Research Article
Development, Characterization, and
In Vitro
Biological
Performance of Fluconazole-Loaded Microemulsions for
the Topical Treatment of Cutaneous Leishmaniasis
Marcela Brito Oliveira,
1Giovana Calixto,
1Márcia Graminha,
2Hugo Cerecetto,
3Mercedes González,
3and Marlus Chorilli
11Department of Drugs and Medicines, School of Pharmaceutical Sciences, UNESP, Rodovia Araraquara-Ja´u, km. 1,
Campus, 14801-902 Araraquara, SP, Brazil
2Department of Clinical Analysis, School of Pharmaceutical Sciences, UNESP, Rodovia Araraquara-Ja´u, km. 1,
Campus, 14801-902 Araraquara, SP, Brazil
3Departamento de Qu´ımica Org´anica, Facultad de Qu´ımica-Facultad de Ciencias, Universidad de la Rep´ublica,
11400 Montevideo, Uruguay
Correspondence should be addressed to Giovana Calixto; [email protected] and Marlus Chorilli; [email protected]
Received 7 September 2014; Revised 18 December 2014; Accepted 20 December 2014
Academic Editor: Sami M. Nazzal
Copyright © 2015 Marcela Brito Oliveira et al. his is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cutaneous leishmaniasis (CL) is a resistant form of leishmaniasis that is caused by a parasite belonging to the genusLeishmania. FLU-loaded microemulsions (MEs) were developed by phase diagram for topical administration of luconazole (FLU) as prominent alternative to combat CL. hree MEs called F1, F2, and F3 (F1—60% 50 M phosphate bufer at pH 7.4 (PB) as aqueous phase, 10% cholesterol (CHO) as oil phase, and 30% soy phosphatidylcholine/oil polyoxyl-60 hydrogenated castor oil/sodium oleate (3/8/6) (S) as surfactant; F2—50% PB, 10% CHO, and 40% S; F3—40% PB, 10% CHO, and 50 % S) were characterized by droplet size analysis, zeta potential analysis, X-ray difraction, continuous low, texture proile analysis, andin vitrobioadhesion. MEs presented pseudoplastic low and thixotropy was dependent on surfactant concentration. Droplet size was not afected by FLU. FLU-loaded MEs improved the FLU safety proile that was evaluated using red cell haemolysis andin vitrocytotoxicity assays with J-774 mouse macrophages. FLU-unloaded MEs did not exhibit leishmanicidal activity that was performed using MTT colourimetric assays; however, FLU-loaded MEs exhibited activity. herefore, these MEs have potential to modulate FLU action, being a promising platform for drug delivery systems to treat CL.
1. Introduction
Leishmaniasis is an anthropozoonosis with natural foci that afects 12 million people worldwide, with approximately 1-2 million new cases occurring every year, and persists in tropical and subtropical regions [1].
It is caused by a parasite belonging to the genus Leishma-nia, in which infection is spread in wild ecotopes by the bite of the infected female sandly (Phlebotomine) in their vertebrate hosts [2–4].
hese parasites can originate two types of diseases: vis-ceral or cutaneous leishmaniases. Although the visvis-ceral form
is more aggressive, the cutaneous leishmaniases (CL) have received considerable attention in recent research because most patients are resistant to current treatments [5].
he irst-line chemotherapy to combat CL for over 60 years is the pentavalent antimonials, which are Sb-(V) com-pounds. Amphotericin B, pentamidine isethionate, milte-fosine, and paromomycin are also available as alternatives therapies, but their use is also limited because of toxicity or high treatment costs [6].
Hence, the searching for alternatives to conventional treatment for CL has been shown to be extremely valuable. One of these alternatives is administration of imidazole
antifungals such as luconazole (FLU), also known as [2-(2,4-diluorophenyl)-1,3-bis (1H-1,2,4-triazol-1-yl) propan-2-ol], that presents moderate lipophilicity (log 0.5), renal excretion of over 80% of the unchanged drug, a half-life of 30 hours, 12% binding plasma protein, and over 90% bioavailability when orally administered [7–11].
FLU can favor the topical treatment of CL, because this drug presents high ainity for keratin prolonging its retention in the skin [12]; moreover FLU acts by inhibiting the P450 cytochrome that blocks the enzyme 14-�-demethylase, which is involved in the synthesis of the parasitic ergosterol mem-brane, thus helping to heal localised lesions [13–17].
he clinical eicacy of FLU against CL was assessed by a randomized, double-blind, placebo controlled trial. For this, a dose of 200 mg daily was administrated for six weeks and the results demonstrated that FLU can be safe and efective to treat the CL [18].
Another study evaluated the eicacy of a cream contain-ing 1% FLU. he results showed that none of the animals was cured by cream [19]. his limitation can be ascribed to the low skin penetration of FLU, because even though studies show that FLU has high ainity for keratin, further studies have reported that high concentration of FLU in the skin is due to ininite dose of FLU, which is not reproduced in the clinical treatment [12]. herefore, it becomes necessary to develop novel topical FLU delivery systems to make viable the clinical treatment.
An interesting and unexplored alternative to overcome these obstacles is to design nanostructured drug delivery systems incorporated with FLU. A lot of attention has been given to micro- and nanostructured surfactants systems, as microemulsions (ME), which are clear, isotropic, and thermodynamically stable colloidal systems obtained sponta-neously by surfactant—cosurfactant combinations [20–23].
ME systems present features, such as (i) controlling drug release; (ii) protecting drug against thermal degradation; (iii) protecting drug against photo bleaching; (iv) reducing the drug dose; (v) minimizing the potential of side efects [24– 28], which make them stand out as a potential topic drug delivery system for CL treatment seeing that the conventional dosage forms such as gels, creams, and ointments against CL have not shown an efective action against all of the Leishma-niaspecies [16]; moreover such formulations presented toxic and irritating, as well as ardency and burning, sensations restricting its use [29].
he other property which makes ME a drug delivery system far superior to conventional systems is its ability to modify the difusional barrier of the skin, improving the partitioning of FLU into the skin [30]; thereby this may increase the FLU leishmanicidal activity.
herefore, the aim of this study was to evaluate the poten-tial of a FLU-loaded ME for the treatment of CL. he for-mulations were structurally characterized using droplet size analysis, zeta potential analysis, X-ray difraction, continuous low, texture proile analysis, andin vitrobioadhesion assays. he safety proile was evaluated using red cell haemolysis and in vitro cytotoxicity assays and the in vitro leishmanicidal activity was evaluated using a MTT colorimetric assay.
2. Materials and Methods
2.1. Materials. Fluconazole was purchased from DEG (Sao Paulo, Sao Paulo, Brazil). Sodium oleate and cholesterol were purchased from Sigma-Aldrich (St Louis, Missouri, USA). Soy phosphatidylcholine was purchased from Lucas Meyer Gmbh & Co (Hamburg, Hamburg, Germany). Phosphate bufer solution was prepared from sodium monohydrogen phosphate and dihydrogen phosphate that were purchased from Merck (Darmstadt, Germany). Polyoxyl-60 hydro-genated castor oil was purchased from Pharma Special (Itape-vi, Sao Paulo, Brazil). Pentamidine isethionate was purchased from Sigma-Aldrich (St Louis, MO, USA).
2.2. Preparation of Formulations. It was constructed as a pseudoternary phase diagram using a transparent bottle that was illed with adequate amounts of cholesterol (oil phase), 50 M of phosphate bufer at pH 7.4 (aqueous phase), and a mixture of surfactants, soy phosphatidylcholine, polyoxyl-60 hydrogenated castor oil, and sodium oleate, in a proportion of 3 : 8 : 6. he mixture was sonicated using Sonic Boom (Sonics, Vibra-Cell, Newtown, Connecticut, USA), which was rated at 220 watts and operated in a discontinuous manner for 20 minutes at intervals of 30 seconds to 1 minute: an ice bath was used throughout the sonication process [31–33].
he formulations were classiied visually as follows: trans-parent viscous systems (TVS), viscous and opaque systems (VOS), opaque liquid systems (OLS), liquid and transparent systems (LTS), and phase separated systems (PS). 1% FLU (w/w) was incorporated by dissolving the drug powder directly in the studied formulations.
2.3. Physicochemical and Structural Characterisation of Systems
2.3.1. Droplet Size Analysis (Dynamic Light Scattering). he droplet size analysis was determined using dynamic light scattering with a ZetaPlus (Brookhaven Instrument Corpo-ration, Holtsville, NY, USA). he samples were diluted and placed in the analysis chamber such that the laser beam passed through the entire length of the dispersions. he system temperature was maintained at 20∘C, the laser wave-length was 532 nm, and the refractive index was consistent with the rate observed for each sample. Ten measurements were made of the diameter and polydispersity index (PDI) of the droplets for each sample in triplicate, and the total duration of the experiment was 5 min [34].
2.3.3. X-Ray Difraction. An X-ray difractometer Siemens D5000 (Siemens, Munich, Bavaria, Germany) with copper radiation monochromatizated by graphite crystal was used to determine the degree of crystallinity or the amorphous state of the samples, the ME components (sodium oleate, soybean phosphatidylcholine, castor oil, polyoxyl-60-hydrogenated, and cholesterol), and FLU. he scanning speed was 0.1 seconds at each 0.050 to 2�∘ranging from 4∘to 70∘[35].
2.3.4. Texture Properties Analysis. Texture proile analyses of the samples were performed using a TA-XTplus texture analyzer (Stable Micro Systems, Surrey, UK) in TPA mode. Ten-gram samples were placed in 50 mL Falcon tubes and centrifuged at 4000 rpm for 10 minutes (Eppendorf 5810R, New York, USA). he samples were placed 10 mm above the analytical probe, which reached a constant speed of 1 mm s−1 by detecting the triggering strength for 2 min, ater which the probe was descended 10 millimetres into the sample. he probe was then returned to the surface at a speed of 0.5 mm s−1, and a second compression was initiated ater 5 seconds. he resulting force-time curve was used to cal-culate various mechanical parameters, such as the hardness, the compressibility, the adhesion, and the cohesiveness. he assays were performed in triplicate at 25∘C [36,37].
2.3.5. Bioadhesion Assay. he bioadhesive force between the pig ears’ skin and the samples was assessed by detachment test using a TA-XTplus texture analyzer (Stable Micro Systems, Surrey, UK). he porcine ear skin was obtained from a local slaughterhouse and cleaned. he injured skins were discarded. he skin was removed from the cartilage with a scalpel and a 400�m thick layer of the stratum corneum and epidermis was separated from the adipose tissue with a dermatome (Nouvag TCM 300, Goldach, USA). hese prepared skins were frozen for the maximum time of four weeks. On the day of the experiment, the skin was thawed in physiological saline solution, containing 0.9% (w/v) NaCl (Merck), at25±0.5∘C for 30 min; then, its hairs were cut with scissors and it was attached to the lower end of a cylindrical probe (diameter 10 mm) with a rubber ring.
he samples were packed into shallow cylindrical vessels and the test started lowering the analytical probe, which contained the skin at a constant speed (1 mm⋅s−1) onto the surface of the sample. he skin and the sample were kept in contact during 60 s and no force was applied during this interval. Ater 60 s, the skin was drawn upwards (0.5 mm⋅s−1) until the contact between the surfaces was broken. he bioad-hesive force of the samples was measured in the maximum detachment force as the resistance to the withdrawal of the probe, what relects the bioadhesion characteristic. Seven replicates were analysed at32 ± 0.5∘C [36,37].
2.3.6. Rheological Study. Continuous low was analysed on a controlled-stress AR2000 (TA Instruments, New Castle, DE, USA) equipped with parallel plate geometry (40 mm diam-eter) and sample gap of 200�m, at32 ± 0.1∘C, in triplicate. Samples of the hydrogel were carefully applied to the lower
plate, ensuring that sample shearing was minimized, and allowed to equilibrate for 3 min prior to analysis.
Continual testing was performed using a controlled shear rate procedure in the range from 0.01 to 100 s−1and back, each stage lasting 120 s, with an interval of 10 s between the curves. he consistency index and low index were determined from the power law described in(1)for a quantitative analysis of low behavior [34]:
� = � ⋅ ��, (1)
where “�” is shear stress; “�” is shear rate; “�” is consistency index; and “�” is low index [36,37].
2.4. In Vitro Biological Assays
2.4.1. Erythrocyte Haemolysis. Before use, freshly collected human blood (O positive) was washed three times with 0.01 M Tris-HCl at pH 7.4, which contained 0.15 M NaCl (Tris-saline). A suspension of 1% (v/v) erythrocytes con-sisting of packed red blood cells resuspended in Tris-saline was prepared. A FLU-loaded ME, a FLU-unloaded ME, and a FLU-free sample were dissolved in Tris-saline to a inal concentration of 27�M. A 1% (v/v) Triton X-100 solution was used as a positive control (which corresponded to 100% lysis). Ater incubation for 1 h at 37∘C, the samples were centrifuged at 3000×g for 2 min. Aliquots of 100�L of the supernatant were transferred to 96-well microplates, and the absorbance was determined at 405 nm using a BioRad Model 3550-UV (USA) microplate reader. he assay was performed in triplicate. he percentage of haemolysis was calculated using the following equation: % haemolysis = (test sample absorbance/absorbance at 100% lysis)×100 [38,39].
Table 1: Rates of the components of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D). Oil phase is cholesterol; aqueous phase is 50 M of phosphate bufer at pH 7.4; surfactant is a mixture of soy phosphatidylcholine, polyoxyl-60 hydrogenated castor oil, and sodium oleate in a proportion of 3 : 8 : 6. LTS: liquid transparent system. VTS: viscous transparent system.
Formulations (% w/w)
F1 F2 F3 F1D F2D F3D
Aqueous phase 60 50 40 60 50 40
Oil phase 10 10 10 10 10 10
Surfactant 30 40 50 30 40 50
Fluconazole — — — 1 1 1
O : S ratio 0,33 0,25 0,20 0,33 0,25 0,20
Phases’ classiication LTS VTS VTS LTS VTS VTS
he cell viability was calculated using the following equation: cell viability (%) = [OD560(sample)/OD5604]×100 [38].
2.4.3. Evaluation of Leishmanicidal Activity. Promastigotes ofL. amazonensisMPRO/BR/1972/M1841-LV-79 strain were maintained at 28∘C in liver-infusion tryptose supplemented with 10% foetal calf serum (FCS). Cultured promastig-otes at the end of the exponential growth phase were seeded at 8 × 106parasites/mL in 96-well lat-bottomed plates (Costar). he F1, F2, F3, F1D, F2D, and F3D for-mulations and FLU were added to the parasite suspen-sion to obtain inal concentrations between 1.6�g/mL and 100�g/mL, which were then incubated at 28∘C for 72 h. he assays were carried out in triplicate. A stock solution of pentamidine isethionate at a concentration of 16.7 mg⋅mL−1 was prepared in sterile, deionised water to serve as a ref-erence drug. he leishmanicidal efects of the samples were assessed using a modiied 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrasodium bromide (MTT) method [35]. he absorbances of the samples were measured at 490 nm [40]. he concentration corresponding to 30% of parasite growth inhibition was expressed as the inhibitory concentration (IC30).
2.5. Statistical Analyses. he data were analysed to obtain the mean and standard deviation and compared using the analy-sis of variance (ANOVA). In the data comparison, a one-way Tukey test was used to assess signiicant diferences between the samples, where values with� < 0.05were considered to be statistically signiicant. he program Origin 7.0 SRO was used in the data treatment.
3. Results and Discussion
3.1. Preparation of Formulations. Figure 1 shows the phase diagram for the system with the characteristic LTS (liquid transparent systems), TVS (transparent viscous systems), PS (phase separation), and VOS (viscous opaque systems) regimes. LTS regime was observed at low oil concentrations (10%) and low surfactant concentrations (10–30%); TVS regime was observed for surfactant concentrations between 20 and 65% and oil concentrations of 10 to 40%; VOS regime was found in the lower central region of the diagram at low oil concentrations of 20–50% and surfactant concentrations
0
10
20
30
40
50
60
70
80
90
100
0
10 20 30 40 50 60 70 80 90 100
0 10 20 30 40 50 60 70 80 90 100
Surfactant
Water Oil
VOS LTS
TVS
PS
F3
F2
F1
Figure 1: Ternary phase diagram. he marked areas represent the following. PS: phase separation; VOS: viscous opaque system; TVS: transparent viscous system; and LTS: liquid transparent system. he points designated F1, F2, and F3 are the studied MEs.
of 10–20%; and the PS regime was found at low water con-centrations (10–30%) and was independent of the surfactant concentration in the formulations.
here were 3 selected formulations of the diagram system, and the rates of the components are showed inTable 1. he oil phase was ixed because the cholesterol interacts with the biological membrane that can lead to increased membrane permeability, aggregation process, enzymatic activity, cell fusion, and the stifness, size, and shape of this membrane. Furthermore, the cholesterol molecule exhibits both polar and nonpolar characteristics and can guide the hydroxyl ion into the aqueous phase and the carbon chain towards the surfactant [41–44].
3.2. Physicochemical and Structural Characterisation of Systems
3.2.1. Droplet Size Analysis (Dynamic Light Scattering).
Table 2: Droplet size, polydispersion index, and zeta potential of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D). Each value represents the mean (±standard deviation) of three replicates.
Formulations Droplet size (nm) PDI Zeta potential (mV)
F1 180.500±0.721 0.174±0.013 −40.000±1.758
F1D 181.200±1.609 0.228±0.012 −38.600±1.967
F2 618.933±28.612 0.615±0.123 −43.000±0.608
F2D 928.833±34.186 0.703±0.079 −45.167±2.316
F3 317.800±9.364 0.686±0.132 −54.567±2.608
F3D 357.633±18.240 0.568±0.083 −55.867±3.044
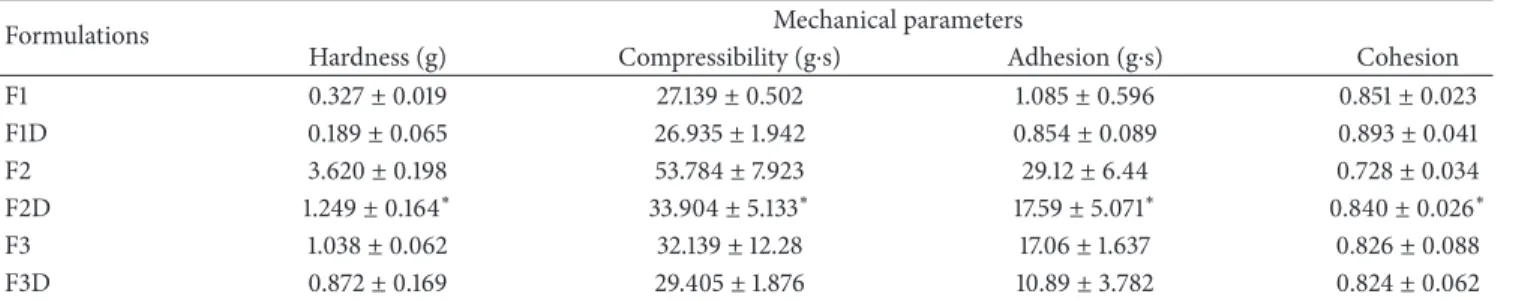
Table 3: Compressibility, hardness, adhesion, and cohesion of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D). Each value represents the mean (±standard deviation) of three replicates at 32∘C.
Formulations Mechanical parameters
Hardness (g) Compressibility (g⋅s) Adhesion (g⋅s) Cohesion
F1 0.327±0.019 27.139±0.502 1.085±0.596 0.851±0.023
F1D 0.189±0.065 26.935±1.942 0.854±0.089 0.893±0.041
F2 3.620±0.198 53.784±7.923 29.12±6.44 0.728±0.034
F2D 1.249±0.164∗ 33.904±5.133∗ 17.59±5.071∗ 0.840±0.026∗
F3 1.038±0.062 32.139±12.28 17.06±1.637 0.826±0.088
F3D 0.872±0.169 29.405±1.876 10.89±3.782 0.824±0.062
∗Signiicant statistical diference compared to the respective control (P<0.05).
the formulations: F1—180.500 to 181.200 nm, F2—618.933 to 928.833 nm, and F3—317.800 to 357.633 nm. he addition of FLU only changed the particle size for the F2 sample, which exhibited a signiicant increase in the droplet diameter following drug addition. his result may be explained by the coalescence of droplets over time because most of the newly formed droplets in the formulation can be found at the surface of the vial to which the drug was added, whereas the droplets with the highest age in the formulation were at the bottom of the storage vessel of the sample. he PDI is an index that relects the relative homogeneity of particle sizes in a sample. he PDI values varied from 0.174 to 0.703 demonstrating a good size distribution of the oil droplets in the ME system, relecting the size homogeneity of the droplets in the bulk ME [45,46].
3.2.2. Zeta Potential Analysis. Table 2 also shows that the zeta potential values for the samples ranged from−38.600 ± 1.967mV to−55.867±3.044mV. he negative charge resulted from the microemulsion surfaces being coated upon contact-ing the liquid because a large repulsion guarantees micro-emulsion stability. his repulsion arises from the negatively charged components of the formulations and ionic forces [47].
Soy phosphatidylcholine and sodium oleate have an ester group from which the sodium atom can dissociate, and the cholesterol structure has a free hydroxyl group. Drug addition did not change the zeta potential values of any of the tested samples.
3.2.3. X-Ray Difraction. X-ray difraction was used to deter-mine whether the materials were crystalline or amorphous in nature. Intense and sharp peaks with narrow halos in the
difraction pattern of a sample are characteristic of crystalline materials; the absence of intense peaks and broad halos are characteristic of amorphous materials [48,49].
Figure 2shows the difractogram obtained for ME com-ponents. he intense and deined difraction peaks for sodium oleate (Figure 2(a)), cholesterol (Figure 2(b)), and luconazole (Figure 2(d)) were crystalline below 10∘ (2�), between 5 and 20∘(2�), and between 5∘and 35∘(2�), respec-tively, that are characteristic of crystalline structures. he difractograms obtained for polyoxyl-60 hydrogenated castor oil (Figure 2(c)) and soy phosphatidylcholine (Figure 2(e)) exhibited low intensity peaks with broad halos; thus, these components are considered amorphous structures [50].
he absence of high peaks inFigure 3shows that the FLU-loaded ME and FLU-unFLU-loaded ME exhibited low intensity peaks with amorphous halos present that is common and desirable characteristic for microemulsion systems [46].
3.2.4. Texture Properties Analysis. he diference in the mechanical characteristics of the FLU-unloaded and -loaded MEs was shown inTable 3.
Hardness, compressibility, and adhesion decreased only when the drug was added to the F2 sample while F1 and F3 were not inluenced by FLU.
he hardness and compressibility are related to the strength of the ME structure under compression and express the applicability of the formulation to the desired site. A low value of these parameters, as obtained for the FLU-loaded MEs, has been reported as an advantage for dermal appli-cation of formulations because this parameter signiicantly afects spreadability of the ME [36,51].
Int
en
sit
y
300
250
200
150
100
50
0
0 10 20 30 40 50 60 70 80
2�∘
(a)
Int
en
sit
y
180
160
140
120
100
80
60
40
20
0
0 10 20 30 40 50 60 70 80
2�∘
(b)
Int
en
sit
y
200
180
160
140
120
100
80
60
40
20
0
0 10 20 30 40 50 60 70 80
2�∘
(c)
Int
en
sit
y
350
300
250
200
150
100
50
0
0 10 20 30 40 50 60 70 80
2�∘
(d)
Int
en
sit
y
0 10 20 30 40 50 60 70 80
2�∘
700
600
500
400
300
200
100
0
(e)
2�∘
In
ten
si
ty (a.u
.)
F1
F1D
0 10 20 30 40 50 60 70 80
(a)
2�∘
In
ten
si
ty (a.u
.)
F2
F2D
0 10 20 30 40 50 60 70 80
(b)
2�∘
In
ten
si
ty (a.u
.)
F3
F3D
0 10 20 30 40 50 60 70 80
(c)
Figure 3: X-ray difraction spectra of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D): (a) F1 and F1D, (b) F2 and F2D, and (c) F3 and F3D.
and relects alterations in product viscosity [51]. In our study, the lowest adhesiveness value was obtained with the F1D classiied as LTS, as demonstrated above.
Finally, cohesiveness parameter shows the degree of dif-iculty in breaking down the MEs internal structure and it is calculated through the ratio of the area under the irst and second immersion. he high cohesiveness values of the ME indicate the ability to prepare homogeneous formulations [37, 51].
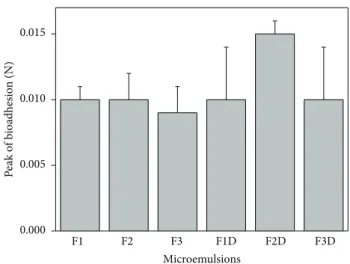
3.2.5. Bioadhesion Assays. Figure 4 shows the peak force corresponding to the maximum force between the probe and the tissue as a function of time. he data shows that the bioadhesion of all of the samples was unchanged by drug addition neither by the composition of the MEs.
he bioadhesive stability of FLU-loaded ME is an impor-tant physicochemical parameter for topical application that allows the interaction of the formulations with supericial
epithelial cells resulting in a closest contact with the biological surface by an extended time and also an increased local gradient of drug concentration in the target site [36,52].
Bachhav and Patravale developed microemulsion based gel for the vaginal delivery of luconazole that showed signiicantly higher antifungal activity as compared to that of commercial product. hus, bioadhesive MEs can improve the performance of the drug [53].
3.2.6. Rheological Study. he low curves of FLU-unloaded MEs and FLU-loaded MEs are presented inFigure 5, in which the relation between the shear rate (Pa) and shear stress (1/s) evidenced that all ME’s exhibited non-Newtonian “shear thinning” pseudoplastic low behaviour.
F1 F2 F3 F1D F2D F3D 0.000
0.005 0.010 0.015
P
ea
k
o
f b
ioadhesio
n (N)
Microemulsions
Figure 4: Peak of bioadhesion (N) of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D). Each value represents the mean (±standard deviation) of at least seven repli-cates. Data were collected at32 ± 0.5∘C. No statistically signiicant diference was detected.
Table 4: Consistency index (�) and low index (�) of FLU-unloaded MEs (F1, F2, and F3) and FLU-loaded MEs (F1D, F2D, and F3D). Each value represents the mean (± standard deviation) of three replicates.
Formulations �(Pa⋅s) �
F1 1.14±0.01 0.718±0.003
F2 1.15±0.04 0.840±0.009
F3 8.44±0.24 0.308±0.007 F1D 1.39±0.02 0.704±0.004 F2D 2.73±0.23 0.673±0.020 F3D 6.27±0.18 0.313±0.007
less than a unity (� < 1). Furthermore, the presence of FLU did not alter the low behaviour of the MEs.
his pseudoplastic behavior may be due to breaking of the organized structures that leads to forming less organized structures such as droplets when the stress is applied to the MEs. It is a desirable attribute for skin pharmaceutical products, because the formulation begins to low easily ater the stress application, leading to a good spreading during application and a formation of a uniform ilm on the skin surface. Ater withdrawal of the stress, the viscosity of formulation increases instantaneously avoiding its outlow [36]. Moreover, �-values decreased with the increase of surfactant concentration, showing a trend of increasing the shear thinning characteristic of the studied systems.
As can be also seen inTable 4, the consistency index (�) increased with the increase of surfactant concentration. hus, the highest surfactant concentration increased the apparent viscosity of the ME that may due to the diferent formation and more organized microstructure [36].
As a further study, the thixotropy of MEs was investigated. As can be seen inFigure 5, a notable thixotropic response was only observed for FLU-unloaded F3 and FLU-loaded
F3, because the descending curve does not overlap with the ascending one.
his phenomenon demonstrated again the crucial role of the surfactant concentration into the ME for its low behaviour. In this case, the increase of surfactant content led to the formation of new structures when the shear rate was increased. herefore, a longer period of time to restore the relaxed molecular coniguration was necessary, what has provided conditions for the appearance of thixotropy.
For the other studied microemulsions, there was a fast recovery of their microstructure showing to be a time-independent structure with the upward and the downward curves overlapped.
3.3. In Vitro Biological Assays. he safety proile was eval-uated using red cell haemolysis and in vitro cytotoxicity assays. Erythrocyte membranes have been studied extensively because erythrocyte cells can be obtained by venous punc-ture, and the membranes are easily isolated by centrifugation. hus, erythrocytes are a good model for drug-membrane interactions and can provide information about changes in lipid composition, enzymes, or other membrane proteins [54].
FLU solutions resulted in the lysis of 4.72 ± 0.98% of the erythrocyte membranes. FLU-unloaded MEs (F1, F2, and F3) resulted in the lysis of1.27 ± 0.52%,1.42 ± 0.43%, and 1.54 ± 0.47% of the erythrocyte membranes, respectively. FLU-loaded MEs (F1D, F2D, and F3D) resulted in the lysis of 3.23±1.24%,3.45±0.46%, and3.54±1.15%, respectively, of the erythrocyte membranes, showing that the ME could decrease the lysis power of FLU. hus, all of the systems showed tolerable erythrocyte haemolysis. Triton X-100, which is a known haemolytic agent, was used as a positive control in the study and showed 100% haemolysis of erythrocytes, thus validating the experiment.
he overall results of the haemolysis study indicated that treatment with the developed lipidic systems was less toxic than previously developed treatments and could be potentially used for therapeutic applications [39].
In vitrocytotoxicity studies were performed using J-774 mouse macrophages as cellular models. he data are shown as a percentage of the cellular viability inFigure 6.
he cell viability results showed that the FLU-free samples did not kill normal macrophages; that is, the cellular viability was greater than 92%. Moreover, the unloaded and FLU-loaded MEs also did not show toxic activity.
herefore, these results showed that these MEs are safety and biocompatibility formulations to eukaryotic cells. Abbas-alipourkabir et al. showed cytotoxicity efects from the adher-ence of particles to cell membranes, particle internalisation, and degradation of products in the cell culture medium or inside the cells. However, the susceptibility of diferent cell types can be diferent for diferent particulate carriers. Sys-tems containing natural lipids should be well-tolerated by living organisms [55,56].
0 20 40 60 80 100 0
10 20 30 40 50 60
St
re
ss ra
te
(1/s)
Shear rate (1/s)
F1 F2
F3
(a)
0 20 40 60 80 100
0 10 20 30 40 50 60
Sh
ea
r str
ess (P
a)
Shear rate (1/s)
F1D F2D
F3D
(b)
Figure 5: (a) Flow rheograms of FLU-unloaded MEs F1 (◼), F2 (�), and F3 (X). (b) Flow rheograms of FLU-loaded MEs F1D (◼), F2D (�),
and F3D (X). Closed symbol represents up curve and open symbol represents down curve. Standard deviations have been omitted for clarity;
however, in all cases, the coeicient of variation of triplicate analyses was less than 10%. Data were collected at32 ± 0.5∘C.
120
100
80
60
40
20
0
FLU F1 F1D F2 F2D F3 F3D
C
ell
ula
r via
b
ili
ty (%)
Formulations
Figure 6: % cellular viability for FLU-free samples, FLU-unloaded MEs (F1, F2, and F3), and FLU-loaded MEs (F1D, F2D, and F3D). No statistically signiicant diference was detected.
amastigote form (which is the form that infects humans by developing inside immune cells, i.e., macrophages) and the promastigote form (the infective vector that develops inside the digestive tract of insects) [57,58].
Even though the amastigote form causes cutaneous lesions to develop in humans, it is not used in screening stud-ies for new drugs or studstud-ies on incorporating the compound into release systems to improve its efectiveness. Instead, the promastigotes ofLeishmaniaare used because they are simple to work with, care for, and grow in a short time, which is an advantage in the large-scale screening of potential new drugs [58–61].
Table 5: IC30values against parasites for luconazole samples and
F1, F2, and F3 formulations with and without luconazole.
Samples IC30(�M)
FLU —
F1 —
F1D 55.62±5.79
F2 —
F2D 42.19±3.12
F3 —
F3D 30.00±2.83∗
∗Signiicant statistical diference compared to the F1D and F2D
formula-tions.
he promastigote form is also used in the triage of new drugs to conirm the action of the active principle and as a surrogate for the amastigote (intracellular macrophage) form because both forms exhibit similar characteristics in metabolic pathways [60].
Antileishmanial assays were performed using FLU, FLU-unloaded ME (F1, F2, and F3), and FLU-loaded ME (F1D, F2D, and F3D) against the promastigote forms ofL. amazo-nensisand the IC30values are shown inTable 5.
here was a reduction in the parasite promastigote forms for FLU-loaded ME, which may suggest that the drug action was potentiated when incorporated into the microemulsions (ME).
with the parasite membrane can increase by reducing the surface tension [62].
Furthermore, the drug penetration into skin is easier when the formulation is thixotropic, as F3D formulation, since the thixotropic characteristic plays a crucial role in the therapeutic eicacy of the pharmaceutical formulations by improving retention time at the administered site besides enhancing the drug systemic bioavailability [63,64].
herefore, the components concentration-deined micro-emulsion and thixotropic formulations can potentiate the antileishmanial treatment.
4. Conclusion
his study demonstrated that it is possible to obtain amor-phous microemulsion prepared with cholesterol as oil phase, 50 M phosphate bufer at pH 7.4 as aqueous phase, a mixture of soy phosphatidylcholine, polyoxyl-60 hydrogenated castor oil, and sodium oleate in a proportion of 3 : 8 : 6 as surfactant. he set of results showed that all microemulsions presented a pseudoplastic low and thixotropy dependent on surfactant concentration. Droplet size was not afected by luconazole drug. FLU-loaded MEs showed antileishmanial activity in thein vitrobiological assays, being that ME with the highest concentration of surfactant in its composition and greater thixotropy demonstrated the major antileishmanial activity. herefore, these MEs should be further investigated since they exhibited promising properties for using as platform for drug delivery systems to treat skin diseases such as cutaneous leishmaniasis.
Conflict of Interests
he authors declared that there is no conlict of interests in this work.
Acknowledgments
his work was inancially supported by the Fundac¸˜ao de Amparo `a Pesquisa do Estado de S˜ao Paulo (FAPESP), Con-selho Nacional de Desenvolvimento Cient´ıico e Tecnol´ogico (CNPq), Programa de Apoio ao Desenvolvimento Cient´ıico (PADC), and the Comisi´on Sectorial de Investigaci´on Cien-t´ıica (CSIC-UdelaR, no. 610, Uruguay).
References
[1] J. Alvar, I. D. V´elez, C. Bern et al., “Leishmaniasis worldwide and global estimates of its incidence,”PLoS ONE, vol. 7, no. 5, Article ID e35671, 2012.
[2] S. R. Reis, L. H. M. Gomes, N. M. Ferreira et al., “Ocorrˆencia de lebotom´ıneos (Diptera: Psychodidae: Phlebotominae) no ambiente peridomiciliar em ´area de foco de transmiss˜ao de leishmaniose tegumentar no munic´ıpio de Manaus, Amazonas,”
Acta Amazonica, vol. 43, no. 1, pp. 121–123, 2013.
[3] P. Desjeux, “Leishmaniasis: current situation and new per-spectives,”Comparative Immunology, Microbiology & Infectious
Diseases, vol. 27, no. 5, pp. 305–318, 2004.
[4] J. Alvar, I. D. V´elez, C. Bern et al., “Leishmaniasis worldwide and global estimates of its incidence,”PLoS ONE, vol. 7, no. 5, Article ID e35671, 2012.
[5] J. J. Shaw and R. Lainson, “Leishmaniasis in Brazil: X. Some observations on intradermal reactions to diferent trypanoso-matid antigens of patients sufering from cutaneous and muco-cutaneous leishmaniasis,”Transactions of the Royal Society of
Tropical Medicine and Hygiene, vol. 69, no. 3, pp. 323–335, 1975.
[6] B. Chawla and R. Madhubala, “Drug targets inLeishmania,”
Journal of Parasitic Diseases, vol. 34, no. 1, pp. 1–13, 2010.
[7] J. Faergemann and H. Laufen, “Levels of luconazole in se-rum, stratum corneum, epidermis-dermis (without stratum corneum) and eccrine sweat,”Clinical and Experimental
Der-matology, vol. 18, no. 2, pp. 102–106, 1993.
[8] S. R. C. J. Santos, E. V. Campos, C. Sanches, D. S. Gomez, and M. C. Ferreira, “Fluconazole plasma concentration measure-ment by liquid chromatography for drug monitoring of burn patients,”Clinics, vol. 65, no. 2, pp. 237–243, 2010.
[9] K. W. Brammer, P. R. Farrow, and J. K. Faulkner, “Pharmacoki-netics and tissue penetration of luconazole in humans,”Reviews
of Infectious Diseases, vol. 12, supplement 3, pp. S318–S326, 1990.
[10] K. Turner, P. Manzoni, D. K. Benjamin, M. Cohen-Wolkowiez, P. B. Smith, and M. M. Laughon, “Fluconazole pharmacokinetics and safety in premature infants,”Current Medicinal Chemistry, vol. 19, no. 27, pp. 4617–4620, 2012.
[11] M. Rahman, M. A. Wahab, and M. S. I. Khan, “Eicacy of pulse therapy of oral luconazole in the treatment of seborrheic der-matitis,”Journal of Dhaka National Medical College & Hospital, vol. 17, no. 2, pp. 25–29, 2012.
[12] A. C. Ayub, A. D. M. Gomes, M. V. C. Lima, C. D. Vianna-Soares, and L. A. M. Ferreira, “Topical delivery of luconazole: in vitro skin penetration and permeation using emulsions as dosage forms,”Drug Development and Industrial Pharmacy, vol. 33, no. 3, pp. 273–280, 2007.
[13] C. Salerno, A. M. Carlucci, and C. Bregni, “Study ofin vitrodrug release and percutaneous absorption of luconazole from topical dosage forms,”AAPS PharmSciTech, vol. 11, no. 2, pp. 986–993, 2010.
[14] E. B. D. Lima, C. Porto, J. O. C. D. Motta, and R. N. R. Sampaio, “Tratamento da leishmaniose tegumentar Americana,”Anais
Brasileiros De Dermatologia, vol. 82, no. 2, pp. 111–124, 2007.
[15] F. E. D. Silva, C. Ziech, G. D. Pavoni, and R. Pasquali, “Desen-volvimento de comprimidos contendo luconazol por com-press˜ao direta,”Latin American Journal of Pharmacy, vol. 28, no. 4, pp. 604–608, 2009.
[16] P. Minodier and P. Parola, “Cutaneous leishmaniasis treatment,”
Travel Medicine and Infectious Disease, vol. 5, no. 3, pp. 150–158,
2007.
[17] N. Shakya, P. Bajpai, and S. Gupta, “herapeutic switching in
Leishmaniachemotherapy: a distinct approach towards
unsat-isied treatment needs,”Journal of Parasitic Diseases, vol. 35, no. 2, pp. 104–112, 2011.
[18] A. A. Alrajhi, E. A. Ibrahim, E. B. De Vol, M. Khairat, R. M. Faris, and J. H. Maguire, “Fluconazole for the treatment of cuta-neous leishmaniasis caused by Leishmania major,”he New
England Journal of Medicine, vol. 346, no. 12, pp. 891–895, 2002.
[19] S. V. Mussi, A. P. Fernandes, and L. A. M. Ferreira, “Comparative study of the eicacy of formulations containing luconazole or paromomycin for topical treatment of infections byLeishmania
(Leishmania) major andLeishmania(Leishmania)
[20] F. K. dos Santos, M. H. Oyafuso, C. P. Kiill, M. P. Dalon-Gremi˜ao, and M. Chorilli, “Nanotechnology-based drug deliv-ery systems for treatment of hyperproliferative skin diseases—a review,”Current Nanoscience, vol. 9, no. 1, pp. 159–167, 2013. [21] T. P. Formariz, M. C. C. Urban, A. Silva Junior et al.,
“Microe-muls˜oes e fases l´ıquidas cristalinas como sistemas de liberac¸˜ao de f´armacos,”Revista Brasileira de Ciˆencias Farmacˆeuticas, vol. 41, no. 3, pp. 301–313, 2005.
[22] R. L. Shinde, A. B. Jindal, and P. V. Devarajan, “Microemulsions and nanoemulsions for targeted drug delivery to the brain,”
Current Nanoscience, vol. 7, no. 1, pp. 119–133, 2011.
[23] A. C. Sintov and L. Shapiro, “New microemulsion vehicle facilitates percutaneous penetration in vitro and cutaneous drug bioavailability in vivo,”Journal of Controlled Release, vol. 95, no. 2, pp. 173–183, 2004.
[24] T. P. Formariz, L. A. Chiavacci, M. V. Scarpa et al., “Structure and viscoelastic behavior of pharmaceutical biocompatible anionic microemulsions containing the antitumoral drug com-pound doxorubicin,”Colloids and Surfaces B: Biointerfaces, vol. 77, no. 1, pp. 47–53, 2010.
[25] G. M. El Maghraby, “Microemulsions as transdermal drug delivery systems,”Current Nanoscience, vol. 8, no. 4, pp. 504– 511, 2012.
[26] A. Oliveira and M. Scarpa, “Microemuls˜oes I: fundamentos te´oricos da formac¸˜ao do sistema microemulsionado,”Infarma, vol. 13, pp. 73–79, 2001.
[27] A. S. Cunha J´unior, S. L. Fialho, L. B. Carneiro, and F. Or´eice, “Microemuls˜oes como ve´ıculo de drogas para administrac¸˜ao ocular t´opica,”Arquivos Brasileiros de Otalmologia, vol. 66, no. 3, pp. 385–391, 2003.
[28] V. A. F. F. M. dos Santos, K. M. Leite, M. da Costa Siqueira et al., “Antiprotozoal activity of quinonemethide triterpenes
fromMaytenus ilicifolia(Celastraceae),”Molecules, vol. 18, no.
1, pp. 1053–1062, 2013.
[29] J. Soto, P. Fuya, R. Herrera, and J. Berman, “Topical paromo-mycin/methylbenzethonium chloride plus parenteral meglu-mine antimonate as treatment for American cutaneous leishma-niasis: Controlled study,”Clinical Infectious Diseases, vol. 26, no. 1, pp. 56–58, 1998.
[30] P. Mura, M. Bragagni, N. Mennini, M. Cirri, and F. Maestrelli, “Development of liposomal and microemulsion formulations for transdermal delivery of clonazepam: efect of randomly methylated�-cyclodextrin,”International Journal of
Pharma-ceutics, vol. 475, no. 1-2, pp. 306–314, 2014.
[31] K. Karim, A. Mandal, N. Biswas et al., “Niosome: a future of tar-geted drug delivery systems,”Journal of Advanced
Pharmaceu-tical Technology and Research, vol. 1, no. 4, pp. 374–380, 2010.
[32] T. P. Formariz, L. A. Chiavacci, V. H. V. Sarmento, C. V. Santilli, E. S. Tabosa do Egito, and A. G. Oliveira, “Relationship between structural features and in vitro release of doxorubicin from biocompatible anionic microemulsion,”Colloids and Surfaces B:
Biointerfaces, vol. 60, no. 1, pp. 28–35, 2007.
[33] P. S. Prestes, M. Chorilli, L. A. Chiavacci, M. V. Scarpa, and G. R. Leonardi, “Physicochemical characterization and rheological behavior evaluation of the liquid crystalline mesophases devel-oped with diferent silicones,”Journal of Dispersion Science and
Technology, vol. 31, no. 1, pp. 117–123, 2009.
[34] J. A. D. Silva, D. P. D. Santana, D. G. C. Bedor et al., “Estudo de liberac¸˜ao e permeac¸˜ao in vitro do diclofenaco de dietilamˆonio em microemuls˜ao gel-like,”Qu´ımica Nova, vol. 32, no. 6, pp. 1389–1393, 2009.
[35] R. Pouliot, L. Germain, F. A. Auger, N. Tremblay, and J. Juhasz, “Physical characterization of the stratum corneum of an in vitro human skin equivalent produced by tissue engineering and its comparison with normal human skin by ATR-FTIR spec-troscopy and thermal analysis (DSC),”Biochimica et Biophysica
Acta: Molecular and Cell Biology of Lipids, vol. 1439, no. 3, pp.
341–352, 1999.
[36] G. Calixto, A. C. Yoshii, H. Rocha e Silva et al., “Polyacrylic acid polymers hydrogels intended to topical drug delivery: prepa-ration and characterization,”Pharmaceutical Development and
Technology, pp. 1–7, 2014.
[37] F. C. Carvalho, G. Calixto, I. N. Hatakeyama, G. M. Luz, M. P. D. Gremi˜ao, and M. Chorilli, “Rheological, mechanical, and bioadhesive behavior of hydrogels to optimize skin delivery systems,”Drug Development and Industrial Pharmacy, vol. 39, no. 11, pp. 1750–1757, 2013.
[38] M. Jumaa, P. Kleinebudde, and B. W. M¨uller, “Physicochemical properties and hemolytic efect of diferent lipid emulsion formulations using a mixture of emulsiiers,” Pharmaceutica
Acta Helvetiae, vol. 73, no. 6, pp. 293–301, 1999.
[39] Z.-R. Huang, “Development and evaluation of lipid nanopar-ticles for camptothecin delivery: a comparison of solid lipid nanoparticles, nanostructured lipid carriers, and lipid emul-sion,”Acta Pharmacologica Sinica, vol. 29, no. 9, pp. 1094–1102, 2008.
[40] S. Muelas-Serrano, J. J. Nogal, R. A. Mart´ınez-D´ıaz, J. A. Escario, A. R. Mart´ınez-Fern´andez, and A. G´omez-Barrio, “In vitro screening of American plant extracts onTrypanosoma cruziand
Trichomonas vaginalis,”Journal of Ethnopharmacology, vol. 71,
no. 1-2, pp. 101–107, 2000.
[41] G. P. Kumar and P. Rajeshwarrao, “Nonionic surfactant vesic-ular systems for efective drug delivery—an overview,” Acta
Pharmaceutica Sinica B, vol. 1, no. 4, pp. 208–219, 2011.
[42] R. Rajera, K. Nagpal, S. K. Singh, and D. N. Mishra, “Niosomes: a controlled and novel drug delivery system,”Biological and
Pharmaceutical Bulletin, vol. 34, no. 7, pp. 945–953, 2011.
[43] N. B. Mahale, P. D. hakkar, R. G. Mali, D. R. Walunj, and S. R. Chaudhari, “Niosomes: novel sustained release nonionic stable vesicular systems—an overview,”Advances in Colloid and
Interface Science, vol. 183-184, pp. 46–54, 2012.
[44] A. Sankhyan and P. Pawar, “Recent trends in niosome as vesic-ular drug delivery system,”Journal of Applied Pharmaceutical
Science, vol. 2, no. 6, pp. 20–32, 2012.
[45] U. Goyal, R. Arora, and G. Aggarwal, “Formulation design and evaluation of a self-microemulsifying drug delivery system of lovastatin,”Acta Pharmaceutica, vol. 62, no. 3, pp. 357–370, 2012. [46] G. B. R. F. da Silva, M. V. Scarpa, G. Rossanezi, E. S. T. do Egito, and A. G. de Oliveira, “Development and characterization of biocompatible isotropic and anisotropic oil-in-water colloidal dispersions as a new delivery system for methyl dihydrojas-monate antitumor drug,”International Journal of Nanomedi-cine, vol. 9, no. 1, pp. 867–876, 2014.
[47] M. Mosca, A. Ceglie, and L. Ambrosone, “Efect of membrane composition on lipid oxidation in liposomes,”Chemistry and
Physics of Lipids, vol. 164, no. 2, pp. 158–165, 2011.
[48] W. L. Lindner, “Characterization of the crystalline, intermediate and amorphous phase in poly(ethylene terephthalate) ibres by X-ray difraction,”Polymer, vol. 14, no. 1, pp. 9–15, 1973. [49] S. V. Canevarolo Jr.,T´ecnicas de caracterizac¸˜ao de pol´ımeros,
Artliber, S˜ao Paulo, Brazil, 2004.
spectroscopy and X-ray powder difraction: a comparative study based on the pre-validation stage results,” Journal of
Pharmaceutical and Biomedical Analysis, vol. 55, no. 5, pp. 1208–
1212, 2011.
[51] A. S. Can, M. S. Erdal, S. G¨ung¨or, and Y. ¨Ozsoy, “Optimization and characterization of chitosan ilms for transdermal delivery of ondansetron,”Molecules, vol. 18, no. 5, pp. 5455–5471, 2013. [52] D. S. Jones, A. D. Woolfson, A. F. Brown, and M. J. O’Neill,
“Mucoadhesive, syringeable drug delivery systems for con-trolled application of metronidazole to the periodontal pocket: in vitro release kinetics, syringeability, mechanical and mucoad-hesive properties,”Journal of Controlled Release, vol. 49, no. 1, pp. 71–79, 1997.
[53] Y. G. Bachhav and V. B. Patravale, “Microemulsion based vag-inal gel of luconazole: formulation, in vitro and in vivo eval-uation,”International Journal of Pharmaceutics, vol. 365, no. 1-2, pp. 175–179, 2009.
[54] S. V. P. Malheiros, N. C. Meirelles, and E. de Paula, “Pathways involved in triluoperazine-, dibucaine- and praziquantel-induced hemolysis,”Biophysical Chemistry, vol. 83, no. 2, pp. 89– 100, 2000.
[55] R. Abbasalipourkabir, A. Salehzadeh, and R. Abdullah, “Cyto-toxicity efect of solid lipid nanoparticles on human breast cancer cell lines,”Biotechnology, vol. 10, no. 6, pp. 528–533, 2011. [56] A. C. Silva, D. Santos, D. C. Ferreira, and E. B. Souto, “Minox-idil-loaded nanostructured lipid carriers (NLC): characteriza-tion and rheological behaviour of topical formulacharacteriza-tions,”
Phar-mazie, vol. 64, no. 3, pp. 177–182, 2009.
[57] M. M. Bastos, N. Boechat, A. T. Gomes et al., “O Uso de Poriri-nas em Terapia Fotodinˆamica no Tratamento da Leishmaniose Cutˆanea,” Revista Virtual de Qu´ımica, vol. 4, no. 3, pp. 257–267, 2012.
[58] A. Weing¨artner, G. Kemmer, F. D. M¨uller et al., “Leishmania promastigotes lack phosphatidylserine but bind annexin V upon permeabilization or miltefosine treatment,”PLoS ONE, vol. 7, no. 8, Article ID e42070, 2012.
[59] A. Ponte-Sucre, J. H. Faber, T. Guider et al., “Activities of naph-thylisoquinoline alkaloids and synthetic analogs against
Lish-mania major,”Antimicrobial Agents and Chemotherapy, vol. 51,
no. 1, pp. 188–194, 2007.
[60] J. L. Siqueira-Neto, O.-R. Song, H. Oh et al., “Antileishmanial high-throughput drug screening reveals drug candidates with new scafolds,”PLoS Neglected Tropical Diseases, vol. 4, no. 5, article e675, 2010.
[61] G. Bringmann, K. homale, S. Bischof et al., “A novel Leishma-nia major amastigote assay in 96-well format for rapid drug screening and its use for discovery and evaluation of a new class of leishmanicidal quinolinium salts,”Antimicrobial Agents and
Chemotherapy, vol. 57, no. 7, pp. 3003–3011, 2013.
[62] S. Magdum Chandrakant, S. Naikwade Nilofar, and R. Shah Rohit, “Preparation and evaluation of luconazole topical microemulsion,”Journal of Pharmacy Research, vol. 2, no. 3, pp. 557–562, 2009.
[63] T. P. Formariz, V. H. V. Sarmento, A. A. Silva-Junior, M. V. Scarpa, C. V. Santilli, and A. G. Oliveira, “Doxorubicin biocom-patible O/W microemulsion stabilized by mixed surfactant containing soya phosphatidylcholine,”Colloids and Surfaces B:
Biointerfaces, vol. 51, no. 1, pp. 54–61, 2006.
Submit your manuscripts at
http://www.hindawi.com
Pain
Research and Treatment
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
World Journal
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation http://www.hindawi.com
Volume 2014
Toxins
Journal ofVaccines
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Antibiotics
Toxicology
Journal of
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Stroke
Research and Treatment
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Drug Delivery
Journal of Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014 Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014 Advances in Pharmacological Sciences
Tropical Medicine
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Medicinal ChemistryInternational Journal of Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Addiction
Journal ofHindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
BioMed
Research International Emergency Medicine International
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Diseases
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Anesthesiology Research and Practice
Scientifica
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Journal of
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014
Pharmaceutics
Hindawi Publishing Corporation
http://www.hindawi.com Volume 2014