geográfica e principais dados de Tabuleiro do Norte – CE.
Fontes: Mapa adaptado do site Wikipédia, 20114. Dados do site do Governo do Estado do Ceará, 20115.
A idade média dos pacientes brasileiros com DG por ocasião do diagnóstico foi de 17±14 anos, com predomínio de pacientes com até 10 anos de idade (43% dos
pacientes), e entre 10 e 20 anos (22% dos pacientes). Não houve diferença significativa da prevalência da DG com relação a sexo e a grande maioria dos
pacientes cadastrados no ICGG (95%) realiza terapia de reposição enzimática com a enzima Imiglucerase (ICGG GAUCHER REGISTRY 2011).
3PEDRO STELIAN, presidente da Associação Nacional de Pacientes com Doença de Gaucher, em
comunicação pessoal, 2011.
4(http://pt.wikipedia.org/wiki/Tabuleiro_do_Norte, acessado em 08 de Maio de 2011). 5
2.3.5 Aspectos clínicos da doença de Gaucher
As manifestações clínicas mais comuns da DG incluem: anemia, trombocitopenia, hepatoesplenomegalia e complicações ósseas (dores, lesões e
crises ósseas, infartos corticais e medulares, expansão medular, osteopenia, osteonecrose e fraturas patológicas). Muitas vezes, porém, o paciente com DG se
encontra assintomático e alterações significativas podem ser descobertas durante o exame físico ou por exames laboratoriais de rotina (KAPLAN; ANDERSSON, et al., 2006; KISHNANI; DIROCCO, et al., 2009). Além disso, por causa da raridade da
doença e da variedade de manifestações clínicas, muitos pacientes são erroneamente diagnosticados ou não diagnosticados (MISTRY;SADAN, et al., 2007).
NORTE:
Pará – 14
Amazonas - 8 Amapá - 6 Acre
-Rondônia - 1
NORDESTE:
Bahia – 22
Ceará –20
Pernambuco - 19 Alagoas - 11
Maranhão –11
R.G do Norte - 10 Paraíba - 7 Sergipe - 4
Piauí - 1
CENTRO-OESTE:
D. Federal
–
14
Goiás
–
10
Mato Grosso 9
Mato G do Sul
–
8
SUDESTE:
São Paulo – 178
Minas Gerais– 84
Rio de Janeiro – 76
Espírito Santo - 12
SUL:
Paraná –45
Rio G do Sul - 25
Santa Catarina - 17 Total em 2011
Brasil: 614 pacientes
Figura 7 - Prevalência da doença de Gaucher, no Brasil.
Existe grande heterogeneidade de fenótipos na DG. Os três principais
fenótipos, entretanto, são: não neuropático (Tipo I, 230800), neuropático agudo (Tipo II, 230900) e neuropático subagudo (Tipo III, 231000) (GRABOWSKI, 2004),
dependendo da presença de manifestações neurológicas ou não. As prevalências estimadas para os Tipos I, II e III são de 91,5%, 1,2%, e 7,3%, respectivamente (ICGG GAUCHER REGISTRY, 2011). O paciente com DG do Tipo I não apresenta
manifestações neurológicas decorrentes da doença como mostra a Tabela 1. A distinção entre os Tipos I e III da DG na infância, quando a criança ainda não teve
nenhuma manifestação neurológica, nem sempre é fácil, embora certos genótipos estejam associados com doença Tipo III juvenil, indicando que determinados pacientes poderão desenvolver anormalidades neurológicas no futuro (BEUTLER;
GRABOWSKI, 2001).
Tabela 1 - Características clínicas dos Tipos da doença de Gaucher.
Características Clínicas Tipo I Tipo II Tipo IIIa Tipo IIIb Tipo IIIc
Início dos sintomas Infância e adulto Primeira Infância Infância Infância Infância
Hepatoesplenomegalia + a ++++ + +++ +++ +
Hiperesplenismo + a ++++ + +++ +++ +
Crises ósseas/fraturas + a ++++ - ++ +++ +
Sintomas
Neurodegenerativos - +++ ++ +
Sobrevida (em anos) 6 a 80 anos ou mais < 2anos décadas 2ª a 4ª décadas 2ª a 4ª décadas 2ª a 4ª
Prevalência étnica Ashkenazi Judeus Pan-étnica Norte da Suécia Pan-étnica Pan-étnica
O Tipo III da DG é subdividido em IIIa, com doença neurológica progressiva
manifestada como mioclonia e demência; IIIb, com doença visceral e óssea agressivas e com manifestações neurológicas limitadas à paralisia horizontal
supranuclear do olhar; e IIIc, com doença neurológica limitada à paralisia horizontal supranuclear do olhar, opacidade da córnea e calcificações de valvas cardíacas, mas, em geral, com pouca doença visceral (BEUTLER; GRABOWSKI, 2001).
A DG é progressiva ao longo do tempo e, se não tratada, pode levar a mortes prematuras provocadas por complicações hemorrágicas, doença hepática, sepse,
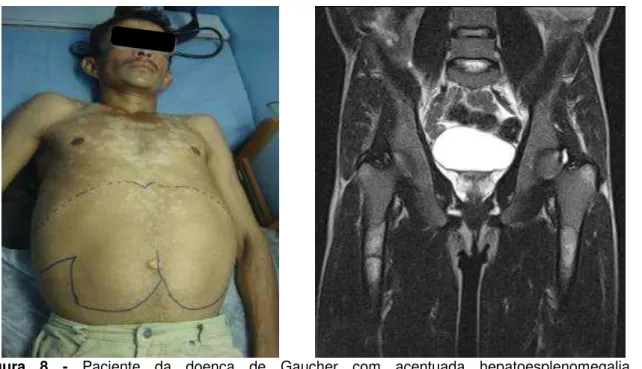
doença pulmonar, hipertensão pulmonar e complicações decorrentes da doença óssea avançada (MISTRY, et al., 2007). Não raro, a DG é diagnósticada no paciente em estado grave (Figura 8), demonstrando que atrasos de diagnóstico podem
resultar em complicações graves atribuíveis à doença. Desse modo, a educação médica é necessária para aumentar a detecção precoce e, quando necessário, o
tratamento dos pacientes com DG (MISTRY, et al., 2007).
Figura 8 - Paciente da doença de Gaucher com acentuada hepatoesplenomegalia e comprometimento ósseo observado na imagem de ressonância magnética dos fêmures.
2.3.6 Diagnóstico da doença de Gaucher
Durante muitos anos, o diagnóstico da DG era baseado somente na histologia,
feito pela presença da célula de Gaucher, uma célula da linhagem monócito-macrófago carregada de lipídios (LEVYLAHAD; ZIMRAN, 1997). Nos dias atuais, o diagnóstico é realizado com maior frequência, pela avaliação da atividade
enzimática da GBA em leucócitos de sangue periférico (BEUTLER; SAVEN,1990; CHARROW; ESPLIN, et al., 1998) ou por cultura de fibroblastos com base na
biopsia de pele ou pela análise molecular das mutações (CHARROW; ESPLIN, et al., 1998; BEUTLER; GRABOWSKI, 2001).
Atividade de GBA baixa (<10% do normal) em leucócitos ou fibroblastos ainda
é o padrão de diagnóstico de doença de Gaucher. As principais vantagens dos testes enzimáticos são relacionadas ao fato de eles serem suficientes para
estabelecer o diagnóstico em indivíduos afetados sem depender do conhecimento da origem étnica ou do conhecimento prévio de uma mutação familiar específica. As limitações dos testes enzimáticos são a labilidade da atividade enzimática, a falta de
correlação entre a atividade enzimática in vitro e o tipo e gravidade da doença, e por eles não permitirem a diferenciação entre heterozigotos e normais (LEVYLAHAD;
ZIMRAN, 1997).
O diagnóstico molecular na DG envolve a identificação de mutações
patogênicas no gene da GBA e é de grande importância, pois algumas mutações podem ser associadas à gravidade do prognóstico, permitindo aconselhamento genético adequado e facilitando a realização dos programas de rastreamento
ABRAHAMOV; ELSTEIN, et al., 1995). As correlações entre o genótipo e o fenótipo
na DG são úteis, principalmente, para a distinção entre as formas neuronopática e não neuronopática. Elas são menos precisas na predição de gravidade dentro de
cada tipo de doença envolvida, em especial, na DG Tipo I que é simultaneamente a mais prevalente e a mais variável forma da enfermidade (LEVYLAHAD; ZIMRAN, 1997).
A principal limitação da análise molecular é quanto à sensibilidade. Como a DG é recessiva, os pacientes devem ter dois alelos com a mesma mutação ou uma
mutação diferente em cada um dos alelos (LEVYLAHAD; ZIMRAN, 1997). Assim, quando comparada com a sensibilidade completa (100%) dos testes enzimáticos, a sensibilidade real da análise de DNA depende de quais mutações a serem
pesquisadas e da frequência combinada dessas mutações em pacientes de um mesmo grupo étnico. Existem três grupos étnicos nos quais há um pequeno número
de mutações, resultando em alta sensibilidade do Teste de DNA para essas mutações: os judeus Ashkenazi, (judeus do leste, de origem europeia) com DG do Tipo I; os suecos de Norrbotten (Região Norte da Suécia), e os árabes de Jenin,
com DG do Tipo III (LEVYLAHAD; ZIMRAN, 1997).
2.3.7 Diagnóstico de heterozigotos para doença de Gaucher
é um diagnóstico de laboratório e é importante por suas implicações reprodutivas
(LEVYLAHAD; ZIMRAN, 1997).
O teste enzimático tem grande utilidade em diagnosticar afetados da DG
(homozigotos), mas não pode distinguir com segurança os indivíduos heterozigotos dos normais, embora os portadores tenham, em média, metade da atividade normal da GBA em leucócitos e fibroblastos. Apesar de várias tentativas de refinar o ensaio
enzimático, cerca de um terço dos portadores apresenta atividade de GBA definida como normal o que leva ao diagnóstico equivocado de não portador, pois há
considerável sobreposição de valores da GBA entre portadores e não portadores de mutação para DG (LEVYLAHAD; ZIMRAN, 1997).
Ao contrário, o Teste de DNA é inequívoco para as mutações testadas.
Portanto, em famílias com mutações conhecidas, o Teste de DNA identifica portadores e não portadores com precisão absoluta. Nas famílias em que uma ou
ambas as mutações são desconhecidas, a detecção de heterozigotos deve basear-se nos testes enzimáticos (que podem resultar em erros por classificar alguns portadores como não-portadores) ou na identificação da mutação familiar por
sequenciamento completo do gene GBA. O sequenciamento do gene da GBA, todavia, em larga escala, é complicado por motivos técnicos, e raramente é
realizado fora do ambiente de pesquisa (LEVYLAHAD; ZIMRAN, 1997).
2.3.8 Avaliação Familiar na doença de Gaucher
O diagnóstico de uma pessoa afetada leva à necessidade da avaliação da
pais e de outras pessoas em risco de serem portadores (irmãos dos pais) ou
afetadas (irmãos do primeiro afetado). Como os pacientes com DG do Tipo I podem ser assintomáticos ou sintomáticos ainda não diagnosticados, é importante orientar
as famílias de que o teste pode revelar outras pessoas afetadas e não apenas portadoras. Isso pode ser vantajoso em pacientes sintomáticos ainda não diagnosticados, pois se beneficiariam com diagnóstico correto e, talvez, o tratamento
(se necessário e disponível), mas também pode estigmatizar indivíduos assintomáticos, como sendo “doentes”, embora possam permanecer assintomáticos
por toda vida, sem necessidade de tratamento (LEVYLAHAD; ZIMRAN, 1997).
2.3.9 Rastreamento Populacional para doença de Gaucher
De um modo geral, um programa de rastreamento seguido por testes
confirmatórios, quando oferecido a populações de risco, é uma poderosa ferramenta de saúde pública. Ele deve ser de relativo baixo-custo, de fácil realização e comprovação, produzindo resultados rápidos, embora não definitivos, além de
determinar quem, dentro de um grupo de baixo risco, é, de fato, de alto risco para determinada patologia (EVANS; GALEN, et al., 2005).
A existência de tratamento eficaz para DG, que proporciona bom controle da doença e que tem impacto positivo na qualidade de vida do paciente (DAMIANO;
PASTORES, et al., 1998; MASEK; SIMS, et al., 1999; GIRALDO; SOLANO, et al., 2005; WEINREB; BARRANGER, et al., 2007), torna relevante a realização de rastreamento em populações de alto risco para o diagnóstico de novos casos e início
Portanto, a análise das atividades de GBA e da quitotriosidase em amostras de
SSPF seguida da realização de exames confirmatórios em leucócitos para os casos suspeitos pode ser útil na triagem para DG em populações reconhecidamente de
risco (CHAMOLES; BLANCO, et al., 2002; CIVALLERO; MICHELIN, et al., 2006). Uma das vantagens para o uso de amostras de SSPF para o diagnóstico de DDL é que essas amostras podem ser transportadas com segurança por longas
distâncias, inclusive pelo correio, em envelopes normais, porque a atividade das enzimas permanece estável em temperatura ambiente por meses (CHAMOLES;
BLANCO, et al., 2002; CIVALLERO; MICHELIN, et al., 2006; MÜLLER; RODRIGUES, et al., 2010). Além disso, a preparação de uma amostra SSPF requer apenas uma pequena quantidade de sangue (MÜLLER, et al., 2010). Desse modo,
essa técnica se reveste da maior importância, sobretudo, em países extensos e com áreas de acesso difícil, onde a coleta regular e a postagem das amostras são fatores
limitantes (CIVALLERO; MICHELIN, et al., 2006).
Questiona-se se o tempo decorrido entre a coleta e a análise da amostra de SSPF produziria ou não alterações nos resultados das atividades enzimáticas na
DG. Os estudos realizados com essa técnica revelaram que a perda mínima de atividade enzimática decorrente do tempo para o transporte das amostras até o
laboratório de análise não impede a diferenciação entre pacientes e controles, pois não há mudanças significativas nas atividades enzimáticas até 21 dias depois de
2.3.10 Tratamento da doença de Gaucher
De um modo geral, o tratamento das DDL se baseia nas seguintes estratégias
e objetivos:
a) terapia de reposição enzimática (TRE) - para suprir a enzima deficiente; b) terapia da redução do substrato (TRS) - para reduzir o acúmulo do substrato
estocado produzido pela deficiência enzimática;
c) terapia de “reforço enzimático” – para melhorar a atividade enzimática;
d) terapia gênica - para suprir uma cópia (ou cópias) com objetivo de repor a função perdida do gene (DESNICK, 2004; FUTERMAN; SUSSMAN, et al., 2004; CONNOCK; BURLS, et al., 2006; CONNOCK; JUAREZ-GARCIA, et al., 2006).
Até o início da década de 1990, o tratamento da DG se limitava a medidas de suporte terapêutico e ao alívio dos sintomas de dores, desconforto respiratório e
alterações hematológicas. A esplenectomia total era considerada um recurso para pacientes com hepatoesplenomegalias volumosas e com comprometimento cardiopulmonar. Após a cirurgia, porém, as dores ósseas continuavam a causar
grande sofrimento aos pacientes. (ZIMRAN; ELSTEIN, et al., 1995).
A imiglucerase, nome comercial CEREZYME®, é um análogo da enzima
humana GBA que é utilizado como TRE para DG. A imiglucerase é produzida por tecnologia de DNA-recombinante com origem em células de ovários de hamster
chinês (GRABOWSKI; BARTON, et al., 1995; ZIMRAN; ELSTEIN, 2003), sendo fabricada pelo laboratório farmacêutico Genzyme, recentemente adquirido pelo laboratório Sanofi-Aventis.6
6 (
A velaglucerase alfa, nome comercial VPRIV®, fabricado pela Shire plc, é uma
enzima lisossômica hidrolítica glucocerebrosido-específica, uma forma recombinante da GBA, indicada como terapêutica de substituição enzimática prolongada para
aqueles que sofrem da DG Tipo I. Velaglucerase alfa tem uma sequência de aminoácidos idêntica à enzima natural e foi aprovada pela Food and Drug Administration (FDA), em 26 de fevereiro de 2010, para uso em crianças e adultos
nos Estados Unidos7.
A taliglucerase alfa, fabricada pela Protalix BioTherapeutics, indicada para TRE
de pacientes com DG, encontra-se disponível para uso no Brasil8, embora não disponha de registro definitivo junto à Agência Nacional de Vigilância Sanitária e não tenha ainda sido aprovada pelo FDA9.
A Terapia de Redução do Substrato (TRS) se baseia na redução do acúmulo do substrato GlcCer pela inibição da enzima glicosilceramida-sintetase, responsável
pelo primeiro passo na síntese da maioria dos glicolipídios, inclusive do GlcCer. O exemplo dessa forma de tratamento é o Miglustate, nome comercial ZAVESCA®, fabricado por Almac Pharma Services Limited. Craigavon, Armagh, Irlanda do Norte
e importado e distribuído por Actelion Pharmaceuticals do Brasil Ltda. Miglustate é indicado para o tratamento oral de pacientes adultos com DG de leve ou de
moderada gravidades para os quais a TRE com imiglucerase não está disponível ou não está indicada, como nos casos de reações adversas graves (ZIMRAN E
ELSTEIN, 2003).
7http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm202288.htm, acesso em 08/05/2011.
8http://portal.saude.gov.br/portal/arquivos/pdf/autorizacao_anvisa_importacao_taliglucerase_alfa.pdf, acessado em 08/05/2011).
A terapia de “reforço enzimático”, usando pequenas moléculas para aumentar a
atividade da enzima GBA mutante, está sendo desenvolvida e apresenta resultados promissores (SAWKAR; CHENG, et al., 2002).
Embora novas drogas e formas de tratamento para DG estejam surgindo nos últimos anos, somente a TRE e TRS estão licenciadas para uso na Europa (BECK, 2007), sendo a TRE o tratamento-padrão para DG visceral (SCHMITZ; POLL, et al.,
2007), por ser bem tolerada, com raros e leves efeitos colaterais (STARZYK; RICHARDS, et al., 2007), devendo ser utilizada por toda vida e não ser interrompida
sem uma cuidadosa monitoração de sua evolução (MISTRY; GERMAIN, 2007). Outras medidas suplementares usadas para alívio das dores ósseas podem melhorar a qualidade de vida dos pacientes, como terapia de suporte, baseada em
analgésicos e antiinflamatórios não esteróides. O alendronato de sódio, por exemplo, quando associado à TRE pode melhorar a osteopenia (WENSTRUP;
BAILEY, et al., 2004).
O elevado custo do tratamento para doenças genéticas raras (para DG, cerca de $200,000/paciente/ano), entretanto, representa um problema de importância
crescente para todos os sistemas de saúde do mundo, especialmente dos países pobres, onde os recursos financeiros devem cobrir despesas com outras doenças
mais prevalentes (GROSS, 2002; BEUTLER, 2006). No Brasil, o tratamento para DG encontra-se custeado pelo Sistema Único de Saúde (SUS), regulamentado pela
Portaria de nº. 449 do Ministério da Saúde, com data de 8 de julho de 2002, que instituiu critérios para o diagnóstico da doença, para inclusão e exclusão de pacientes para tratamento, esquemas terapêuticos, de monitorização do tratamento
3 OBJETIVO GERAL
O propósito desta dissertação é analisar o rastreamento populacional para DG
em Tabuleiro do Norte - CE - Brasil, realizado durante o período de 01 junho de 2007 a 31 de maio de 2008, após a avaliação das atividades das enzimas GBA e quitotriosidase em SSPF, seguida de análise enzimática da GBA em leucócitos e
quitotriosidase no plasma.
3.1 OBJETIVOS ESPECÍFICOS
Conhecer o perfil enzimático dos participantes quanto à medida das atividades
enzimáticas da GBA e quitotriosidase em amostras de SSPF e suas correlações com a análise das atividades enzimáticas da GBA em leucócitos e quitotriosidase
plasmáticas.
Encaminhar os participantes cujas medidas da atividade da GBA forem compatíveis com DG para avaliação clínica e de exames complementares.
Realizar a análise molecular dos indivíduos cujas atividades enzimáticas da GBA em leucócitos forem compatíveis com DG.
4 METODOLOGIA
O rastreamento populacional é uma parte do Estudo Populacional da Doença
de Gaucher em Tabuleiro do Norte-CE, aprovado pelo Comitê de Ética em Pesquisa do Hospital Geral César Cals de Oliveira, Fortaleza - CE, em 01 de setembro de 2006, sob número 054/2006.
Este estudo é do tipo corte transversal, realizado durante o período 01 de junho de 2007 a 31 de maio de 2008, no Centro de Referência dos Erros Inatos do
Metabolismo de Tabuleiro do Norte (CREIM), envolvendo 740 indivíduos de ambos os sexos, com idade variando de um ano a 85 anos, utilizando amostras de SSPF para rastreamento da DG,
A amostragem foi não aleatória, constituída de voluntários, sem ressarcimento de despesas ou recompensas financeiras aos participantes. A adesão dos indivíduos
foi motivada pelo trabalho intensivo de Educação em Saúde realizado nas escolas municipais, nas unidades básicas de saúde e através das emissoras de rádio locais e regionais. Palestras educativas, com duração de 60 minutos, foram realizadas
antes de cada coleta, utilizando vídeos, cartazes e revistas. Os critérios de inclusão dos indivíduos no estudo foram:
a) ser residente na cidade e ter ascendência de famílias de Tabuleiro do Norte - CE;
b) ter participado de pelo menos uma sessão educativa para DG Gaucher realizada no CREIM;
Embora não representasse um critério de exclusão, os parentes de primeiro
grau (pais, filhos e irmãos) dos atuais pacientes não participaram do estudo, pois haviam sido investigados por ocasião do diagnóstico da doença em seus
componentes familiares.
O tamanho da amostra foi calculado utilizando a fórmula para populações infinitas. Fixaram-se o nível de significância de 5% e um erro amostral de 3,6%. Em
razão da ausência de dados prévios, a proporção de 50% foi adotada para cálculo da amostra, porque esse valor implica tamanho máximo de amostra.
4.1 DESENHO DO ESTUDO
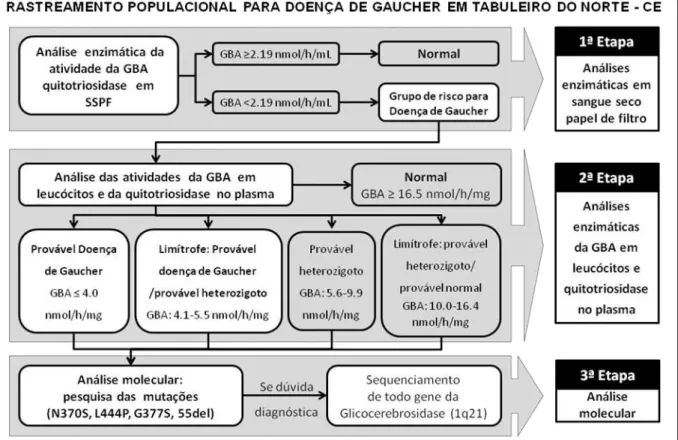
O referido estudo foi idealizado em duas etapas (Figura 9):
a) análise enzimática da atividade da GBA e quitotriosidase em amostras
de SSPF;
b) análise da atividade da GBA nos leucócitos e quitotriosidase no plasma de indivíduos selecionados pela primeira etapa;
A opção de realizar a triagem com suporte em ensaios enzimáticos, em vez da análise molecular isoladamente, se deu pelo desconhecimento do perfil genético
dessa população e pelas limitações de infraestrutura da região.
O estudo da análise molecular das mutações dos indivíduos cujas atividades
enzimáticas da GBA em leucócitos foram compatíveis com afetados ou portadores de mutações da DG foi aprovada pelo Comitê de Ética em Pesquisa do Hospital Geral César Cals, no dia 05 de setembro de 2008, e obteve aprovação da Comissão
realizado o rastreamento das mutações da GBA mais prevalentes no Brasil (N370S,
L444P, G377S e 55del) para os indivíduos selecionados..
GBA: β-glucocerebrosidase; SSPF: sangue seco em papel de filtro. Figura 9. Desenho do Estudo.
Fonte: Dados do autor, ano 2008.
O sequenciamento do DNA do gene responsável pela produção da GBA de todos os indivíduos com “provável DG" (GBA em leucócitos <4,1 nmol/h/mg de
4.2 EXAMES LABORATORIAIS
4.2.1 Avaliação das Atividades Enzimáticas em Sangue Seco em Papel de Filtro
Para preservar a identidade dos voluntários, as amostras de SSPF foram codificadas, utilizando as letras iniciais do nome, data de nascimento e sexo do
participante, como exemplo: RGC03091960M.
As análises das atividades da GBA e da quitotriosidase em SSPF foram
realizadas no Laboratório dos Erros Inatos do Metabolismo do Hospital das Clínicas de Porto Alegre - RS (HCPA), utilizando a técnica desenvolvida por Civallero, 2006 (MICHELIN et al., 2006).
Amostras de 2ml de sangue venoso periférico foram coletadas do antebraço de cada voluntário, sem necessidade de jejum. A punção venosa para coletar amostras de sangue foi preferida à punção de “ponta-de-dedo” por proporcionar maior
quantidade de sangue e permitir a preparação de amostras mais homogêneas e de melhor qualidade para análise. O sangue coletado foi colocado em tubos de ensaio
com heparina e homogeneizado. Após a completa homogeneização, uma pequena quantidade do sangue foi aspirada e gotas foram pingadas dentro de quatro círculos
delimitados no papel de filtro (903 Protein Saver Card, Whatman Inc., USA). Logo após, as amostras foram secadas ao ar livre por quatro horas e acondicionadas,
Pequenos discos de 3 mm de diâmetro, contendo aproximadamente 3,6µL de
sangue, foram destacados das amostras de SSPF e encubados a 37ºC com substrato (4-methylumbeliferyl-h-d-glucoside and
4-Methylumbeliferyl-b-D-N-N0-N00-triacetylchitotrioside) e diluentes apropriados. As amostras foram centrifugadas e submetidas à análise fluorométrica (nanomoles do substrato hidrolizado/h/mL de sangue) (CIVALLERO; MICHELIN, et al., 2006).
Os laudos identificados apenas pelo código foram decodificados pelo investigador e entregues individualmente aos participantes, que tiveram a
oportunidade de esclarecer suas dúvidas ao final de cada etapa do estudo.
Figura 10 - Etapas de preparação das amostras de SSPF para análise enzimática. Fonte: Fotos obtidas pelo autor, ano 2007.
4.2.2 Avaliação das atividades da β-glucocerebrosidase em leucócitos e da quitotriosidase no plasma
Para confirmação da suspeita de DG, foram realizadas medidas das atividades
da GBA em SSPF foram menores do que 2,19 nmol/h/mL. Amostras de 10 ml de
sangue venoso do antebraço, sem necessidade de jejum, foram coletadas em tubos de ensaio com heparina para serem centrifugados em centrífuga refrigerada. Dessa
forma, os leucócitos foram separados do plasma, congelados a - 300C por 24 horas e enviados em gelo seco, por via aérea, para análise enzimática da atividade da GBA em leucócitos e quitotriosidase no plasma no laboratório do HCPA. O material
chegou ao seu destino no prazo máximo de 48 horas e em excelente estado de conservação. As atividades da GBA em leucócitos e da quitotriosidase no plasma foram avaliadas conforme técnicas descritas por Peters, Coyle et al. (PETERS;
COYLE, et al., 1976) e de Hollak, Van Weely et al., respectivamente (HOLLAK; WEELY, et al., 1994).
4.2.3 Análise Molecular das Mutações
De início, as amostras de DNA dos indivíduos com maiores suspeitas da DG (GBA em leucócitos<5,6 nmol/h/mg de proteínas) foram extraídas de material
colhido por pequena escovação da mucosa bucal (face interna da bochecha). O rastreamento das mutações (N370S, L444P, G377S e 55del) foi realizado no
Departamento de Genética e Biologia Evolutiva da Universidade de São Paulo (São Paulo, Brasil), utilizando o método descrito por Richards et al. (1993). O DNA foi extraído e amplificado conforme técnica de Polymerase Chain Reaction (PCR) ou
Reação em Cadeia da Polimerase com primers específicos para cada mutação. Em seguida, foi feita a digestão do produto de PCR com enzima de restrição (para cada
os fragmentos foram submetidos à eletroforese em gel de policrilamida a 12%
(N370S e G377S) ou eletroforese em gel de agarose a 2% (L444P e 33del) (ROZENBERG; ARAUJO, et al., 2006).
Para esclarecer dúvidas diagnósticas após o rastreamento das quatro mutações mencionadas (excluir a possibilidade da presença de outras mutações), amostras de sangue dos indivíduos com provável DG (atividade GBA≤4,0 nmol/h/mg
de proteína) foram encaminhadas para sequenciamento do DNA da região do gene codificador da GBA (1q21) no Laboratório de Desenvolvimento Molecular do
Departamento de Pediatria da Universidade de Washington, Estados Unidos da América. Para isso, foi usado sangue seco em papel de filtro FTA Classic Card (fabricado pela Whatman - Whatman), por permitir uma análise eficaz das mutações
e pela facilidade de envio de amostras para centros especializados a longas distâncias, conforme descrito por Devost e Choy (DEVOST; CHOY, 2000). O
sequenciamento do DNA realizado é capaz de identificar até cerca de 95% das mutações, entretanto, muitas vezes, não pode identificar deleções ou rearranjos que interferem com as ligações dos sítios nos primers.
4.2.4 Análise dos Dados
Os dados coletados foram submetidos à análise descritiva (distribuição de
quitotriosidase (em SSPF e no plasma). O nível de significância estatística foi
estabelecido em 5% (p <0,05).
Para análise estatística, os indivíduos foram inicialmente divididos em dois
grupos, com base em atividade da GBA em SSPF:
a) Indivíduos em risco para DG (GBA<2,19nmol/ml/h); b) Normais (GBA≥2,19 nmol/ml/h).
A seguir, os indivíduos foram classificados em cinco categorias (“provável doença de Gaucher”, “limítrofe entre provável DG/provável heterozigoto”, “provável heterozigoto”, “limítrofe entre provável heterozigoto/normal” e “normal”) de acordo
com os níveis de atividade da GBA adotados como parâmetros pelo laboratório de referência. Esses paramétricos laboratoriais foram obtidos pela análise enzimática
de pessoas previamente diagnosticadas, por estudo molecular, como homozigotos, heterozigotos e normais para DG. As sobreposição de valores das atividades da
5 RESULTADOS
O rastreamento para DG em Tabuleiro do Norte-CE envolveu 740 voluntários,
assintomáticos e sem parentesco em primeiro grau com os sete pacientes do Município, durante o período de um ano.
Os dados relacionados às atividades da GBA e da quitotriosidase em SSPF
foram separados por categorias (sexo, faixa etária, níveis de atividade de GBA e de quitotriosidase) e estão resumidos no APÊNDICE B. Houve maior participação de
mulheres no estudo (496 mulheres ou 67%), embora as atividades educativas tenham sido direcionadas indistintamente para homens e mulheres. Atribuiu-se a maior participação das mulheres por elas serem, provavelmente, as mais
interessadas nas questões relacionadas à saúde. A idade dos participantes variou de um ano a 85 anos, com média de 31,4±19,2 anos. O Gráfico 1 mostra a diferença
entre a idade das mulheres (média= 33,3±18,4 anos) e a dos homens (média= 27,4±20,2 anos) foi significativa (p<0,01; IC99%).
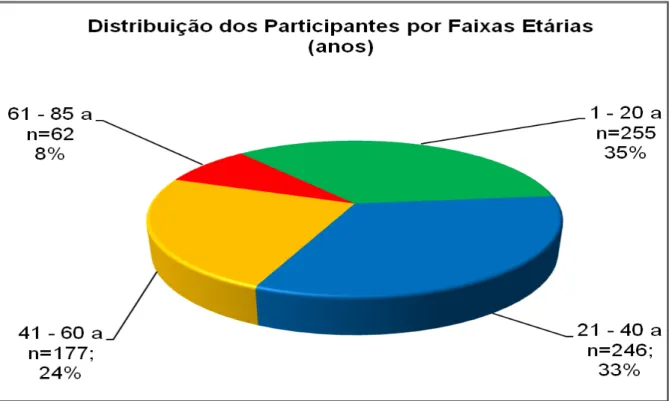
Para fins de análise de dados, os investigados foram agrupados em quatro
faixas etárias: I) 1 a 20 anos; II) 21 a 40 anos; III) 41 a 60 anos; e IV) 61 a 85 anos. Observou-se diferença significativa entre o número de participantes das quatro
faixas etárias (p<0.001; IC99%), com maior participação dos indivíduos mais jovens
(faixas etárias: I, 35%; II, 33%). A participação de pessoas de mais de 61 anos,
Gráfico 1 - Correlação entre sexo e idade dos participantes. Fonte: Dados coletados pelo autor, ano 2008.