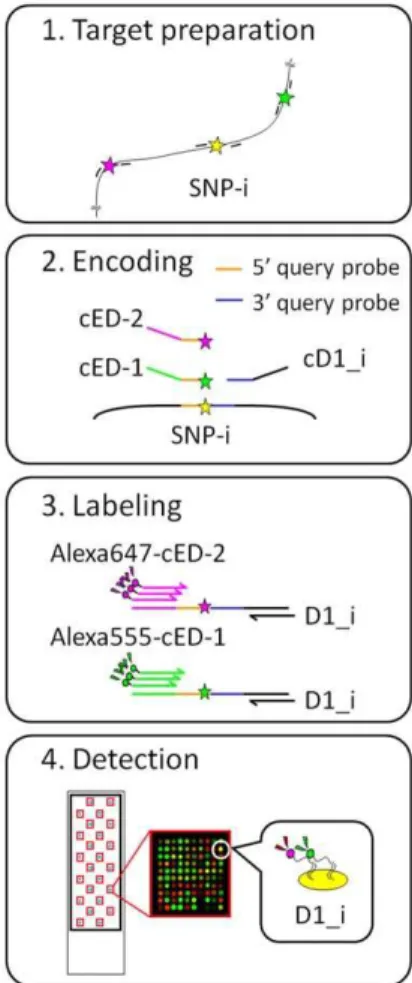
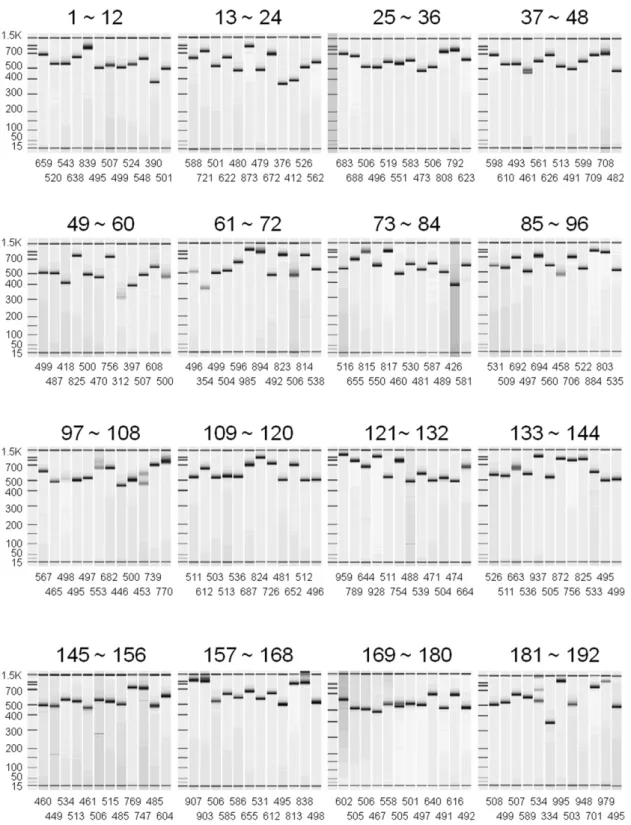
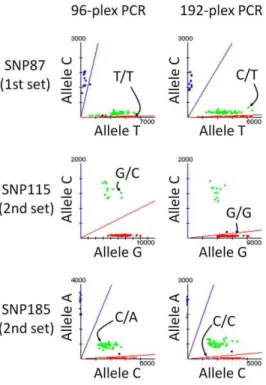
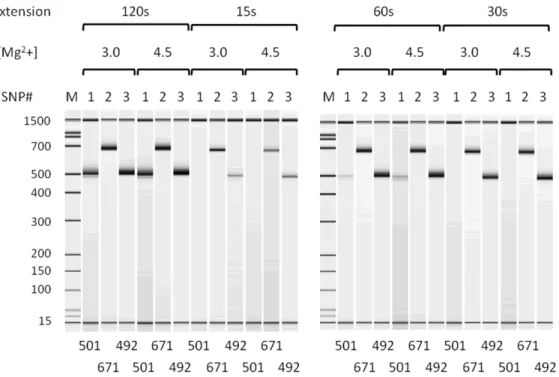
Highly parallel and short-acting amplification with locus-specific primers to detect single nucleotide polymorphisms by the DigiTag2 assay.
Texto
Imagem
Documentos relacionados
Este primeiro contato com o público-alvo permitiu que se conhecesse o estado nutricional, o consumo alimentar e os níveis plasmáticos de zinco destas crianças, fornecendo
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
Building on the present day knowledge of structural differences between the host and the parasite LDH, we have isolated and cloned Theileria parva lactate dehydrogenase ( Tp
Este relatório relata as vivências experimentadas durante o estágio curricular, realizado na Farmácia S.Miguel, bem como todas as atividades/formações realizadas
The objective of the research was to identify single nucleotide polymorphisms (SNPs) of the lysozyme gene in a Chinese well-known native chicken breed (Langshan chicken) and
Esta concavidade não é evidente na melodia canónica, tanto na sua estrutura intervalar como na articulação das notas limite, tal como podemos observar pela linha
uma formação objetiva de cultura que encerra, ao mesmo tempo, elementos históricos, sociais e racionais, aí intervindo, portanto, não apenas fatores reais (natureza humana,
Como afirma Monteiro (1999) assiste-se, assim, à progressiva medicalização da infância e, consequentemente, da maternidade, a que não será também alheio o aumento do
