MESTRADO PROFISSIONAL EM ENGENHARIA DE MATERIAIS
ANA MARIA THIAGO DE SOUZA
AVALIAÇÃO DE TOXICIDADE DA PSEUDOBOEMITA PARA LIBERAÇÃO CONTROLADA DE FÁRMACOS
AVALIAÇÃO DE TOXICIDADE DA PSEUDOBOEMITA PARA LIBERAÇÃO CONTROLADA DE FÁRMACOS
Dissertação apresentada ao Programa de Mestrado Profissional em Engenharia de Materiais da Universidade Presbiteriana Mackenzie, como requisito parcial à obtenção do título de Mestre Profissional em Engenharia de Materiais.
ORIENTADOR: PROF. DR. ANTONIO HORTENCIO MUNHOZ JÚNIOR CO-ORIENTADORA: PROFª DRª ROBERTA MONTERAZZO CYSNEIROS
S729a Souza, Ana Maria Thiago de
Avaliação de toxicidade da pseudoboemita para liberação controlada de fármacos. / Ana Maria Thiago de Souza – São Paulo, 2013.
88 f.: il.; 30 cm.
Dissertação (Mestrado Profissional em Engenharia de Materiais) - Universidade Presbiteriana Mackenzie - São Paulo, 2013.
Orientador: Prof Dr. Antonio Hortêncio Munhoz Júnior Bibliografia: p. 71-79
AVALIAÇÃO DE TOXICIDADE DA PSEUDOBOEMITA PARA LIBERAÇÃO CONTROLADA DE FÁRMACOS
Dissertação apresentada ao Programa de Mestrado Profissional em Engenharia de Materiais da Universidade Presbiteriana Mackenzie, como requisito parcial à obtenção do título de Mestre Profissional em Engenharia de Materiais.
Aprovada em 22/02/2013:
BANCA EXAMINADORA
____________________________________ Prof. Dr. Antonio Hortêncio Munhoz Júnior
Universidade Presbiteriana Mackenzie
____________________________________ Profª. Drª. Leila Figueiredo de Miranda
Universidade Presbiteriana Mackenzie
____________________________________ Prof. Dr. Leonardo Gondim de Andrade e Silva
À Universidade Presbiteriana Mackenzie e à Escola de Engenharia por me proporcionar um excelente curso de pós-graduação.
Ao Dr. Antônio Hortêncio Munhoz Júnior, minha gratidão por ter sido um orientador persistente, paciente, amigo e por me incentivar e ajudar a concluir esta jornada em que entrei nas escuras, fugindo um pouco de meu campo habitual de trabalho e pesquisas.
À Drª Roberta Cysneiros, minha co-orientadora, estou grata pelas sugestões, colaboração e por compartilhar seus conhecimentos.
À minha querida amiga, veterinária Drª Raquel de Assis Sirvente, agradeço por pela sinceridade, pela amizade e apoio neste momento difícil pelo qual eu e minha família estamos passando. Obrigada pelos nossos cafés diários e pelos suportes na fase experimental deste trabalho.
Ao Prof. Dr. Roberto Rodrigues Ribeiro, à Drª Miriam Ribeiro, Drª Paulete Romoff, Profª Raquel Cymrot e à Cinthia Indy Tamayose, obrigada pela colaboração.
Ao corpo técnico: farmacêuticos Eduardo Fuzaro, Karoline de Oliveira e Bruna Kogici; às biólogas Iara Cabral, Natália Barom (a Nati), Iara Araújo e Ana Paula Leite; aos funcionários Domingos Sávio Mariano da Silva (o Dô), Oliveiro Augusto (o Oliva), Liuba, Cabral, Luis Henrique e ao Evaldo Lourenço, obrigada pelo auxílio para a realização deste trabalho. Com certeza, este não seria possível sem a ajuda de vocês.
Às amigas do laboratório (Fabiane, Nailliw, Adêlisandra, Babi e Profª Paola Lupianhes), obrigada pela força.
As pesquisas em nanotecnologia avançaram consideravelmente nos últimos 30 anos, possibilitando o aparecimento de novos materiais com aplicações que atendem aos interesses de diversos setores. Na área da saúde, a indústria farmacêutica tem utilizado os materiais nanoestruturados para desenvolver novos medicamentos e veiculadores de fármacos. A aplicação das nanopartículas oferece inúmeras vantagens comparativamente ao sistema convencional de liberação, tais como: liberação controlada e progressiva dos fármacos a partir da degradação da matriz, nível plasmático efetivo com menor flutuação ao longo do tempo, menor risco de toxicidade, maior penetração pelas barreiras biológicas e direcionamento específico aos alvos teciduais. Neste trabalho, o material utilizado como liberador de fármaco foi a pseudoboemita. Esta é um oxi-hidróxido de alumínio e consiste em uma estrutura similar a boemita. Com sua produção via processo sol-gel, a partir de reagentes de alta pureza, foi possível obter pseudoboemita de pureza elevada, com área específica elevada e com total ausência de contaminantes, tornando-a, portanto, promissora para aplicações na liberação controlada de fármacos. Em vista da possível utilização da pseudoboemita como excipiente para liberação controlada de fármacos, este trabalho teve como objetivo determinar a sua toxicidade aguda (50mg/kg, 300mg/kg e 2000mg/kg) e. subaguda (1000mg/kg) em ratos machos (Wistar rats).A metodologia consistiu em testes de toxicidade a partir das avaliações
e análises dos parâmetros bioquímicos e histopatológicos resultantes de sua administração. Para finalizar, a pseudoboemita foi testada in vivo como possível liberador controlado do
fármaco aciclovir. Em ambos os testes, a administração não determinou mortalidade nos grupos e na avaliação histopatológica dos fígados dos animais também não foram observadas qualquer alterações na integridade tecidual. As leituras realizadas com cromatografia líquida de alta eficiência mostraram que ocorreu absorção do aciclovir para circulação sistêmica nos animais que receberam a pseudoboemita com aciclovir. O aciclovir administrado com a pseudoboemita foi absorvido pelo trato gastrointestinal. Os resultados demonstraram que a pseudoboemita tem baixa toxicidade em altas doses em curto prazo e se enquadra na categoria de atóxico; a ausência de alumínio nas amostras de plasma enfatiza a ausência de absorção do alumínio para a circulação sistêmica e o aciclovir presente no plasma dos ratos permitiu confirmar que este está presente na circulação sistêmica dos animais mesmo após as dessorções da pseudoboemita.
The nanotechnology research progressed significantly over the past 30 years, allowing for the development of new materials with applications that meet the interests of various sectors. In health, the pharmaceutical industry has used nanostructured materials to develop the release of drugs. Application of the nanoparticles offers numerous advantages compared to conventional release, such as progressive and controlled release of drugs from the matrix degradation, effective plasma levels with less fluctuation over time, less risk of toxicity, biological penetration through the barriers and targeting to specific tissue targets. In this study, the material used in drug delivery is pseudoboehmite. This is an oxyhydroxide of aluminum and consists of a structure similar to boehmite. With its production by sol-gel process, starting from high purity reagents, it is possible to obtain high-purity pseudoboehmite with high specific area and with total absence of contaminants, making it therefore promising for applications in controlled release of drugs. In view of the possible use of pseudoboehmite as carriers for controlled drug delivery, this study aimed to determine its acute (50mg/kg, 300mg/kg and 2000mg/kg) and. subacute toxicity (1000mg/kg) in male rats (Wistar rats). The methodology consisted of toxicity tests from the evaluations and analyzes of the biochemical and histopathological parameters resulting from its administration. Finally, the pseudoboehmite was tested in vivo as possible releasing controlled drug acyclovir. In both tests, the administration has not determined mortality in groups and histopathological evaluation of the livers of the animals were not observed any changes in tissue integrity. These readings showed there to absorption of the acyclovir systemic circulation in animals receiving the pseudoboehmite. The readings were taken with a high performance liquid chromatography showed that there was absorption of acyclovir systemic circulation in animals receiving the pseudoboehmite with acyclovir. Acyclovir administered with pseudoboehmite was absorbed from the gastrointestinal tract. The results showed that the pseudoboehmite has low toxicity at high doses of short and falls under the category of nontoxic; the absence of aluminum in the plasma samples emphasizes the absence of aluminum absorption into the systemic circulation and acyclovir present in the plasma of rats allowed confirm that this is present in the systemic circulation of animals even after the steps of the desorption of pseudoboehmite.
Fluxograma 1 O trajeto e a representação das etapas envolvidas na liberação e absorção de um fármaco contido na fórmula farmacêutica... 21
Desenho 1 Representação esquemática da estrutura epitelial gastrointestinal e suas possíveis rotas de transporte de fármacos... 23
Gráfico 1 Relação entre concentração de fármaco por intervalo de tempo... 25
Desenho 2 Representação esquemática de nanocápsulas e nanoesferas poliméricas... 28
Desenho 3 Representação esquemática de bicamada lipídica e lipossoma... 28
Desenho 4 Representação esquemática de um
dendrímero... 29
Desenho 5 Representação esquemática de uma
micela... 30
Quadro 1 Resumo das etapas de obtenção de materiais cerâmicos a partir do processo sol-gel...
34
Esquema 1 Esquema ilustrativo do processo de gelatinização (transição
sol-gel)... 35
Esquema 2 Representação da estrutura da
pseudoboemita... 40
Microfotografia 1
Microscopia eletrônica de varredura da amostra da pseudoboemita... 41
Gráfico 2 Perfil de liberação do aciclovir pela pseudoboemita em um período de 2 horas... 42
Quadro 2 Preparação da pseudoboemita... 49 Esquema 3 Estrutura química do fármaco aciclovir... 49 Fotografia 1 Equipamento semi-automatizado utilizado para leituras das amostras de
plasma... 53
Fotografia 2 Equipamento utilizado para leituras de sódio, potássio e alumínio das amostras de plasma... 53
Fotografia 3 Equipamento (CLAE) utilizado para detecção de aciclovir no plasma dos ratos...
oral de dose única de pseudoboemita... 58
Quadro 3 Análise histopatológica da toxicidade oral aguda da pseudoboemita nos grupo controle e experimental... 62
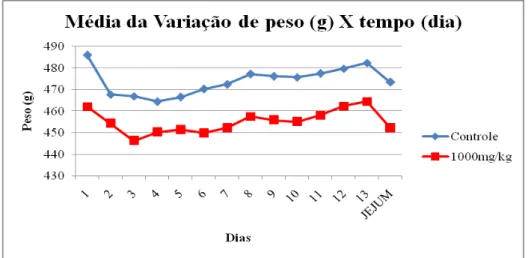
Gráfico 4 Ganho de peso corporal em ratos Wistar rats, macho, após o tratamento
oral com doses repetidas de
pseudoboemita... 64
Tabela 1 Características de um excipiente ideal... 19 Tabela 2 Fatores que exercem influências sobre a biodisponibilidade dos
fármacos... 20
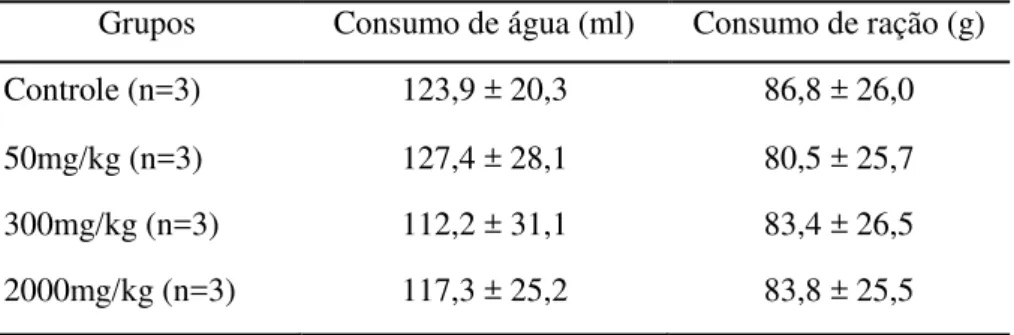
Tabela 3 Nanotecnologia, materiais e forma nanoestrutural... 27 Tabela 4 Distribuição dos 15 ratos, dose de ensaio e via de administração... 51 Tabela 5 Consumo de ração e água (14 dias) em ratos tratados com água destilada e
pseudoboemita, via oral, na toxicidade aguda (Média ± DP, n= número de animais)... 57
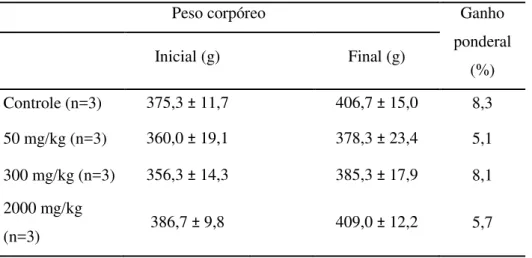
Tabela 6 Evolução de peso absoluto de ratos controle e tratados com pseudoboemita. Dados expressos em Média ± DP. N=número de animais... 58
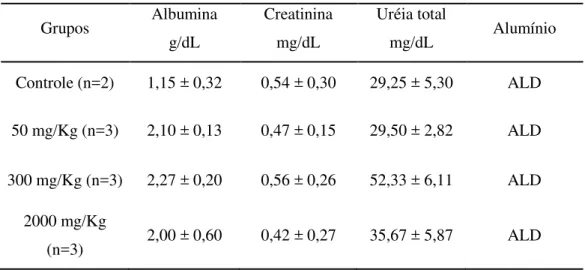
Tabela 7 Valores bioquímicos de ratos tratados com pseudoboemita,via oral, na toxicidade aguda (Média ± DP)... 60
Tabela 8 Valores bioquímicos (glicemia) de ratos tratados com pseudoboemita,via oral, na toxicidade aguda (Média ± DP)... 60 Tabela 9 Consumo de ração e água (14 dias) em ratos tratados com água destilada
pseudoboemita, via oral, na toxicidade aguda (Média ± DP, n= número de animais)... 64
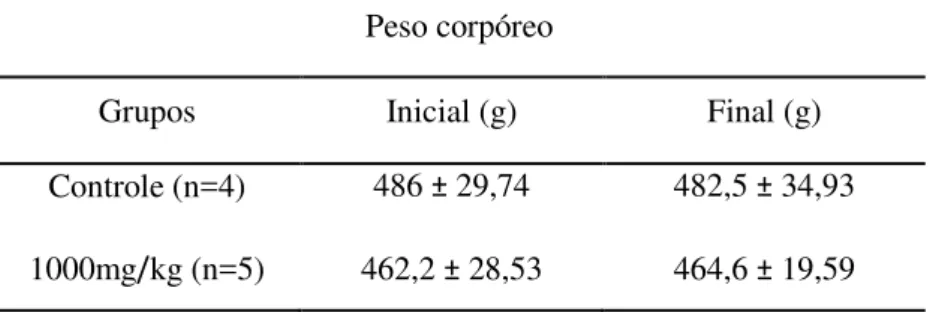
Tabela 10 Evolução de peso absoluto de ratos controle e tratados com pseudoboemita e água destilada. Dados expressos em Média ± DP, n=número de animais... 65
Tabela 11 Valores bioquímicos de ratos tratados com pseudoboemita via oral, na toxicidade subaguda (Média ± DP)... 66 Tabela 12 Valores bioquímicos (glicemia) de ratos tratados com pseudoboemita, via oral,
na toxicidade subaguda (Média ± DP)... 67 Tabela 13 Valores de tempo de retenção e áreas de picos dos cromatogramas de aciclovir
ABC Associação Brasileira de Cerâmica
ACV Aciclovir
AIDS Síndrome da Imunodeficiência Adquirida
ALCAP Alumino Calcium Phosphorous Oxide Ceramics
ALD Abaixo do Limite de Detecção
ANVISA Agência Nacional de Vigilância Sanitária
AZT Azidothymidine
BET Área superficial específica
CEUA Comitê de Ética no Uso de Animais
CLAE Cromatografia Líquida de Alta eficiência
DL50 Dose Letal 50% DRX Difração de raios-X
DSC Calorimetria Exploratória Diferencial
DTA Análise Térmica Diferencial
EDTA Ethylenediamine tetraacetic acid
FDA Food and Drugs Administration
HA Hidroxilapatite
HIV Human Immunodeficiency Virus
MEV Microscopia Eletrônica de Varredura
OECD Organization for Economic Cooperation and Development
TCPL Tricalcium phosphate-lysine
TCP Tricálcio de fosfato
TG Análise Termogravimétrica
TGI Trato gastrointestinal
UPM Universidade Presbiteriana Mackenzie
UV-vis Espectroscopia no ultravioleta-visível
1 INTRODUÇÃO... 15
1.1 OBJETIVO GERAL... 16
1.2 OBJETIVO ESPECÍFICO... 16
1.3 JUSTIFICATIVA... 17
1.4 METODOLOGIA... 17
2 REFERÊNCIAL TEÓRICO... 18
2.1 EXCIPIENTE... 18
2.2 VIAS DE ADMINISTRAÇÃO E FATORES FISIOLÓGICOS QUE INFLUENCIAM NA ABSORÇÃO DE FÁRMACOS... 19
2.3 LIBERAÇÃO CONTROLADA DE FÁRMACOS... 24
2.4 SISTEMAS DE LIBERAÇÃO NANOESTRUTURADOS... 26
2.5 MATERIAIS CERÂMICOS E CERÂMICA AVANÇADA... 31
2.2.1 Cerâmicos... 32
2.2.2 Cerâmica fina / avançada... 32
2.2.3 Processo sol-gel... 33
2.6 CERÂMICA COMO EXCIPIENTE FARMACÊUTICO E SISTEMA LIBERADOR DE DROGAS... 36
2.7 PSEUDOBOEMITA... 40
2.8 TOXICIDADE... 43
2.8.1 Toxicidade Oral Aguda – DL50... 44
2.8.2 Toxicidade Oral Subaguda... 45
2.8.3 Parâmetros bioquímicos... 46
2.8.4 Parâmetros histopatológicos... 46
3 MATERIAIS E MÉTODOS... 48
3.1 MATERIAIS... 48
3.1.1 Animais... 48
3.1.2 Pseudoboemita... 48
3.2.1 Estabelecimento das doses de ensaio e administração... 50
3.2.1.1 Toxicidade oral aguda... 50
3.2.1.2 Toxicidade oral subaguda... 51
3.2.2 Determinação da DL50 – toxicidade oral aguda e subaguda... 52
3.2.3 Obtenções das amostras (plasma) – toxicidade oral aguda e subaguda... 52
3.3.4 Avaliação histopatológica – toxicidade oral aguda e subaguda... 54
3.3.5 Variáveis avaliadas... 54
3.3.6 Determinação da presença do aciclovir no plasma sanguíneo... 54
3.3.7 Análise Estatística... 56
4 RESULTADOS E DISCUSSÃO... 57
4.1 Toxicidade oral aguda... 57
4.1.1 Avaliação da toxicidade oral aguda... 57
4.1.2 Avaliação bioquímica da toxicidade oral aguda... 59
4.1.3 Avaliação macroscópica e histopatológica – toxicidade oral aguda... 61
4.2 Toxicidade oral subaguda... 63
4.2.1 Avaliação da toxicidade oral subaguda... 63
4.2.2 Avaliação bioquímica da toxicidade oral subaguda... 65
4.2.3 Avaliação macroscópica e histopatológica – toxicidade oral subaguda... 67
4.3 Quantificação de aciclovir no plasma dos ratos... 68
5 CONCLUSÃO... 70
REFERÊNCIAS BIBLIOGRÁFICAS... 71
1 INTRODUÇÃO
As pesquisas em nanotecnologia avançaram consideravelmente nos últimos 30 anos, possibilitando o aparecimento de novos materiais com aplicações que atendem aos interesses de diversos setores. As aplicações dessa ciência são diversas e tem sido atuada em destaque na medicina moderna nos campos de terapias, diagnoses e imagens (PARVEEN; MISRA; SAHOO, 2012). Na área da saúde, a indústria farmacêutica tem utilizado os materiais nanoestruturados para desenvolver novos medicamentos e veiculadores de fármacos. A aplicação das nanopartículas oferece inúmeras vantagens comparativamente ao sistema convencional de liberação, tais como: liberação controlada e progressiva dos fármacos a partir da degradação da matriz, nível plasmático efetivo com menor flutuação ao longo do tempo, menor risco de toxicidade, maior penetração pelas barreiras biológicas e direcionamento específico aos alvos teciduais (COUVREUR; VAUTHIER, 2006; MUNHOZ
JR. et al., 2006; SAKATA, 2007, MUNHOZ JR. et al., 2010).
Esses novos medicamentos têm sido utilizados no tratamento de várias patologias como câncer, HIV (Human Immunodeficiency Virus), leishmaniose, malária, dentre
outras, de maneira mais eficaz e com menor toxicidade. (COUVREUR; VAUTHIER., 2006; ORIVE et al., 2005; MUNHOZ JR. et al., 2010).
Atualmente, uma gama de carregadores de fármacos tem sido utilizada para liberação controlada. Esses carregadores podem ser formados por diversos materiais, como os poliméricos, os lipídicos, os protéicos, dentre outros.
No presente estudo, o carregador utilizado como possível controlador de liberação de fármacos é a cerâmica fina do tipo pseudoboemita. Sua produção e obtenção foram realizadas no Laboratório de Engenharia de Materiais, na Universidade Presbiteriana Mackenzie (MUNHOZ JR. et al., 2006).
A pseudoboemita é um oxi-hidróxido de alumínio e consiste em uma estrutura similar a boemita (MUNHOZ JR. et al., 2006). A diferença entre essas duas estruturas
envolve características relacionadas ao tamanho de suas células unitárias: a célula unitária da pseudoboemita é maior do que a célula unitária da boemita. Essa diferença ocorre em razão da incorporação de maior quantidade de água na estrutura da pseudoboemita (KLOPROGGE et al., 2006).
orgânico-inorgânicos), no qual ocorrem as reações de hidrólise e condensação do precursor para a formação de partículas de tamanho coloidal (sol) e posterior formação da rede tridimensional (gel). A grande vantagem deste processo é a obtenção de materiais com características e propriedades pré-planejadas, sendo possível, portanto, a realização de controle de suas características físicas e químicas quanto a estequiometria, porosidade, estrutura cristalina e tamanho das partículas (MUNHOZ JR. et al., 2006). Por meio da análise
dessas, observou-se que a pseudoboemita é constituída por uma superfície altamente porosa e viável como sistema de liberação controlada dos fármacos.
Utilizando esse processo, a partir de reagentes de alta pureza, é possível obter pseudoboemita de pureza elevada, com área específica elevada e com total ausência de contaminantes, tornando-a, portanto, promissora para aplicações como na liberação controlada de fármacos (MUNHOZ JR. et al., 2006; MUNHOZ JR. et al., 2010).
Novicks (2009) e MUNHOZ JR. et al. (2010) demonstraram através de
análises in vitro, que a pseudoboemita formada pelo precursor nitrato de amônio e obtida pelo
processo sol-gel é altamente porosa e viável como sistema de liberação controlada dos fármacos Aciclovir e Atenolol. A interação dessas moléculas farmacêuticas com o substrato pseudoboemita foi analisada por espectroscopia de absorção no infra-vermelho, análise térmica diferencial (DTA), termogravimétria (TG) e espectroscopia no ultravioleta-visível (UV-vis). A pseudoboemita foi caracterizada por microscopia eletrônica de varredura (MEV), por difração de raios-X e a solução do fármaco após a adsorção foi estudada via cromatografia líquida de alta eficiência (CLAE).
1.1 OBJETIVO GERAL
Determinar a toxicidade da pseudoboemita como excipiente para liberação controlada de fármacos.
1.2 OBJETIVO ESPECÍFICO
1.3 JUSTIFICATIVA
Atualmente, uma gama de carregadores de fármacos tem ocupado posição de destaque no âmbito da micro e nanotecnologia. E as nanoestruturas constituídas por materiais cerâmicos representam um avanço significativo nas ciências farmacêuticas e na produção de medicamentos mais eficazes e menos tóxicos (ROY et al., 2003; PAUL; SHARMA, 2003;
MUNHOZ JR. et al., 2006, SILVEIRA, 2007; NOVICKS, 2009; MUNHOZ JR. et al., 2010).
Novicks (2009), Munhoz Jr. et al. (2010) e MARTINS et al (2012)
demonstraram através de análises in vitro, que a pseudoboemita obtida pelo processo sol-gel é
viável como sistema de liberação controlada dos fármacos Aciclovir, Atenolol e Glucantime. Portanto, para dar continuidade no estudo deste material nanoparticulado há necessidade de determinar sua toxicidade.
1.4METODOLOGIA
O desenvolvimento da parte teórica deste trabalho consistiu em amplo levantamento bibliográfico e estudos de pesquisas publicadas em sites acadêmicos na internet, artigos científicos, anais de congressos, teses, dissertações, manuais e livros.
Com relação aos procedimentos experimentais, a metodologia consistiu na realização de testes de toxicidade oral aguda e subaguda da pseudoboemita em roedores a partir das avaliações e análises dos parâmetros bioquímicos e histopatológicos resultantes de sua administração.
E para finalizar, a pseudoboemita foi testada in vivo como possível liberador
2 REFERENCIAL TEÓRICO
Segundo o Glossário de Denominações Comuns Brasileiras (ANVISA, 2012), os excipientes são substâncias que, em concentrações presentes em algumas formas farmacêuticas, apresentam inocuidade química, microbiológica e ausência de atividade farmacológica.
2.1EXCIPIENTES
Em diversos medicamentos, sejam esses sólidos, líquidos ou semi-sólidos, ocorre a presença de excipientes que têm a função de somente diluir, dissolver ou avolumar os princípios ativos em suas respectivas formas farmacêuticas. Esses estão inseridos na concepção tradicional de excipiente. Entretanto, ocorre também outra função mais importante, que é a melhoria do desempenho desses princípios ativos dos fármacos, em termos de assegurar sua estabilidade, biodisponibilidade (liberação e farmacocinética), aceitabilidade, facilidade de administração de um ou mais princípios ativos, direcionamento e absorção em sítios específicos (PIFFERI; SANTORO; PEDRANI, 1999; LIRA, 2004).
A melhoria do desempenho desses princípios ativos tornou os excipientes componentes essenciais dos fármacos e mundialmente aceitos. Para tal, há uma necessidade de qualificar cuidadosamente e detalhadamente as propriedades químicas e físicas dos excipientes. Além disso, para que um excipiente seja considerado ideal, este deve possuir as características descritas na Tabela 1 (PIFFERI; SANTORO; PEDRANI, 1999).
A pseudoboemita estudada no presente trabalho é um excipiente modificador da liberação de fármacos (NOVICKS, 2009; MUNHOZ JR. et al., 2010), que controla a
Tabela 1: Características de um excipiente ideal. Fármaco-toxicologicamente inativo
Quimicamente e fisicamente inerte à droga
Compatível com outros componentes da formulação Incolor e sem sabor
Alta fluidez
Alta compressibilidade
Disponibilidade mundial e econômico Garantia de qualidade
Fácil armazenamento
Desempenho consistente com fármaco específico
Fonte: Pifferi, Santoro; Pedrani (1999).
2.2 VIAS DE ADMINISTRAÇÃO E FATORES FISIOLÓGICOS QUE INFLUENCIAM A ABSORÇÃO DE FÁRMACOS
Segundo Aulton (2005), as vias de administração podem ser classificadas em via oral, retal, parenteral (subcutânea, intramuscular e intravenosa), tópica e respiratória. A via intravenosa oferece um acesso direto para a circulação sistêmica e o total da dose administrada fica disponível no plasma para ser distribuído para outros tecidos do corpo e para o(s) sítio(s) de ação do fármaco. Outras vias exigirão uma etapa de absorção antes que o fármaco chegue à circulação sistêmica.
A via oral é a mais popular, por ser uma via natural, e conveniente, porque a produção de suas formas farmacêuticas é relativamente fácil. Além disso, essas formas não precisam ser esterilizadas, são compactas e podem ser produzidas em grande quantidade (AULTON, 2008; FLORENCE; ATTWOOD, 2003; SANT et al., 2012).
físico-químico é apenas um fator parcial e não sua representação completa de fatores que afetam a biodisponibilidade dos fármacos no organismo (FLORENCE; ATTWOOD, 2003).
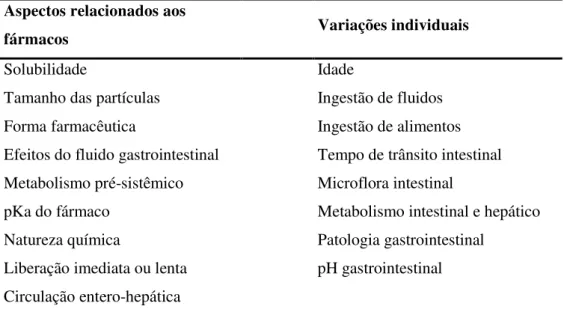
Tabela 2: Fatores que exercem influências sobre a biodisponibilidade dos fármacos. Aspectos relacionados aos
fármacos Variações individuais
Solubilidade Idade
Tamanho das partículas Ingestão de fluidos
Forma farmacêutica Ingestão de alimentos
Efeitos do fluido gastrointestinal Tempo de trânsito intestinal Metabolismo pré-sistêmico Microflora intestinal pKa do fármaco
Metabolismo intestinal e hepático Natureza química
Patologia gastrointestinal Liberação imediata ou lenta pH gastrointestinal Circulação entero-hepática
Fonte: Moura; Reyes (2002).
Fluxograma 1: O trajeto e a representação das etapas envolvidas na liberação e absorção de um fármaco contido na forma farmacêutica.
Fonte: adaptado de Aulton (2008).
A primeira fase, a biofarmacêutica, é compreendida pelos processos que ocorrem com o medicamento a partir de sua administração, incluindo as etapas de liberação e dissolução do princípio ativo. Esta é a fase em que o fármaco está disponível para absorção e em que os fatores físico, físico-químicos e forma farmacêutica são indispensáveis para que o medicamento possa ser liberado na quantidade e velocidade adequadas ao objetivo terapêutico pretendido (STRORPIRTIS et al., 2009; MOURA; REYES, 2002).
seja, é a fase definida como estudo quantitativo dos processos de absorção, distribuição, eliminação (biotransformação e excreção) dos fármacos ou dos metabólitos (MOURA; REYES, 2002).
A absorção é definida como uma série de processos pelos quais uma substância química externa de um ser vivo penetra em seu organismo sem lesão traumática, atravessando as diversas membranas biológicas, como o epitélio gastrointestinal, endotélio vascular e membranas citoplasmáticas, até alcançar a corrente sanguínea (OGA; CAMARGO; BATISTUZZO, 2008;SPINOSA; GÓRNIAK; PALERMO-NETO, 2008).
O processo de distribuição consiste na transferência reversível de um agente tóxico da circulação geral para os diferentes tecidos. A extensão da distribuição de uma substância tóxica ou medicamento do sangue para os tecidos depende das seguintes variáveis (SPINOSA; GÓRNIAK; PALERMO-NETO, 2008):
i. Hidrossolubilidade: compostos hidrossolúveis apresentam pouca disposição no tecido adiposo ou sistema nervoso central e são distribuídos por toda a água corpórea;
ii. Ligação com as proteínas plasmáticas: os agentes que se ligam às proteínas plasmáticas apresentam redução na distribuição nos tecidos e são retidos na circulação;
iii. Ligação com as proteínas teciduais: os agentes com alta afinidade por proteínas teciduais apresentam uma distribuição mais extensa e
iv. Lipossolubilidade: os agentes lipossolúveis ficam concentrados nos tecidos adiposos e no sistema nervoso central. Neste último, devido ao alto conteúdo de lipídeos, membranas citoplasmáticas e retículo endoplasmático das células.
Ainda na fase farmacocinética, após a distribuição ocorrem os processos de biotransformação e excreção.
A biotransformação consiste na transformação química desses agentes externos dentro do organismo, visando favorecer a sua eliminação e/ou inativação: toda substância química absorvida pelo trato gastrointestinal passa pelo fígado por meio da veia porta, onde é biotransformado para, posteriormente alcançar o resto do organismo, ou seja, a biotransformação elimina uma substância química externa do organismo convertendo-o em um metabólito. Após essa biotransformação, ocorre a excreção por excretas, como fezes e urina, ou até mesmo pelo ar expirado (SPINOSA; GÓRNIAK; PALERMO-NETO., 2008).
de um fármaco pelo receptor (BARREIRO; FRAGA, 2001) e seu efeito fisiológico no organismo (SILVA, 1994).
O trato gastrointestinal (TGI) atua como compartimento que processa mecânico-quimicamente o alimento ingerido, além de seu revestimento atuar como barreira contra patógenos e subprodutos da digestão. O TGI consiste em um tubo muscular, com aproximadamente 6m de comprimento e diâmetro variável, e estende-se da boca até o ânus. Apresenta quatro principais regiões anatômicas: esôfago, intestino delgado, intestino grosso e cólon. A superfície do lúmen é bastante rugosa, o que aumenta sua superfície de absorção. E ao contrário ao que se imagina, a absorção de fármacos é muito pequena no estômago e essa ocorre principalmente no intestino delgado, em razão de sua maior área de superfície de absorção (AULTON, 2008).
O transporte de fármacos e nutrientes pelo epitélio, no TGI, ocorre através de duas rotas: a transcelular (dividida em difusão passiva simples, transporte mediado por carregadores e endocitose) e a paracelular (TAO; DESAI, 2007). (Desenho1).
Desenho 1: Representação esquemática da estrutura epitelial gastrointestinal e suas possíveis rotas de transporte de fármacos.
2.3 LIBERAÇÃO CONTROLADA DE FÁRMACOS
Nos últimos anos, estudos sobre sistemas de liberação de fármacos têm sido intensamente estudados. Estes sistemas compreendem a utilização de vetores que permitem a veiculação de uma determinada droga ao sítio de ação específico, a otimização de sua velocidade e regime de dosagem. Dentre esses, os amplamente abordados são compreendidos por sistema de Liberação Modificada (modificação química) e as de Liberação Controlada (micropartículas compostas por substâncias como proteínas, lipídios, carboidratos ou polímeros sintéticos, nanopartículas de polímeros, sistema de liberação de peptídeos e proteínas, sistemas biodegradáveis sistemas osmóticos e regulados, como o magnético e ultra-som) (LANGER, 1990; PILLAI; DHANIKULA; PANCHAGNULA, 2001; TERENCE, 2002; SCHAFFAZICH; GUTERRES, 2003; SAHOO; LABHASETWAR, 2003; ORIVE et al., 2004; COUVREUR; VAUTHIER, 2006; STORPIRTIS et al., 2009; MALLIPEDDI;
ROHAN, 2010).
Os sistemas de liberação não são, no entanto, limitados à medicina e farmacologia. Também vem sendo utilizados, por exemplo, em coleiras anti-pulgas em animais de estimação, pesticidas, fertilizantes, cosméticos entre outros.
Segundo Terence (2002), um sistema de liberação “ideal” deve proporcionar a
liberação do fármaco exclusivamente no seu sítio de ação biológica e deve ser capaz de modular o intervalo de administração, a velocidade de liberação e duração do efeito. Isso deve-se pela necessidade de minimizar a quantidade necessária de fármaco para a obtenção do efeito terapêutico e para alterar a disponibilidade intencional de produtos formulados para liberar o fármaco lentamente durante horas, dias, semanas ou anos.
O sistema de liberação controlada vem sendo utilizado com diversas drogas para uma grande gama de tratamentos de doenças graças a modificações e evoluções das tradicionais pílulas aos sofisticados sistemas de liberação controlada. E quando utilizados em medicina, oferecem as seguintes vantagens (LANGER, 1990; TERENCE, 2002):
i. Maior eficácia terapêutica com liberação progressiva e controlada de fármacos, a partir da degradação da matriz;
ii. Controle da liberação do agente ativo, diminuindo a toxicidade e aumentando o tempo de permanência na circulação;
iv. Natureza dos veículos variáveis sem ocorrência de instabilidade e decomposição do fármaco;
v. Conveniente por necessitar de menor número de doses; vi. Economia, no caso de medicamentos de alto custo;
vii. Tanto substâncias hidrolíficas quanto lipolíficas podem ser incorporadas.
O Gráfico 1é uma representação esquemática para interpretação das aplicações de dosagens simples excessiva, simples repetidas e por sistema de liberação controlada de fármaco. A relação entre a concentração plasmática (μg/ml) de fármaco por intervalo de
tempo (horas), quando não aplicada a tecnologia de liberação controlada, é representada pelas curvas azul e verde. Estas atingem picos e declínios críticos, causando toxicidade ou ineficácia do fármaco no organismo quando em nível superior ou inferior à faixa terapêutica.
Gráfico 1: Relação entre concentração plasmática de fármaco por intervalo de tempo.
Fonte: Terence (2002).
Através da curva representada de cor vermelha para a liberação controlada do agente ativo, este permanece em uma concentração eficaz durante um período de tempo maior, de maneira controlada e constante. Isto é descrito como cinética de liberação de ordem zero.
2.4 SISTEMAS DE LIBERAÇÃO NANOESTRUTURADOS
A nanotecnologia farmacêutica é uma área que está diretamente envolvida no desenvolvimento, caracterização e aplicação de sistemas terapêuticos em escala micro ou nanométricas. Com o propósito de direcionar e controlar a liberação de fármacos em sítios de ação específicos, essa área tem sido um grande foco de pesquisas nas últimas décadas (VAAGE et al., 1995; SENIOR, 1998; CHERIAN; RANA; JAIN, 2000; SAHOO;
LABHASETWAR, 2003 ; LAMPRECHT et al, 2001; BRIGGER; COURRIER; BUTZ;
VANDAMME, 2002; GROOT et al., 2003; SAHOO; LABHASETWAR., 2003; ROY et al.,
2003; KOZIARA et al., 2004; PARK et al., 2004; HUGHES, 2005; MUNNHOZ et al., 2006;
SAKATA et al., 2007; CHOI; OH; CHOY., 2009; NOVICKIS, 2009; CHENG et al., 2010;
CHITHRANI et al., 2010; MALLIPEDDI; ROHAN., 2010; MUNHOZ JR. et al., 2010).
Os nanossistesmas oferecem algumas vantagens como proteção do fármaco contra a degradação, liberação do fármaco à sítios de ação específicos e liberação de moléculas biológicas como proteínas, peptídeos e oligonucleotídeos. A eficiência dessa liberação para diferentes alvos no organismo está diretamente relacionada com o tamanho das partículas, ou seja, é o tamanho dos carregadores que permite a penetração nas células e a liberação intracelular do fármaco sem ocorrer risco de degradação extracelular (DESAI, 1997;
MALLIPEDDI; ROHAN., 2010). São classificados de acordo com a forma estrutural e com o
Tabela 3: Nanotecnologia, materiais e forma nanoestrutural. Tecnologia de liberação
de drogas Materiais Forma nanoestrutural
Biológico Lipídeos, peptídeos, ác. Nucleicos, polissacraídeos, vírus
Lipossomas, nanotubos, nanopartículas
Polimérico Poli (ác. glicólico), poli (ác. láctico), poli (etileno-glicol) entre outos.
Vesículas, esferas, nanopartículas, micelas, dendrímeros.
Base de silicone Silicone Poros, nanopartículas
Base de carbono Carbono Nanotubos,
nanopartículas Metálico Ouro, prata, platina, paládio Nanotubos,
nanopartículas
Fonte: Hughes (2005).
Os principais materiais utilizados para nanosistemas carregadores para a distribuição de fármacos no organismo são os lipossomas e nanopartículas. Estas se referem a
sistemas carregadores que apresentam diâmetro inferior a 1μm e as duas diferentes estruturas,
a nanoesfera e nanocápsula (Desenho 2), as quais diferem entre si segundo a composição e organização estrutural (PUISIEUX et al., 1994; SCHAFFAZICK; GUTERRES, 2003;
SAHOO; LABHASETWAR, 2003; ORIVE et al., 2004): as nanoesferas constituem sistemas
em que o fármaco encontra-se disperso homogeneamente no interior de uma matriz polimérica e as nanocápsulas constituem um invólucro, geralmente polimérico, disposto ao redor de um núcleo diferenciado, que pode estar dissolvido e/ou adsorvido à parede polimérica.
As nanopartículas poliméricas apresentam aplicações promissoras, principalmente, na vetorização de anticancerígenos, proteínas, peptídeos, vacinas tumorais e administração de antibióticos pela via oral (SCHAFFAZICK; GUTERRES, 2003).
Desenho 2: Representação esquemática de nanocápsulas e nanoesferas poliméricas: a). fármaco dissolvido no núcleo oleoso das nanocápsulas; b). fármaco adsorvido à parede polimérica das nanocápsulas; c). fármaco retido na matriz polimérica das nanoesferas; d). fármaco disperso molecularmente na matriz polimérica das nanoesferas.
Fonte: Schaffazick; Guterres (2003).
Os lipossomas são nanoestruturas biológicas formadas por vesículas esféricas artificiais produzidas a partir de bicamadas (lamelas) de fosfolipídios e colesterol (Desenho 3). Possuem centros aquosos que podem ser utilizados para encapsular drogas hidrofóbicas e hidrofílicas, e podem também ligar-se a anticorpos, que atuam como transportadores para sítios de ação específicos. Suas propriedades variam substancialmente de acordo com a composição lipídica, tamanho e método de preparo (BARZA; STUART; SZOKA, 1987; LASIC; VALLNER; WORKING, 1999; SAHOO; LABHASETWAR , 2003; ORIVE et al.,
2004; ORIVE et al., 2005; BATISTA et al., 2007; MERTINS, 2008; MALLIPEDDI;
ROHAN., 2010).
Além das nanopartículas e lipossomas, outros nanosistemas carregadores também são utilizados para a distribuição de fármaco no organismo: os dendrímeros, miscelas, nanotubos e cerâmica nanoparticulada (SAHOO; LABHASETWAR, 2003; ORIVE
et al., 2004; ORIVE et al., 2005; HUGHES, 2005; MALLIPEDDI; ROHAN, 2010).
Os dendrímeros são nanoestruturas formadas pela presença de cadeias e múltiplas ramificações a partir de um núcleo, promovendo uma aparência arbórea. Suas moléculas poliméricas são fundamentadas na aplicação de progressões matemáticas para a síntese orgânica (Desenho 4) e, como tal, possuem topologia molecular bem definida. Possuem arquitetura tridimensional, globular bem definida e com alto grau de ramificações. Os dendrímeros são preparados por meio de uma síntese tridimensional repetitiva e como resultado deste procedimento controlado, eles têm sido comparados a polímeros monodispersos. Esses polímeros dendrimétricos diferem dos polímeros clássicos pelo seu grau de ramificação, funcionalização terminal e monodispersão (MONTANARI; MONTANARI; PILÓ-VELOSO, 1998; VOGTLE; GESTERMANN; HESSE, 2000; GROOT
et al., 2003; SAHOO; LABHASETWAR, 2003; ORIVE et al., 2004; ORIVE et al., 2005;
HUGHES, 2005; TAMILVANAN, 2008; MALLIPEDDI; ROHAN, 2010).
a). b).
Desenho 4: a). Representação esquemática de um dendrímero. b). Representação esquemática do crescimento dendrimétrico.
Fonte: Montanari; Montanari; Piló-Veloso (1998); Sahoo; Labhasetwar (2003).
encapsuladas, cuja principal vantagem é a preparação de medicamentos parenterais contendo ativos de baixa hidrossolubilidade. Seu sistema é útil para administração sistêmica de drogas insolúveis em água e tem sido extensamente estudada como carregadores de drogas (KATAOKA; HARADA; NAGASAKI, 2001; ADAMS; LAVASANIFAR; KWON, 2003; NISHIYAMA; KATAOKA, 2003; ROY et al., 2003; VILLANOVA; ORÉFICE, 2010).
Desenho 5: Representação esquemática de uma micela. Fonte: Adaptado de Mertins (2008).
As matérias-primas metálicas como prata, ouro, platina e paládio também têm sido produzidas como nanopartículas para algumas aplicações farmacêuticas. Dentre essas, a prata nanoparticulada tem recebido considerável atenção como agente antibacteriano (SONDI; SALOPEK-SONDI, 2004) e antiviral, incluindo hepatite B (LU; SUN; CHEN, 2008) e HIV in vitro (SUN et al., 2005; LARA et al., 2010).
Uma das desvantagens da utilização da prata para essas aplicações está relacionada ao seu potencial tóxico. Alguns estudos têm demonstrado os efeitos citotóxicos e cumulativos dessas nanopartículas em vários órgãos como pulmões, rins, cérebro e fígado (BRAYDICH-STOLLE et al., 2005; KIM et al., 2008; ASHARANI et al., 2009). Estudos
similares com outras nanopartículas inorgâncicas como ouro e titânio tem indicado toxicidade quanto a danificação do DNA e apoptose celular (GOODMAN et al., 2004).
(JAIN et al., 1998 apud SAHOO; LABHASETWAR , 2003; CHERIAN et al., 2000 apud
SAHOO; LABHASETWAR, 2003), devido a possibilidade de controlar suas microestruturas, como a área específica e porosidade, além de não possuir impurezas metálicas ou inorgânicas em função de se conhecer os reagentes que as originaram via polimerização inorgânica (MUNHOZ JR. et al., 2006; NOVICKIS, 2009).
Dentre as vantagens das cerâmicas nanoparticuladas na ciência moderna, destacam-se (SCHIMIDT, 1994; SAHOO; LABHASETWAR, 2003; MUNNHOZ et al.,
2006):
i. Facilidade de produção: diversas técnicas podem ser utilizadas para sua produção (por precipitação química, processo sol-gel, atrito mecânico e outras). A síntese por métodos químicos via sol-gel é a mais estudada e permite um controle estequiométrico do produto nanoestruturado;
ii. Influência na fuga da nanopartícula do sistema retículo-endotelial pelo seu tamanho nanométrico (SER – sistema constituído por células dotadas de capacidade fagocitária e que desempenham funções de defesa contra corpos estranhos no organismo);
iii. Ausência de aumento volumétrico e deformação de porosidade com as mudanças de pH;
iv. Proteção contra desnaturação induzida por pH externo, temperatura e ação de enzimas;
v. Silica, alumina e titânia compatíveis com sistemas biológicos;
vi. Fácil modificação de suas superfícies, podendo ser conjugada a uma variedade de anti-corpos para orientação aos locais desejados de ação.
2.5 MATERIAIS CERÂMICOS E CERÂMICA AVANÇADA
O termo “cerâmica” é oriundo da palavra grega keramiko, cujo significado é
“matéria prima queimada”, ou seja, matéria com propriedades que são atingidas por meio de
2.5.1 Cerâmicos
Os materiais cerâmicos são compostos inorgânicos constituídos por elementos metálicos e não metálicos em proporções adequadas. São essencialmente ligados entre si por ligações químicas iônicas ou covalentes e oriundos de variadas composições químicas formadas, na maioria das vezes, por óxidos nitretos, carbetos e silicatos.
Os cerâmicos são geralmente duros, frágeis, bons isolantes térmicos e elétricos, além de possuírem elevada temperatura de fusão e grande estabilidade química (CALLISTER, 2008).
Segundo a Associação Brasileira de Cerâmica (2012), os materiais cerâmicos são classificados em:
i. cerâmica vermelha;
ii. materiais de revestimento (placas cerâmicas); iii. cerâmica branca;
iv. materiais refratários; v. isolantes térmicos; vi. fritas e corantes; vii. abrasivos;
viii. vidros, cimento e cal; ix. cerâmica fina ou avançada.
2.5.2 Cerâmica fina ou avançada
O termo “cerâmica fina” ou “cerâmica avançada” tem sido utilizado para
designar cerâmicas com funções específicas e utilizadas em aplicações de alta tecnologia (CALLISTER, 2008).
A diferença da cerâmica avançada da cerâmica comum está relacionada aos processos de síntese para suas produções: para a síntese de pós cerâmicos para a produção da cerâmica avançada é necessária a utilização de matérias-primas altamente puras, de composição química rigorosamente controlada e rigoroso processo de sinterização, enquanto a cerâmica comum pode ser obtida por simples prensagem e sinterização de matérias primas não refinadas, geralmente encontradas na Natureza (MAZALI, 2005).
homogeneidade química, minimização na formação de aglomerados, controle e estabilização da fase cristalina desejada (MAZALI, 2005; MUNHOZ JR. et al, 2006). E dentre as diversas
técnicas de síntese desses pós, o processo sol-gel é o mais estudado (SCHIMDT, 1994; MUNHOZ JR. et al., 2006).
2.5.3 Processo sol-gel
Os métodos desenvolvidos para a produção de nanoparticulados incluem grande variedade de métodos físicos, químicos e mecânicos. E dentre essas diversas técnicas utilizadas para a síntese de pós nanoparticulados de cerâmicas, o processo sol-gel é o mais utilizado e estudado (SCHIMDT, 1994; MUNHOZ JR. et al., 2006).
O processo sol-gel (PSG) consiste na metodologia de preparação de materiais, partindo-se de precursores moleculares e, como o nome indica, envolve a formação de uma suspensão de partículas muito finas de tamanho coloidal dispersa num líquido (sol) e sua transformação numa rede contínua (gel) (RING, 1996). O termo sol é geralmente empregado
para definir uma dispersão de partículas coloidais (dimensão entre 1 e 100nm) estáveis em um fluido, enquanto o termo gel pode ser constituído como um sistema formado pela estrutura
rígida de partículas coloidais (gel coloidal) ou cadeias poliméricas (gel polimérico) que imobiliza a fase líquida nos seus interstícios (HIRATSUKA; SANTILLI; PILCINELLI, 1995).
Quadro 1: Resumo das etapas de obtenção de materiais cerâmicos, a partir do processo sol-gel, empregando precursores alcóxidos, onde o M é um cátion metálico, R é uma cadeia orgânica, n e x são números inteiros diferentes de zero.
Fonte: Mazali (2005).
Esquema 1: Ilustração do processo de gelatinização (transição sol-gel): a). formação do gel particulado (coloidal); b). formação de gel polimérico.
Fonte: Hiratsuka; Santilli; Pilcinelli (1995).
O método sol-gel proporciona algumas vantagens na preparação dos materiais, pois permite realizar sínteses em baixas temperaturas, evitando a decomposição térmica das espécies imobilizadas, além de reduzir o risco de contaminação e perda de componentes voláteis. Além disso, outra grande vantagem deste processo é a obtenção de materiais com características e propriedades pré-planejadas, sendo possível, portanto, a realização de controle de suas características físicas e químicas quanto a estequiometria, porosidade, estrutura, textura e tamanho dos poros (MUNHOZ JR. et al., 2006). Essas vantagens
2.6 CERÂMICA COMO EXCIPIENTE FARMACÊUTICO E SISTEMA LIBERADOR DE FÁRMACOS
As primeiras aplicações e uso de materiais cerâmicos em seres humanos foram relatados na medicina e odontologia em 1920 como biorreparadores de tecido ósseo. E uma vez considerados biocompatíveis, esses biomateriais foram também testados nas últimas décadas como excipientes e como sistema de liberação de fármacos (PAUL; SHARMA, 2003). Muitos materiais têm sido utilizados em formulações farmacêuticas como excipientes: sílica, caulim, esmectitas, argilas minerais, alguns silicatos, como o talco e outros (HERMOSÍN et al., 1981; GALAN; LISO; FORTEZA, 1985; CARRETERO, 2002; LIRA,
2004; AGUZZI et al, 2007; DUREJA; PHARAM; MADAN, 2007).
Dentre os diferentes materiais, as argilas minerais são muito atraentes como carregadores e suportes para substâncias com propriedades farmacêuticas, devido sua capacidade de adsorver e desorver moléculas orgânicas (HOYO; VICENTE; RIVES, 2001; SOARES, 2003).
Alguns tipos de argilas fibrosas e lamelares são utilizadas com a finalidade de diminuir a viscosidade dos produtos (caráter tixotrópico), facilitando a administração (VISERAS; LOPEZ-GALINDO, 1999) e exibem outras propriedades desejáveis para produtos utilizados como desintegrantes, aglutinantes e diluentes. Essas argilas incham na presença de água e formam géis em baixa concentração, sendo quimicamente inertes e estáveis em ampla faixa de temperatura (WAI; DEKAY; BANKER, 1966; SOARES, 2003).
Outra argila utilizada como excipiente de fármaco é a bentonita. Esta pertence a família de esmectitas e também possui propriedade tixotrópicas que a torna ideal como agente suspensor em medicamentos (VISERAS; LOPEZ-GALINDO, 1999).
A bentonita é um material promissor para sua utilização como excipiente funcional em comprimidos, devido a sua capacidade de formar géis em concentrações baixas por intumescimento em água, sendo apropriada como agente ligante e desintegrante (SOARES, 2003; LIRA, 2004). Sua presença no TGI como excipiente de fármaco é de grande utilidade devido ao decréscimo na biodisponibilidade dos fármacos administrados oralmente e ação contínua decorrente a velocidade de adsorção controlada, assegurando os níveis plasmáticos eficientes por longos períodos de tempo (SOARES, 2003).
O material argiloso caulim também pode ser utilizado como excipiente farmacêutico. Esse é utilizado como veículo adsorvente e diluente em produtos farmacêuticos e pesticidas, além de proporcionar volume em cosméticos e ração animal (GOMES da SILVA, 2007).
No início da década de 80, Bajpai et al. (1988) deram início ao estudo da
cerâmica como sistema de liberação controlada de proteínas, polipeptídeos e hormônios. Desenvolveram a ALCAP (“Alumino Calcium Phosphorous Oxide Ceramics”), biocompatível e não tóxica capaz de liberar esteróides continuadamente por 12 meses. Com baixo custo e fácil produção a ALCAP, juntamente com o tricálcio de fosfato (TCP), também foi testada in vitro, como liberador controlado do fármaco Azidothymidine (AZT), droga
administrada em pacientes com AIDS e que causa efeitos colaterais graves, incluindo efeitos tóxicos sobre a medula óssea (BENGHUZZI; BARBARO; BAJPAI, PAUL; SHARMA, 2003).
A ZCAP (“Zinc Calcium Phosphorous Oxid Ceramic”), também desenvolvida por Bajpai (1988), foi testada em ratos diabéticos. Foram inseridos nesses animais implantes subcutâneos com cápsulas de ZCAP contendo insulina e medições de glicemia no sangue foram realizadas durante 6 semanas. A normoglicemia nesses animais foram mantidas por 3 semanas, determinando, portanto, o uso da ZCAP como possível dispositivo de liberação controlada de insulina para diabéticos (ARAR; BAJPAAI., 1992). Além disso, o uso da
ZCAP pode trazer vantagens devido a liberação de traços de zinco de sua estrutura, gerando melhoria no processo de cicatrização de lesões (PAUL & SHARMA, 2011).
A hidroxilapatita (HA) é uma cerâmica similar aos ossos e dentes humanos. Suas propriedades físicas, químicas e biológicas a tornaram materiais implantáveis e substitutos ósseos aprovados pela Food and Drugs Administration (FDA) e que a torna
possível como sistema liberador de drogas local, nos ossos (KUNDU et al., 2010), e drogas
anti-cancêr (UCHIDA et al., 1992; PAUL & SHARMA, 2011).
Com relação às cápsulas biocerâmica TCPL (“Tricalcium phosphate-lysine”),
parecia mostrar mudanças fisiológicas nos órgãos reprodutivos (ZIZZI et al., 1999; PAUL &
SHARMA, 2011).
Em sistemas de liberação de drogas, cerâmicos a base de fosfato de cálcio também estão sendo pesquisados, mas limitados ainda à liberação in vitro utilizando como
fármacos os hormônios de crescimento, antibióticos e quimioterápicos (GUICHEUX et al.,
1997; ITOZAKU et al., 1998; YAYLAOGLU et al. 1999).
Silveira (2007) utilizou a anortita, um vitrocerâmico biocompatível, como aplicação de veículo de fármaco in vitro. Esta apresenta propriedades de materiais
inteligentes, ou seja, sofrem degradação de acordo com a variação de pH do meio em que se encontra, possui características compatíveis a sistemas reservatórios e é capaz de armazenar substâncias no interior de suas câmaras. A anortita e o fármaco teste (Prolopa – Levodopa + Benzerazida) foram submetidas a submersão em soluções tampão fosfato de pH 3,5 e 7,4 e a leitura de adsorbância mostrou que o fármaco acondicionado no interior das cápsulas de anortita foi liberado, apontando a viabilidade da realização de pesquisas que venham validar esse biomaterial como sistema de liberação de fármacos.
Roy et al. (2003) desenvolveram uma cerâmica nanoparticulada modificada a
base de sílica como carregador de drogas fotossensíveis para aplicações em terapia fotodinâmica. Esta terapia consiste no tratamento de lesões e destruição de células cancerígenas e pré-cancerígenas por meio de reações fotoquímicas em que há a combinação da incidência do laser com a droga fotossensível.
Novicks (2009) e Munhoz Jr. et al. (2010) avaliaram a cerâmica
nanoparticulada do tipo pseudoboemita in vitro, frente a adsorção e dessorção de dois tipos de
moléculas farmacêuticas (Aciclovir e Atenolol). Este excipiente foi analisado por Espectroscopia de absorção no infra-vermelho, por Análise térmica diferencial (DTA), Termogravimétria (TG) e Espectroscopia no ultravioleta-visível (UV-vis), além de ser caracterizado por microscopia eletrônica de varredura (MEV), por difração de raios-X (DRX), medição da área específica pelo método de BET (área superficial específica) e ter a solução do fármaco com a pseudoboemita após a adsorção estudada via cromatografia de alta eficiência (CLAE). Determinaram que as interações entre os fármacos e a pseudoboemita são resultados da adsorção no caso do atenolol e aumento de solubilidade no caso do aciclovir. E ainda, por meio da MEV, determinaram que a pseudoboemita sintetizada apresentava alta porosidade, havendo expectativas futuras para seu uso como sistema liberador de fármaco.
dos comprimidos, por prensagem direta a seco. Os comprimidos foram caracterizados por DRX, Calorimetria Exploratória Diferencial (DSC), TG e MEV. Foi também obtido o perfil de liberação do medicamento através de UV-vis para simulação in vitro. Os resultados
permitiram observar que não houve reação entre o fármaco e o excipiente. Além disso, foi também observada a constância dos níveis plasmáticos in vitro (MARTINS et al., 2012).
O único adjuvante aprovado em humanos é o hidróxido de alumínio (GOTO et al., 1993; MOTA; LIMA; MELO, 2006; ROWE; SHESKEY; OWEN, 2006). Durante os
últimos oitenta anos, algumas formulações de adjuvantes têm sido desenvolvidas, sendo que poucas foram testadas em triagens clínicas e a maioria nunca foi aceita para a vacinação, em razão da toxicidade e aos efeitos adversos (GUPTA; SIBER, 1995).
O termo adjuvante originou-se da palavra adjuvare que significa ajudar. Os
adjuvantes consistem, então, em qualquer material capaz de promover ou ocasionar uma elevada e prolongada reposta imune a um antígeno, além de direcionar essa reposta imune para uma resposta protetora, evitando a doença. São imunopotencializadores, podendo ser compostos naturais ou sintéticos (MOREIN et al., 1996; CHIN; GIL, 1998; AUDIBERT;
LISE, 2003). Seu uso é importante em vacinas, quando o antígeno possui baixa imunogenicidade. Sua utilidade é dependente de sua segurança e capacidade em estimular a imunidade por longos períodos (CHIN; GIL, 1998).
O hidróxido de alumínio é também classificado como boemita pobremente cristalina ou pseudoboemita (GOTO et al., 1993; ROWE; SHESKEY; OWEN, 2006). No
entanto, a pseudoboemita é um oxi-hidróxido de alumínio, enquanto que o Al (OH)3 é um hidróxido de alumínio. São, portanto, substâncias diferentes.
O adjuvante de hidróxido de alumínio induz o aumento da permeabilidade vascular associado a efeito tóxico sobre macrófagos. Tem sido largamente utilizado na prática de vacinas como a do tétano e hepatite B, e em uso veterinário para vacinas parenterais nos Estados Unidos e Europa (GOTO et al., 1993; ROWE; SHESKEY; OWEN, 2006), além de
ter sido utilizado em vacinas experimentais anti-HIV, aumentado a produção específica de anticorpos (HART et al., 1990; RESENDE et al., 2004). É um material naoparticulado, com
área superficial de 550 m2/g (JOHNSTON; EISE; FRY, 1991).
2.7 PSEUDOBOEMITA
A pseudoboemita consiste em uma cerâmica sintética avançada com características de composição, morfologia e granulometria desejadas. O processo sol- gel é um dos processos de obtenção desse material nanoparticulado (MUNHOZ JR. et al., 2006;
MOROZ et al., 2006; MUNHOZ JR. et al., 2010).
Esse material nanoparticulado é uma estrutura similar a boemita (MUNHOZ
JR. et al, 2006). A diferença entre essas duas estruturas envolvem características relacionadas
ao tamanho de suas células unitárias: a célula unitária da pseudoboemita é maior do que a célula unitária da boemita. Essa diferença ocorre devido a incorporação de maior quantidade de água na estrutura da pseudoboemita (KLOPROGGE et al., 2006).
O Esquema 2 apresenta a estrutura da pseudoboemita, segundo Moroz et al.
(2006).
Esquema 2: Representação da estrutura da pseudoboemita. Fonte: Moroz et al. (2006).
Novicks (2009) e MUNHOZ JR. et al. (2010) realizaram microscopias da
O processo sol-gel consiste em um método de preparação de cerâmicas a partir de precursores moleculares e, como o nome indica, envolve a formação de uma suspensão de partículas muito finas dispersas num líquido (sol) e sua transformação numa rede contínua (gel) (RING, 1996). Neste processo, é possível realizar a obtenção de materiais com características e propriedades desejadas como porosidade, estrutura cristalina e tamanho das partículas, devido a possibilidade de controlar todas as etapas, desde o precursor até o produto final (MACEDO, 2006; MUNHOZ JR. et al., 2006).
Micrografia 1: Microscopia eletrônica de varredura de amostra da pseudoboemita.
Fonte: Munhoz jr. et al. (2010).
Além do processo sol-gel, Moroz et al. (2006), sintetizou essa pseudoboemita
com outros dois métodos diferentes a fim de estudar as estruturas das mesmas e concluiu que a pseudoboemita de maior área específica foram aquelas preparadas a partir de nitrato de alumínio e hidróxido de amônio.
Com a finalidade de quantificar a liberação do fármaco aciclovir com a utilização da pseudoboemita como excipinte, Novickis (2009) e Munhoz jr. et al. (2010)
realizaram ensaios de adsorção em um dissolutor com fluido gastrointestinal (pH=1 e pH=7) e determinaram a cinética de liberação do fármaco pelo comprimido, através da construção da curva de liberação por espectroscopia no ultra-violeta visível (UV-vis) (Gráfico 2). Foram observadas boas estabilidades de concentração entre os valores de 510 e 480 μg/mL.
Gráfico 2: Perfil de liberação do aciclovir pela pseudoboemita em um período de 2 horas. Concentração do aciclovir determinado por espectroscopia no ultra-violeta visível (UV-vis).
Fonte: Munhoz Jr. et al. (2010).
Novicks (2009) e Munhoz Jr. et al (2010) demonstraram por meio de análises in vitro, que a pseudoboemita formada por esses precursores e obtida via polimerização
inorgânica de baixo custo (processo sol-gel) é viável como sistema de liberação controlada dos fármacos Aciclovir e Atenolol. Martins et al. (2012) também verificaram esta potencial
continuidade no estudo deste material nanoparticulado há necessidade agora de determinar a toxicidade da pseudoboemita.
Todo o processo de produção para obtenção da pseudoboemita foi realizado no Laboratório de Engenharia de Materiais da Universidade Presbiteriana Mackenzie.
2.8 TOXICIDADE
A toxicologia é a ciência que estuda os efeitos adversos ou tóxicos em organismos vivos decorrentes das interações desses com substâncias químicas. Estes efeitos são definidos como alterações anormais ou nocivas ao organismo após a exposição a um determinado elemento tóxico, podendo ser de maior gravidade gerando sua morte ou de menor gravidade, podendo ocasionar alterações no consumo de alimentos, variações no peso corpóreo ou órgãos, alterações anátomo-patológicas e bioquímicas. A toxicologia é capaz, portanto, de quantificar, identificar e determinar quais os níveis toleráveis de toxicante num organismo (MOREAU; SIQUEIRA, 2010; MORAES; SZNELWAL; FERNÍCOLA, 1991; OGA; CAMARGO; BATISTUZZO, 2008).
A toxicidade, segundo Oga et al. (2008), compreende a capacidade inerente e
potencial de uma determinada substância química de provocar efeitos nocivos em organismos vivos. Sua avaliação envolve a intensidade do efeito adverso (letal, moderada e leve), a concentração/dose e a duração da exposição (agudo, subagudo e crônico). E para se conhecerem esses efeitos de uma determinada substância em um organismo, há a necessidade de determinar a relação dose-resposta que conduz ao cálculo da DL50 (Dose Letal de 50% dos animais), realizar testes toxicológicos e determinar informações preliminares da substância a ser estudada.
A relação dose-resposta compreende a relação entre a exposição a o espectro de efeitos tóxicos observados. E com a curva formada por essa relação é possível calcular a Dose Letal 50% e a dose mínima para produzir uma resposta detectável numa população teste. No Brasil, a Resolução 1/78 (Diário Oficial 17/10/78) do Conselho Nacional de Saúde, estabelece 5 ensaios de toxicidade: aguda, sub-aguda, crônica, teratogenicidade, embriotoxicidade e estudos especiais (estudos de comportamento, carcinogenicidade e outros).
diretamente ligadas à toxicidade. Dentre essas, as informações fundamentais são: odor, cor, ponto de fusão e ebulição, densidade, viscosidade, solubilidade e volatilidade (OGA; CAMARGO; BATISTUZZO., 2008).
2.8.1 Toxicidade Oral Aguda – DL50
A toxicidade aguda é definida como os efeitos que ocorrem em um período curto após a administração de uma dose ou doses múltiplas durante o período de 24 horas. A dose única é utilizada para determinar a potência de um composto a ser testado, enquanto as doses múltiplas são utilizadas para avaliar o efeito cumulativo desse composto. Seu teste é seguido de observação de sinais e sintomas de toxicidade, assim como a morte dos animais durante o período de 14 dias após a administração (OGA; CAMARGO; BATISTUZZO.,
2008; SPINOSA; GÓRNIAK; PALERMO-NETO., 2008; ANVISA, 2010).
Os principais objetivos desse teste são:
i. Avaliar a toxicidade intrínseca dos agentes tóxico ou substância química; ii. Avaliar a suscetibilidade das espécies;
iii. Identificar os órgãos alvos;
iv. Promover informações para o delineamento e seleção dos níveis de dose para estudo mais prolongados (toxicidade crônica).
O teste de toxicidade aguda é um pré-requisito para agências reguladoras dos países, como a FDA e Angência Nacional de Vigilância Sanitária (ANVISA), para a
aprovação de novos fármacos aditivos alimentares, produtos domésticos químicos industriais e outros. E para a realização deste teste, é necessário conduzí-lo de acordo com protocolos internacionais preconizados por organizações como a Organization for Economic
Cooperation and Development: OECD (VALADARES, 2006; OGA; CAMARGO;
BATISTUZZO., 2008;ANVISA, 2010).
Para o estudo da toxicidade aguda da pseudoboemita em roedores, serão seguidos as diretrizes do Guia OECD 423 (OECD, 2001), pois seus protocolos são de grande aceitação pelos órgãos reguladores e atendem os requisitos de experimentação e critérios de bem-estar animal.
dose disponível. Devem ser utilizados 3 animais por dose e tem como princípios identificar a menor dose que cause mortalidade, determinar a faixa estimada de DL50 e identificar sinais de toxicidade.
Os parâmetros a serem avaliados são mortalidade, sinais clínicos, variação de peso corporal, modificação no consumo de ração e água, latência, duração e reversibilidade da toxicidade, investigações anátomo e histopatológica.
2.8.2 Toxicidade Oral Subaguda
Os testes de toxicidade subaguda são realizados para a obtenção e caracterização do perfil toxicológico de uma determinada substância química, após exposições repetidas. Tem como objetivos (OGA; CAMARGO; BATISTUZZO., 2008;
ANVISA, 2010):
i. Determinar a maior dose da substância química que não produz efeito tóxico; ii. Identificar os órgãos alvos afetados;
iii. Determinar os efeitos tóxicos;
iv. Estabelecer se esses efeitos são reversíveis;
v. Prover dados sobre dosagens para estudo de toxicidade crônica.
Nesse ensaio de toxicidade subaguda também envolve as avaliações anatomopatológica e histopatológica de todos os animais no final da experimentação. Caso um animal ficar doente, moribundo ou morrer durante a experimentação, ele deverá ser sacrificado e necropsiado, e seus órgãos retirados para análises.
Para a realização desse teste é adotado o protocolo internacional preconizado pela Organização para Cooperação Econômica e Desenvolvimento – Organization for
Economic Cooperation and Developmente: OECD, no qual são seguidas diretrizes do Guia
407 (OECD, 1995). Seus protocolos são de grande aceitação pelos órgãos reguladores e atendem requisitos de experimentação e critérios de bem-estar animal.
As doses em estudos de administração repetidas são estabelecidas de acordo com as informações obtidas no teste de toxicidade aguda. A duração mínima desse teste deve ser um período de 2 semanas (14 dias), sendo comumente realizado em 28 dias.