Tese apresentada à Faculdade de Medicina da Universidade de São Paulo para obtenção do Título de Doutor em Ciências
Programa de Nefrologia
Orientadora: Dra. Rosa Maria Affonso Moysés
Aos meus queridos pais Sandra e José Carlos,
que sempre acreditaram e me fizeram acreditar.
Ao meu marido Ramon,
Esta tese é resultado do esforço e colaboração de várias pessoas
que, direta ou indiretamente, são por ela responsáveis. Ela não seria
possível, em primeiro lugar, sem o apoio e incentivo de meus pais, exemplos
de integridade e generosidade que sempre nos proporcionaram acesso ao
conhecimento e um ambiente propício ao desenvolvimento pessoal. A eles,
devo a lição da persistência e a certeza de que seria capaz. Agradeço por
sua dedicação e suporte constantes. Amo vocês.
Também não seria possível sem Ramon, pela coragem em mudar de
cidade e mergulhar nesta aventura que iniciamos juntos. Agradeço por
apoiar meu crescimento profissional e por tornar mais fácil este caminho
que, certamente, seria muito mais difícil sem seu carinho, amor e bom
humor.
Agradeço ainda à minha irmã Tatiane, que com talentos tão diversos
dos meus, me ensinou a valorizar as diferenças. A ela e ao meu cunhado
Cláudio, agradeço por nos darem minha linda sobrinha Bárbara, que trouxe
alegria à nossa família.
Aos avós Lourdes e José (in memorian), pela paciência e cuidado
comigo. À avó Maria (in memorian), pelos bons momentos. Aos tios e
primos, pela convivência agradável. Aos meus sogros Cleuza e Sérgio e à
Valéria, por permitirem uma integração familiar tão harmoniosa. Às amigas
Catarina e Graciela, pelo apoio e companheirismo.
paciência, incentivo e acompanhamento próximo ao longo de todo este
período. À Dra. Vanda, criadora do grupo, pela acolhida calorosa e por ser,
além de referência técnica, exemplo de ser humano.
À Sílvia Titan, minha admiração e gratidão pela disponibilidade e
auxílio inestimável na análise estatística e revisão do texto.
Ao Dr. Raul Dias Santos, por viabilizar a realização do projeto no
Instituto do Coração e pela valiosa colaboração.
Meus agradecimentos sinceros à Luciene e Fabiana, pelas dosagens
de FGF-23 e pela prontidão em ajudar sempre.
Ao Dr. Rui Toledo e todos os funcionáros da Pós-Graduação da
Nefrologia, pela qualidade do trabalho desenvolvido e auxílio aos
pós-graduandos.
Ao Dr. Carlos Eduardo Rochitte e equipe, pela análise dos Escores de
Agatston; Drs. Luis Antônio Machado César e Eulógio Martinez por
permitirem a inclusão de seus pacientes e ao Dr. Pedro Lemos e equipe,
pelas análises referentes à cineangiocoronariografia.
À Dra. Maria Goretti Penido, grande incentivadora.
Por fim, agradeço a todos os profissionais do LIM-16 pela convivência
amistosa e aos colegas de pós-graduação e amigos Juliana, Bia, Luciene,
Rodrigo Bueno, Rodrigo Azevedo, Melani, Daniela e Patrícia, que tornaram
“A mente que se abre a uma nova idéia
jamais voltará ao seu tamanho original.”
Essa tese está de acordo com:
Referências: adaptado de International Committee of Medical Journal Editors (Vancouver)
Universidade de São Paulo. Faculdade de Medicina. Serviço de Biblioteca e Documentação. Guia de apresentação de dissertações, teses e monografias. Elaborado por Anneliese Carneiro da Cunha, Maria Julia de A. L. Freddi, Maria F. Crestana, Marinalva de Souza Aragão, Suely Campos Cardoso, Valéria Vilhena. 2a ed. São Paulo: Serviço de Biblioteca e Documentação; 2005.
LISTA DE ABREVIATURAS E SIGLAS LISTA DE FIGURAS
LISTA DE TABELAS RESUMO
SUMMARY
1. INTRODUÇÃO... 2
1.1CALCIFICAÇÃO VASCULAR... 3
1.1.1 Calcificação vascular e morbimortalidade ... 3
1.1.2 Fisiopatologia da calcificação vascular ... 5
1.1.3 Métodos de imagem para avaliação da calcificação vascular ... 10
1.2METABOLISMO MINERAL... 11
1.2.1 Homeostase do fósforo... 11
1.2.1.1 Paratormônio ... 12
1.2.1.2 Vitamina D ... 15
1.2.1.3 Fibroblast Growth Factor 23... 18
1.2.1.4 Klotho... 23
1.2.2 Homeostase do cálcio ... 26
1.3METABOLISMO MINERAL E MORTALIDADE/MORBIDADE CARDIOVASCULAR... 27
1.3.1 Fósforo e doenças cardiovasculares ... 27
1.3.1.1 Fósforo e calcificação vascular... 31
1.3.2 Vitamina D e doenças cardiovasculares ... 32
1.3.3 PTH e doenças cardiovasculares ... 36
1.3.4 FGF-23 e doenças cardiovasculares... 37
2. OBJETIVOS... 41
2.1 OBJETIVO PRINCIPAL... 41
2.2 OBJETIVOS SECUNDÁRIOS... 41
3. CASUÍSTICA E MÉTODOS... 43
3.1DEFINIÇÃO DOS FATORES DE RISCO... 44
3.2ANÁLISES BIOQUÍMICAS... 45
3.3ESCORE DE CALCIFICAÇÃO CORONARIANA (ESCORE DE AGATSTON) ... 45
3.4CINEANGIOCORONARIOGRAFIA E ESCORES ASSOCIADOS... 46
3.4.1 Escore de Friesinger... 46
3.4.2 Escore de Gensini... 47
3.6ANÁLISE ESTATÍSTICA... 47
3.6.1 Cálculo do tamanho amostral ... 47
3.6.2 Tratamento e testes estatísticos... 48
3.7 RECURSOS FINANCEIROS... 49
4. RESULTADOS ... 51
4.1FATORES PREDITORES DA PRESENÇA DE CALCIFICAÇÃO CORONARIANA... 54
4.2FATORES PREDITORES DA PRESENÇA DE DOENÇA CORONARIANA OBSTRUTIVA... 63
4.2.1 Efeito das variáveis analisadas sobre o Escore de Gensini... 63
4.2.2 Efeito das variáveis analisadas sobre o Escore de Friesinger... 72
4.2.3 Efeito das variáveis analisadas sobre a presença de obstrução coronariana superior a 50%... 76
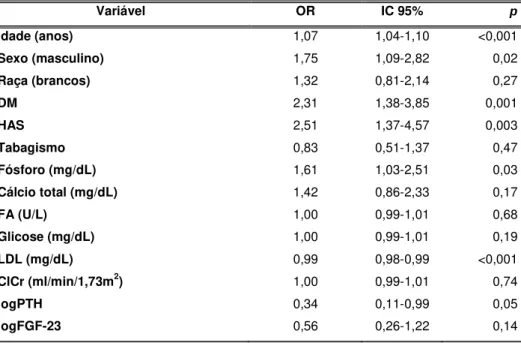
4.3ANÁLISE DAS VARIÁVEIS DETERMINANTES DO FÓSFORO SÉRICO... 81
4.4ANÁLISE DAS VARIÁVEIS DETERMINANTES DO FGF-23 SÉRICO... 84
4.5ANÁLISE DAS VARIÁVEIS DETERMINANTES DO PTH ... 86
5. DISCUSSÃO... 90
6. CONCLUSÃO ... 105
LISTA DE ABREVIATURAS E SIGLAS
25(OH)vitD 25 hidroxi-vitamina D
BMP Bone morphogenetic protein
Ca Cálcio
CaR Receptor sensível a cálcio
ClCr Clearance de creatinina
CML Células musculares lisas
CV Calcificação vascular
DAC Doença arterial coronariana
DCV Doenças cardiovasculares
DM Diabetes mellitus
DMP-1 Proteína matriz da dentina-1
DP Desvio padrão
DRC Doença renal crônica
EA Escore de Agatston
EBCT Tomografia computadorizada por emissão de elétrons
FAPESP Fundação de Amparo à Pesquisa do Estado de São Paulo
FGF-23 Fibroblast Growth Factor 23
FGFR Receptor para os Fibroblast Growth Factor´s
FMUSP Faculdade de Medicina da Universidade de São Paulo
HAS Hipertensão arterial sistêmica
HDL Lipoproteína de alta densidade
HR Hazard ratio
HVE Hipertrofia ventricular esquerda
IAM Infarto agudo do miocárdio
ICC Insuficiência cardíaca congestiva
IL Interleucina
InCor Instituto do Coração da FMUSP
INF-γ Interferon-γ
IMC Índice de massa corpórea
IQR Intervalo interquartil
KL Klotho
LDL Lipoproteína de baixa densidade
MCP-1 Monocyte chemoattractant protein 1
MDRD Modification of Diet in Renal Disease
MEPE Fosfoglicoproteína extracelular
MGP Proteína de matriz Gla
MMP Metaloproteinase de matriz
MSTC Tomografia computadorizada helicoidal
Na Sódio
NF-κB Fator Nuclear Kappa B
OPG Osteoprotegerina
OR Odds ratio
P Fósforo
PAD Pressão arterial diastólica
PAS Pressão arterial sistólica
PHEX Gene regulador do fósforo com homologias às
endopeptidases no cromossomo X
PPi Pirofosfato inorgânico
PTH Paratormônio
PTHR Receptor de paratormônio
RANK Receptor Ativador do Fator Nuclear Kappa B
RANKL Ligante do Receptor Ativador do Fator Nuclear Kappa B
sFRP-4 Secreted frizzled-related protein-4
TC Tomografia computadorizada
TGF-β Transforming growth factor β
TNF-α Fator de necrose tumoral α
VDR Receptor de vitamina D
Vit D Vitamina D
LISTA DE FIGURAS
Figura 1 – Fatores que participam da regulação da calcificação vascular...9
Figura 2 – Regulação da homeostase do fósforo...26
Figura 3 – Efeito da hiperfosfatemia sobre as células vasculares lisas...32
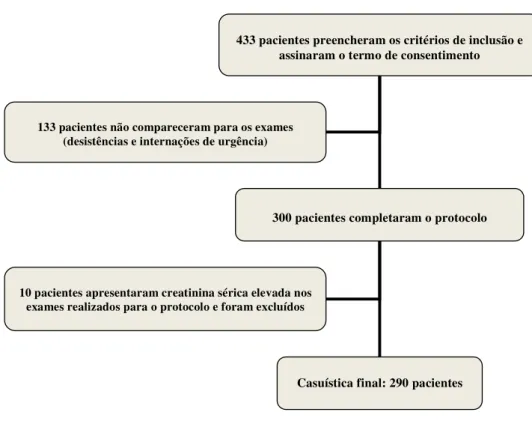
Figura 4 – Processo de inclusão dos pacientes no protocolo...51
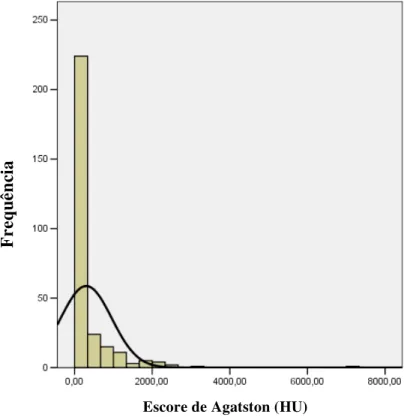
Figura 5 – Distribuição do Escore de Agatston na população estudada...54
Figura 6 – Distribuição dos níveis séricos de fósforo entre os grupos divididos de acordo com o Escore de Agatston...60
LISTA DE TABELAS
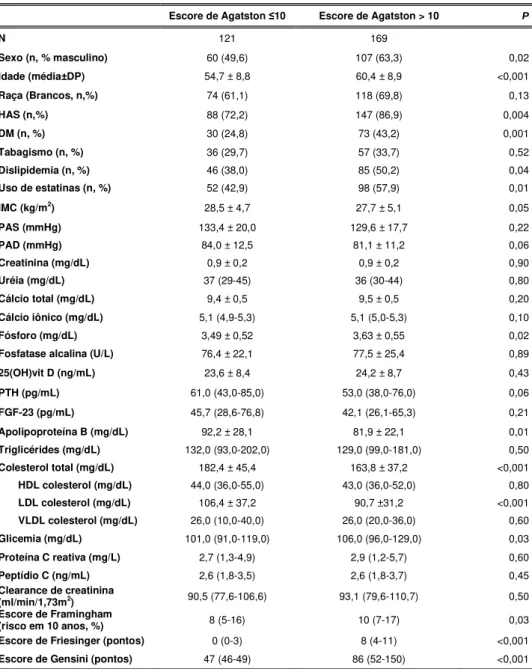
Tabela 1 - Características clínicas e escores da população estudada...52 Tabela 2 - Parâmetros laboratoriais da população estudada...53 Tabela 3 - Análise univariada dos fatores preditores do Escore de Agatston de acordo
com o modelo binomial negativo com excesso de zeros ...56 Tabela 4 - Análise multivariada dos fatores preditores do Escore de Agatston de acordo
com o modelo binomial negativo com excesso de zeros...57 Tabela 5 - Características clínicas e laboratoriais de acordo com o Escore de Agatston...59 Tabela 6 - Regressão logística univariada sobre o risco de apresentar o Escore de
Agatston maior que 10 HU...61 Tabela 7 - Modelos de regressão logística multivariada sobre o risco de apresentar o Escore de Agatston maior que 10 HU...62 Tabela 8 - Características dos grupos divididos de acordo com os tercis do Escore de
Gensini ... ...65 Tabela 9 - Correlações entre o Escore de Gensini e variáveis contínuas...66 Tabela 10 - Fatores associados ao logGensini na análise de regressão linear
univariada...67 Tabela 11 - Fatores associados ao logGensini na análise de regressão linear
multivariada...68 Tabela 12 - Modelos de regressão linear multivariada: avaliação do efeito do FGF-23 sobre o Escore de Gensini...69 Tabela 13 - Regressão logística univariada sobre o risco de apresentar Escore de Gensini no terceiro tercil vs primeiro+segundo tercis...70
Tabela 14 - Modelos de regressão logística multivariada: avaliação do fósforo como fator de risco para Escores de Gensini no terceiro tercil ...71 Tabela 15 - Modelos de regressão logística multivariada: avaliação do FGF-23 como fator de risco para Escores de Gensini no terceiro tercil...71 Tabela 16 - Características dos grupos divididos de acordo com a mediana do Escore de Friesinger...73 Tabela 17 - Regressão logística univariada sobre o risco de apresentar Escore de
Friesinger superior à mediana...74 Tabela 18 - Regressão logística multivariada: efeito ajustado do fósforo sobre o risco de
apresentar Escore de Friesinger superior à mediana...75 Tabela 19 - Regressão logística multivariada: efeito ajustado do FGF-23 sobre o risco de
apresentar Escore de Friesinger superior à mediana...76 Tabela 20 - Características dos grupos divididos de acordo com a presença de
obstrução coronariana acima de 50%...78 Tabela 21 - Regressão logística univariada sobre o risco de apresentar obstrução
coronariana superior a 50%...79 Tabela 22 - Regressão logística multivariada: efeito ajustado do fósforo sobre o risco de
Tabela 23 - Regressão logística multivariada: efeito ajustado do FGF-23 sobre o risco de
apresentar obstrução coronariana superior a 50%...80
Tabela 24 - Características dos grupos divididos de acordo com o tercil de fósforo sérico...82
Tabela 25 - Correlações entre o fósforo sérico e demais variáveis...83
Tabela 26 - Análise de regressão linear multivariada stepwise sobre o fósforo sérico...84
Tabela 27 - Características dos grupos divididos de acordo com o tercil de FGF-23 sérico...85
Tabela 28 - Correlações entre FGF-23 e demais variáveis...86
Tabela 29 - Características dos grupos de acordo com o tercil de PTH...87
Tabela 30 - Correlações entre o PTH e demais variáveis...88
Resumo
Cancela ALE. Avaliação da relação entre metabolismo mineral e doença arterial coronariana em pacientes com função renal preservada [tese] São Paulo: Faculdade de Medicina, Universidade de São Paulo; 2011.
INTRODUÇÃO: Os níveis séricos de fósforo (P) têm sido associados a doenças cardiovasculares e mortalidade em pacientes com doença renal crônica e na população geral. Estudos in vitro demonstram que altas concentrações de fósforo extracellular são capazes de induzir calcificação vascular e disfunção endotelial. O Fibroblast Growth Factor 23 (FGF-23) é um hormônio fosfatúrico e foi relacionado à presença de aterosclerose em pacientes idosos. OBJETIVO: O objetivo deste estudo foi investigar as relações entre P, FGF-23 e outros atores do metabolismo mineral e a ocorrência de doença arterial coronariana em pacientes com função renal preservada. MÉTODOS: Duzentos e noventa pacientes clinicamente estáveis com indicação de cineangiocoronariografia eletiva e clearance de creatinina superior a 60 ml/min/1.73 m2 foram submetidos à Tomografia Computadorizada Multislice para avaliação da calcificação coronariana e coleta de sangue para dosagens bioquímicas. A calcificação coronariana foi quantificada através do Escore de Agatston (EA) e os Escores de Friesinger e Gensini foram calculados para quantificar a obstrução coronariana. RESULTADOS: A média de idade dos pacientes foi 58,1± 9,3 anos, 81% eram hipertensos e 35,5% diabéticos. Os pacientes foram divididos em grupos de acordo com o EA utilizando-se como ponto de corte o valor de 10 Unidades Hounsfield (HU). O P sérico foi maior no grupo de pacientes com EA > 10 HU (3,63 0,55 vs 3,49 0,52mg/dL; p=0,019). Cada 1 mg/dL de elevação no P sérico associou-se a um aumento de 92% no risco de apresentar o EA > 10HU [Odds Ratio (OR) =1,92, CI 1,56-3,19; p=0,01]. Quando os pacientes foram divididos de acordo com a mediana do Escore de Friesinger (4 pontos), o grupo com valores superiores à mediana apresentou P sérico maior (3,6 0,5 vs. 3,5 0,6 mg/dl; p=0,04) e FGF-23 menor (mediana 40,3 pg/mL intervalo interquartil 24,1-62,2 vs. 45,7 pg/mL intervalo interquartil 31,7-76,1; p=0,01) quando comparado àquele com valores menores ou iguais a 4. Pacientes no tercil mais alto do escore de Gensini também apresentaram P sérico mais elevado que os demais (p<0,05). Nas análises de regressão logística uni e multivariadas, cada 1 mg/dL de elevação no P sérico implicou em um aumento de 74% no risco de apresentar o Escore de Friesinger superior à mediana (OR 1,74, CI 1,06-2,88; p=0,03) e o FGF-23 sérico foi preditor negativo do Escore de Friesinger (OR 0,26, CI 0,11-0,63; p=0,002) Os níveis séricos de cálcio e paratormônio não mostraram associação com a presença de doença coronariana. CONCLUSÃO: Em pacientes com suspeita de doença arterial coronariana e função renal preservada, o fósforo sérico foi preditor da presença de calcificação e obstrução coronariana e houve uma associação negativa entre o FGF-23 sérico e a presença de obstrução coronariana.
Cancela ALE. Evaluation of the relationship between mineral metabolism and coronary artery disease in patients with preserved renal function. [thesis]. Faculty of Medicine, University of Sao Paulo, SP (Brazil); 2011.
INTRODUCTION: Serum phosphorus (P) has been associated with cardiovascular diseases and mortality in chronic kidney disease patients and in the general population. In vitro studies suggest that excessive phosphorus induces vascular calcification and endothelial dysfunction. Fibroblast growth factor 23 (FGF-23) is a phosphaturic hormone and has been correlated to atherosclerosis in the community. AIM: This study intended to investigate the associations between P, FGF-23 and other mineral metabolism players and coronary artery disease in patients with preserved renal function. METHODS: Two-hundred ninety patients with a creatinine clearance higher than 60ml/min/1,73m2 undergoing elective coronary angiography were submitted to Multislice Computed Tomography in order to evaluate coronary calcification and blood was collected for biochemical analyses. Coronary artery calcification was quantified using the Agatston Score (AS). Friesinger (FS) and Gensini Scores (GS) were calcutalet to quantify coronary obstruction. RESULTS: Considering the whole population, mean age was 58.1±9.3 anos, 81% were hypertensive and 35.5% were diabetics. Patients were divided according to AS using the value of 10 Hounsfield Units (HU) as the cutoff.point. Serum phosphorus was higher in patients with an AS > 10HU when compared to the group with an AS 10 HU (3.63 0.55 vs 3.49 0.52mg/dL, p=0.019). Each 1 mg/dL of elevation in the serum phosphorus implied a 92% additional risk of presenting an AS > 10 HU [Odds Ratio (OR) =1.92, CI 1.56-3.19; p=0.01]. Patients were also divided using the median Friesinger score (4 points) as the cutoff value. Serum phosphorus was higher (3.6 0.5 vs. 3.5 0.6 mg/dl, p=0.04) and intact FGF-23 was lower (median 40.3 interquartile range 24.1-62.2 pg/mL vs. 45.7 interquartile range 31.7-76.1 pg/mL, p=0.01) in the FS > 4 group. Patientis in the higher Gensini Score tertile presented elevated serum phosphorus when compared to the other groups (p<0,05). In the uni and multivariate logistic regression analyses, a rise of 1 mg/dL of serum phosphorus carried a 74% increase in the risk of having a FS higher than 4 (OR 1.74, CI 1.06-2.88; p=0.03) and FGF-23 was a negative predictor of FS (OR 0.26, CI 0.11-0.63; p=0.002). Serum calcium and parathormone were not associated with the presence of coronary artery disease. CONCLUSIONS: In patients with suspected coronary artery disease and preserved renal function, phosphorus was predictive of both coronary artery calcification and obstruction. There was a negative association between FGF-23 and coronary obstruction.
1. INTRODUÇÃO
As doenças cardiovasculares (DCV) são consideradas a principal
causa de morte nos países desenvolvidos (1). No Brasil, 300.000 óbitos, ou
32% do total, foram atribuídos a este tipo de evento no ano de 2006 (2).
Apesar dos avanços obtidos nas últimas décadas no controle de condições
de risco tradicionalmente estabelecidas como diabetes, hipertensão e
dislipidemia, a incidência de DCV permanece estável (1,2). Paralelamente,
surgem novos fatores antes desconhecidos que parecem estar envolvidos
na fisiopatologia da doença aterosclerótica, responsável, em última
instância, pela ocorrência de eventos clínicos.
Marcadores bioquímicos do metabolismo mineral e ósseo,
principalmente o fósforo (P) sérico e seus moduladores, foram associados já
há algum tempo à morbimortalidade cardiovascular na população de
portadores de doença renal crônica (DRC) (3) e mais recentemente na
população geral (4). O mecanismo fisiopatológico subjacente a estas
associações provavelmente relaciona-se ao envolvimento do fósforo no
processo de calcificação vascular (5) que, por sua vez, está intimamente
relacionado à aterosclerose. Curiosamente, levantamentos populacionais
americanos mostram que a ingestão de fósforo é superior aos níveis
recomendados pelos órgãos reguladores e tende a aumentar
proporcionalmente ao uso de produtos industrializados que contêm
fósforo sobre diversos sistemas começam a ser estudados e possuem uma
dimensão ainda desconhecida.
1.1 Calcificação vascular
1.1.1 Calcificação vascular e morbimortalidade
A presença de calcificação vascular (CV) está associada à ocorrência
de eventos cardiovasculares, sejam eles fatais ou não. Uma metanálise
realizada em 2009 incluiu 30 estudos prospectivos que utilizaram diversos
métodos de avaliação da calcificação vascular (radiografias, ecocardiograma
e tomografia computadorizada). Após uma média de 10,1 anos de
seguimento de 218.080 pacientes, a presença de calcificação em um destes
métodos conferiu uma razão de chances (odds ratio) de 4,62 para
mortalidade por qualquer causa e 3,94 para mortalidade cardiovascular (7).
Em uma coorte que seguiu mais de 25.000 pacientes por uma média de 6,8
anos, os riscos relativos de morte aumentaram progressivamente de 2,2 a
12,5 vezes para escores de calcificação coronariana de 11 a 100 até
escores maiores que 1000 unidades Hounsfield quando comparados a
escores de 0 (8). O valor prognóstico da presença de calcificação
coronariana se mantém entre as diferentes raças (9).
As diretrizes da American Heart Association em conjunto com o
American College of Cardiology de 2007 reforçam a importância destes
doença coronariana, principalmente em pacientes assintomáticos com risco
cardiovascular intermediário e em pacientes com sintomas atípicos (10).
A calcificação vascular e a aterosclerose são processos relacionados
e ocorrem simultaneamente do ponto de vista histopatológico. Estudo que
avaliou 723 segmentos coronarianos encontrou correlação entre a presença
de cálcio e a área da placa aterosclerótica. Os autores observaram que a
ausência de calcificação não excluía a possibilidade de haver placas, mas
que a probabilidade de haver doença obstrutiva era proporcional ao
conteúdo arterial de cálcio (11). A presença da calcificação vascular também
instabiliza a placa aterosclerótica e aumenta o risco de ruptura (12).
Agatston e cols, no estudo em que descreveram o escore usado
atualmente, avaliaram 584 pacientes divididos em dois grupos: o primeiro
grupo era composto por 109 pacientes com história de infarto agudo do
miocárdio (IAM) ou mais de 50% de estenose luminal à angiografia (grupo
aterosclerose clínica) e o segundo grupo não tinha história de doença arterial
coronariana (DAC). O primeiro grupo, como esperado, apresentou maior
prevalência de placas calcificadas que o grupo sem DAC; um escore de
calcificação maior que 300 apresentou uma sensibilidade de 74% e uma
especificidade de 81% para a detecção de aterosclerose clínica (IAM ou
estenose luminal > 50%). O valor preditivo negativo do escore de cálcio igual
1.1.2 Fisiopatologia da calcificação vascular
Existem quatro tipos de calcificação cardiovascular: calcificação
aterosclerótica, calcificação da camada média arterial, calcificação valvar e
arteriolopatia urêmica calcificante(14). As duas primeiras são quase sempre
concomitantes e contribuem para a ocorrência de eventos clínicos. A
calcificação valvar compartilha fatores de risco com os dois primeiros tipos,
enquanto a arteriolopatia urêmica calcificante ocorre em pacientes
portadores de doença renal crônica e usuários de varfarin e possui
mecanismos diversos das demais categorias de calcificação (15).
A aterosclerose é um processo predominantemente inflamatório do
qual participam macrófagos, linfócitos, mediadores inflamatórios, como as
interleucinas (IL) 1, 4 e 6, interferon-γ (INF-γ) e fator de necrose tumoral-α
(TNF-α), adiponectina e a lipoproteína de baixa densidade (LDL) em sua
forma oxidada (14). Esses fatores favorecem a proliferação das células
endoteliais, apoptose celular, necrose, calcificação e a formação da placa
aterosclerótica (16). A calcificação aterosclerótica, como o próprio nome diz,
ocorre na camada íntima vascular dentro da placa aterosclerótica e sua
extensão reflete a gravidade do processo (17). Histopatologicamente
apresenta-se de forma excêntrica, com distribuição irregular e obstrução do
lúmen vascular (15). As estruturas se assemelham àquelas da ossificação
endocondral, além de haver a mineralização do núcleo lipídico com áreas
A calcificação da camada média, também conhecida como
arteriosclerose de Mönckeberg, é bastante frequente em pacientes
diabéticos, portadores de DRC e idosos (15). Esse tipo de calcificação
compartilha as características da ossificação não-endocondral ou
intramembranosa (18). O acometimento da camada média reduz a
complacência vascular, aumenta a pressão de pulso e o trabalho miocárdico
e, através destes mecanismos, contribui para a ocorrência de eventos
cardiovasculares (15).
Nosso entendimento da fisiopatologia da calcificação vascular evoluiu
muito nos últimos anos. Inicialmente considerava-se a calcificação um
evento meramente passivo, decorrente da interação físico-química entre
cálcio e fósforo e sua consequente deposição sob a forma de hidroxiapatita
na parede vascular. Recentemente, porém, várias evidências sugerem que
se trata de um processo ativo, regulado biologicamente (18). Sabe-se que
ocorre uma espécie de osteogênese dentro da parede vascular, que pode
ser endocondral ou membranosa, e que isto só é possível devido à
transformação fenotípica das células musculares lisas vasculares em células
com características semelhantes às dos osteoblastos (18). Estas passam a
expressar fatores de regulação do tecido ósseo e proteínas estruturais
ósseas, como o fator de transcrição conhecido como Cbfa-1, responsável
pela diferenciação osteoblástica, a fosfatase alcalina, a osteopontina, as
BMP´s (bone morphogenetic proteins), a proteína de matriz Gla (MGP),
RANK/RANKL (Receptor Ativador do Fator Nuclear Kappa B/Ligante do
(17). Além disto, células semelhantes a osteoclastos também foram
observadas nas placas calcificadas e teriam a função de reabsorver parte do
mineral depositado (18).
A regulação do processo de calcificação vascular é feita através do
balanço entre inibidores e ativadores da transição fenotípica que resulta na
produção de células semelhantes aos osteoblastos. As BMP´s 2 e 4
participam ativamente via Msx2 e Wnt (19) e sua produção é estimulada pela
hiperglicemia (20). A presença de concentrações elevadas de fósforo no
meio extracelular também age sobre as células musculares lisas dos vasos,
efeito que será descrito com mais detalhes posteriormente (5, 21).
Entre os inibidores deste processo destacam-se a MGP e a fetuína,
que evitam a precipitação dos cristais de hidroxiapatita ligando-se a eles e,
assim, facilitam sua eliminação pelas células (19). Ambas são ainda
antagonistas do efeito osteogênico das BMP´s (19).
Estudos sobre a regulação da calcificação tecidual têm destacado o
papel de moléculas pertencentes à família do TNF: o RANK, seu ligante
(RANKL) e a OPG. O RANKL, uma proteína transmembrana, está presente
na superfície dos osteoblastos. Quando o RANKL se liga ao RANK presente
na superfície dos precursores dos osteoclastos, estas células se diferenciam
e passam a reabsorver o tecido ósseo. Se, por outro lado, a OPG presente
no meio extracelular se liga à RANKL, não ocorre a diferenciação
osteoclástica e a reabsorção óssea é inibida (22). Assim, o balanço entre
RANKL-OPG influencia a remodelação e a massa óssea (23). Além disso,
imunológicas (24) e parece participar da regulação da calcificação vascular
(22, 23). Ratos knock-out para OPG desenvolvem, além de redução de
massa óssea, calcificação arterial média difusa (23).
Os níveis séricos de OPG já foram correlacionados com a gravidade
da lesão coronariana observada à angiografia (24) e com o aumento de
eventos cerebrovasculares (25). Também já foi demonstrada associação
entre níveis elevados de OPG e progressão acelerada de calcificação
vascular em pacientes dialíticos (26). Ainda não se sabe se a elevação da
OPG é parte do mecanismo que leva à calcificação vascular ou uma
tentativa de auto-regulação e limitação do processo.
A complexidade das vias responsáveis pela calcificação vascular
pode ser estimada pelo grande número de mediadores inflamatórios,
hormônios e outras proteínas sabidamente envolvidas, conforme demonstra
IFN-γ:Interferon γ, IL-6: Interleucina 6, MMP: Metaloproteinase de matriz, TNF: Fator de necrose tumoral, HDL:Lipoproteína de alta densidade, RANKL: Ligante do receptor ativador do fator nuclear κ B , OPG:
Osteoprotegerina, PPi: Pirofosfato inorgânico, vit D: Vitamina D, PTH: Paratormônio, FGF-23: Fator de crescimento de fibroblastos-23, BMP: Proteína morfogenética óssea, MGP: Proteina Gla de matriz
FONTE: Adaptado de Sage e cols (18) FGF-23
Klotho
↓ Vitamina K
Metabolismo mineral
↑BMP2/4
↓BMP7 MGP Runx2, Sox9 Wnt-βcatenina Colágenos tipo I e II
Transformação fenotípica Inflamação IFN-γ IL-6 MMP´s Macrófagos Degradação da elastina
Cristais minerais
Fetuína A Ca Leptina TNF HDL RANKL OPG
P PPi Vit D Insulina Glicose PTH
1.1.3 Métodos de imagem para avaliação da calcificação vascular
Os principais métodos de imagem utilizados para avaliar a presença
de calcificações vasculares são: radiografia simples, ultrassonografia e
tomografia computadorizada (27). Nenhum deles, no entanto, é capaz de
localizar a camada vascular em que se encontra a calcificação, ou seja, se
na íntima ou na camada média.
Apenas calcificações extensas são visualizadas na radiografia
comum, o que torna este método pouco sensível. A ultrassonografia é
bastante útil no diagnóstico de calcificações valvares. Seu uso para a
detecção de depósitos vasculares, no entanto, é limitado, uma vez que o
ultrasom intravascular, apesar de muito sensível e específico (28), é invasivo
e indisponível na maioria dos centros.
A tomografia computadorizada (TC), inicialmente realizada por
emissão de elétrons (EBCT) e mais recentemente a TC helicoidal (MDTC),
ganha progressivamente mais espaço e tornou-se o método mais popular na
avaliação da calcificação vascular coronariana. Isto foi possível devido ao
desenvolvimento de escores como o de Agatston (13), capazes de fornecer
uma quantificação da área calcificada que permite acompanhar a progressão
da doença e avaliar os efeitos de diferentes intervenções. Já foi
demonstrada correlação entre a área de cálcio detectada pela EBCT e a
área da placa aterosclerótica na análise histopatológica (29). Além do seu
valor preditivo em relação a eventos clínicos, o escore de calcificação
cintilografia miocárdica (30) e de estenose coronariana significativa
detectada tanto à cineangiocoronariografia (31, 32) quanto à
angiotomografia (33).
1.2 Metabolismo mineral
1.2.1 Homeostase do fósforo
O fósforo (P) é um elemento fundamental em diversos processos
celulares, principalmente de geração e transferência de energia e
mineralização óssea. Participa ainda da estrutura da membrana celular, DNA
e RNA e vários outros mediadores intracelulares. No entanto, dos cerca de
600 gramas de fósforo presentes no organismo humano, 85% encontram-se
depositados na matriz extracelular óssea (34).
A concentração sérica de fósforo em adultos com função renal normal
varia de 3,0 a 4,5 mg/dL e apresenta um ritmo circadiano próprio, com
valores mais baixos pela manhã e mais elevados à noite, além de variar de
acordo com sexo, idade, ingesta proteica e taxa de crescimento (34). O P
está presente no soro principalmente sob a forma inorgânica (PO4) e pode
ser dividido em três frações: iônica (55%), ligada a proteínas (10%) e ligada
a cátions como sódio, cálcio e magnésio (35%) (35). A homeostase do
fósforo depende da absorção intestinal, da excreção urinária e do balanço
O transporte de fósforo através das membranas das células intestinais
e tubulares é feito por co-transportadores sódio/fósforo (Na/Pi) regulados
biologicamente por hormônios.
O fósforo é encontrado na maioria dos grupos de alimentos e
aproximadamente 800-1400 mg de fósforo são ingeridos ao dia. Destes,
60% são absorvidos no intestino delgado, principalmente no jejuno. A
absorção de P no intestino ocorre tanto por mecanismo difusional, passivo,
pela via paracelular, quanto de forma ativa, mediada pelo cotransportador
Na/Pi tipo 2b e é regulada pelo calcitriol (34).
Aproximadamente 85% do fósforo sérico é filtrado através dos
glomérulos e apenas 12 a 13% são excretados pela urina. Mais de 80% da
reabsorção tubular de fósforo é realizada no túbulo proximal pelos
cotransportadores Na/Pi tipos 2a e 2c presentes na membrana apical das
células tubulares. Hipercalcemia, depleção do volume extracelular e redução
da ingestão de fósforo aumentam a reabsorção proximal de fósforo,
enquanto hipocalcemia, PTH e FGF-23 promovem a fosfatúria pela redução
do número de cotransportadores Na/Pi na membrana celular (35).
1.2.1.1 Paratormônio
O PTH é produzido pelas células principais das glândulas
paratireóides e o principal estímulo para sua produção é a redução da
concentração extracelular de cálcio. Além da hipocalcemia, outros estímulos
são o aumento do fósforo extracelular e a redução na concentração sérica
séricas de fósforo e cálcio agindo principalmente no intestino, rins e tecido
ósseo. (35)
A molécula do paratormônio é composta por 84 aminoácidos. Além da
forma intacta, diversos fragmentos são produzidos tanto pelas paratireóides
quanto pelo metabolismo sistêmico do PTH. O fragmento aminoterninal
contém 34 aminoácidos (1-34) e é responsável por suas funções biológicas
conhecidas, pois é a porção que se liga ao receptor específico para PTH
(PTHR1). Os fragmentos carboxiterminais englobam os últimos 50
aminoácidos e não são capazes de ativar o PTHR1. Por este motivo,
tradicionalmente não se atribuía nenhuma função biológica a estes
peptídios, considerados inativos. Entretanto, foi demonstrada a presença de
um receptor específico para os fragmentos C-terminais e, em algumas
circunstâncias de acúmulo destes fragmentos, como na insuficiência renal,
sua importância biológica pode ser maior do que se pensava anteriormente
(36).
A administração de PTH promove a liberação de cálcio e fósforo do
osso pelos osteoclastos e, em situações de exposição prolongada ao PTH,
como no hiperparatireodismo, ocorre um aumento no número de
osteoclastos e da reabsorção óssea. No rim, o paratormônio exerce três
funções principais: estimula a reabsorção de cálcio e a síntese de calcitriol e
inibe a reabsorção de fósforo (35).
A ação fosfatúrica do PTH ocorre através da inibição da reabsorção
do fósforo tanto no túbulo proximal quanto no distal. O mecanismo de ação é
da produção e internalização dos cotransportadores Na/Pi 2a (e
provavelmente 2c), diminuindo a reabsorção tubular (35).
O cálcio extracelular é o principal regulador da síntese de PTH e o
faz através do receptor sensível a cálcio (CaR). A presença de hipocalcemia
estimula a produção e secreção do PTH, além de promover a proliferação
das células da paratireóide. A relação PTH-Ca iônico pode ser representada
por uma curva signoidal inversa que reflete a grande resposta do PTH a
pequenas variações do cálcio sérico. As células paratireóides são capazes
de aumentar a secreção de PTH em segundos, o que ajuda a manter os
níveis de cálcio sérico dentro de uma estreita faixa de referência (35).
O fósforo, por outro lado, promove a produção do PTH e,
provavelmente, também sua secreção. O mecanismo sensor de fósforo nas
células paratreóides, no entanto, ainda não está esclarecido (35). O calcitriol
age ativando o receptor nuclear para vitamina D (VDR) e reduz a secreção
de PTH.
O Fibroblast Growth Factor 23 (FGF-23) influencia de diversas formas
a regulação do PTH. Antes considerado um regulador positivo da produção
de PTH por seu efeito inibitório sobre a síntese de vitamina D ativa (37), hoje
se sabe que o FGF-23 reduz a produção de PTH agindo diretamente sobre
as paratireóides através de seu receptor (38). Indiretamente, a fosfatúria
resultante do aumento de FGF-23 pode reduzir a concentração sérica de
fósforo e inibir a secreção do PTH. Por outro lado, se os níveis de FGF-23
mantiverem-se cronicamente elevados, há uma diminuição significativa da
a depender da situação considerada, a interação FGF-23-paratireóide pode
variar e resulta do balanço entre suas ações diretas e indiretas. (39).
1.2.1.2 Vitamina D
Os seres humanos obtêm vitamina D basicamente através da
produção endógena de vitamina D3 pela pele e, em menor grau, pela
ingestão de alimentos que contêm naturalmente os precursores D3 ou D2 ou
que foram enriquecidos artificialmente. Na pele, a vitamina D3 é gerada pela
conversão fotolítica do 7-dehidrocolesterol induzida pela exposição aos raios
UV. Estas formas não possuem atividade biológica e, para que sejam
capazes de ativar o receptor de vitamina D (VDR), precisam sofrer duas
hidroxilações que ocorrem no fígado e no rim (35).
No fígado, as vitaminas D2 e D3 sofrem a primeira hidroxilação, no
carbono 25, reação que produz a 25-hidroxi-vitaminaD (25(OH)vitD), que
circula no sangue ligada à proteína carreadora de vitamina D e outras
proteínas. O nível sérico da 25(OH)vitD é utilizado clinicamente como
indicador do status de vitamina D, uma vez que é proporcional à exposição
solar e ingesta de vitamina D2 e D3 (35, 40).
O último passo na ativação da vitamina D é a segunda hidroxilação,
desta vez no carbono 1, catalisada pela enzima 1α-hidroxilase presente
principalmente nas células tubulares renais. O resultado é a formação da
1,25(OH)2vitD ou calcitriol, que é responsável pelos efeitos conhecidos da
menor que o de 25(OH)vitD, da ordem de pg/mL, o que torna sua dosagem
pouco prática do ponto de vista clínico. Ainda nos túbulos renais existe uma
segunda enzima que utiliza a 25(OH)vitD como substrato, a 24-hidroxilase,
capaz de produzir um metabólito inativo, a 24,25(OH)2vitD (35).
A atividade da 1α-hidroxilase é amplamente regulada. PTH e FGF-23
possuem efeitos opostos sobre a produção renal de calcitriol, estimulando e
inibindo o processo, respectivamente. A hipocalcemia aumenta a atividade
da 1α-hidroxilase diretamente no rim e indiretamente através do estímulo à
produção de PTH pelas paratireóides. A sobrecarga de fósforo, por outro
lado, reduz o calcitriol sérico tanto indiretamente pelo aumento da secreção
de FGF-23 quanto de forma direta (35, 41).
O calcitriol pode ser considerado um hormônio, pois age em várias
células do organismo através de um receptor nuclear próprio, o VDR. Sua
função mais importante parece ser auxiliar na manutenção dos níveis séricos
de cálcio dentro dos limites fisiológicos através da integração entre tecido
renal, onde é produzido, e paratireóides, intestino e osso (35).
No intestino, o calcitriol aumenta o transporte de cálcio e fósforo. Nas
paratireóides, inibe a produção de PTH em um mecanismo de
retroalimentação, aumenta a sensibilidade celular ao cálcio e à própria
vitamina D ativa promovendo a expressão dos receptores CaR e VDR e
impede a proliferação celular. No rim, ocorre o aumento da produção dos
canais TPRV5, responsáveis pelo transporte de cálcio pelos túbulos distais
em resposta ao calcitriol. Por fim, a presença de níveis adequados de
apropriado entre os processos de reabsorção e formação óssea, além de
aumentar a expressão de FGF-23 pelas células ósseas em um mecanismo
de feedback (35, 42) .
Na presença de níveis baixos de 25(OH)vitD e, consequentemente,
de 1,25(OH)2vitD, o cálcio sérico cai e ativa os CaR das glândulas
paratireóides, aumentando a produção de PTH. Este mecanismo fisiológico
levou os primeiros estudiosos do assunto a considerar inadequados níveis
séricos de 25(OH)vitD abaixo dos quais o PTH se eleva, o que ocorre a
partir dos 30 ng/mL. A deficiência de vitamina D é definida como níveis
inferiores a 20 ng/mL enquanto insuficiência éd definida como níveis entre
20 e 30 ng/mL (40). Os hábitos alimentares ocidentais e o uso cada vez mais
freqüente de bloqueadores solares tornaram a deficiência de vitamina D um
problema crescente em diversas populações (43), incluindo a população
brasileira (44).
A conversão da 25(OH)vitD para 1,25(OH)2D não acontece apenas
nas células renais, pois vários outros tecidos expressam a 1α-hidroxilase. O
receptor da vitamina D também foi encontrado em outras células que não
eram consideradas, a princípio, alvos da ação do calcitriol, como
cardiomiócitos, células musculares lisas vasculares e várias células do
sistema imune (41). Assim, o calcitriol não se presta apenas ao controle do
metabolismo mineral, mas participa também de outros processos, como
controle da proliferação celular e imunomodulação (45).
de neoplasias como o câncer de cólon (46) e mama (47), embora esta
associação não tenha sido demonstrada para outros tipos de tumores, como
o câncer de próstata (48). Além disso, estoques inapropriados de vitamina D
favorecem o aparecimento de doenças auto-imunes como diabetes tipo 1
(49), esclerose múltipla (50), artrite reumatóide, lupus eritematoso sistêmico
e doença de Chron (51). Os efeitos da vitamina D sobre o sistema
cardiovascular serão descritos adiante.
1.2.1.3 Fibroblast Growth Factor 23
O Fibroblast Growth Factor-23 (FGF-23) é um hormônio identificado
recentemente que pertence à família dos FGF´s, mais especificamente à
subfamília FGF-19, e é composto por 251 aminoácidos (52). O aumento da
atividade do FGF-23, seja por superexpressão do gene responsável ou pela
diminuição de seu metabolismo, é responsável pelo aparecimento de
condições que cursam com hipofosfatemia secundária ao aumento da
excreção renal de fósforo, como o raquitismo hipofosfatêmico autossômico
dominante (ADHR) (53) e a osteomalácia induzida por tumor (TIO) (52). A
partir desta observação, pesquisas in vitro e in vivo progressivamente
esclareceram o papel do FGF-23 no metabolismo do fósforo e da vitamina D
e sua descoberta foi fundamental para a compreensão atual da homeostase
do fósforo. Assim, o FGF-23 parece ser o principal membro da família das
“fosfatoninas”, termo cunhado para designar a classe de substâncias
(sFRP-4) e pela fosfoglicoproteína extracelular (MEPE), entre outras (54, 55).
O FGF-23 é produzido principalmente no tecido ósseo pelos
osteoblastos e osteócitos em resposta ao aumento na concentração de
fósforo extracelular e também em menor quantidade por outros tecidos como
fígado e rim (56). Além do fósforo sérico, o calcitriol estimula a produção do
FGF-23 de maneira dose-dependente (57, 58) e o tratamento com calcitriol
aumenta os níveis séricos de FGF-23 (59, 60).
A resposta ao aumento do conteúdo de fósforo na dieta foi
inicialmente demonstrada tanto em animais saudáveis (61,62) quanto na
presença de insuficiência renal (63). Em humanos, o FGF-23 se eleva
apenas após uma sobrecarga mantida de fósforo na dieta. Nishida e cols
não conseguiram demonstrar alteração aguda em seu nível sérico após a
ingesta de uma única refeição rica em fósforo (64). Já Burnett e cols (65)
estudaram 66 indivíduos saudáveis randomizados para receber dietas com
diferentes concentrações de fósforo por 5 dias. O FGF-23 intacto sérico
aumentou em média 23% após a sobrecarga dietética de fósforo e houve
redução de 9% após restrição de fósforo. Provavelmente um aumento mais
prolongado na ingestão de fósforo seja necessário para estímular à
produção de FGF-23.
Ao contrário dos demais componentes da família dos FGF´s, o
FGF-23 atua de maneira sistêmica através de receptores, principalmente o
FGFR1, que possuem o Klotho (KL) como cofator (66). Seu órgão alvo
principal é o rim, onde aumenta a excreção de fósforo. O local exato de ação
diminuir a reabsorção de fósforo no túbulo proximal através da inibição da
expressão dos cotransportadores Na/Pi2a e Na/Pi2c (67), ativando a via da
MAP-quinase (68). Ainda no túbulo proximal, inibe a enzima 1α-hidroxilase,
responsável pela produção da forma ativa da vitamina D e aumenta a
atividade da 24-hidroxilase (69).
Além de agir no rim, o FGF-23 também inibe a produção de PTH
pelas paratireóides, cujas células expressam FGFR e klotho (37,38). Esta
ação direta do FGF-23 é antagônica ao efeito indireto causado pela
supressão da 1-α-hidroxilase; o resultado final sobre a concentração de PTH
depende, portanto, do balanço entre estas duas ações aparentemente
contrárias. Por outro lado, o FGF-23 também parece ser regulado pelo PTH.
Neves e cols (70) demonstraram que, em animais nefrectomizados, os níveis
de FGF-23 eram maiores naqueles que receberam altas doses de PTH,
sendo esse o primeiro estudo a revelar um efeito independente do PTH nos
níveis de FGF-23, achados que foram posteriormente confirmados em ratos
sem disfunção renal (71). O PHEX (gene regulador do fósforo com
homologias às endopeptidases no cromossomo X) e a DMP-1 (proteína
matriz da dentina-1) agem inibindo a produção de FGF-23 pelo osteócitos
por mecanismos ainda incertos (72).
Animais deficientes em FGF-23 apresentam, além de hiperfosfatemia,
alterações ósseas, calcificações ectópicas e hipoglicemia (73). Estas
alterações são revertidas após a deleção do receptor de vitamina D (VDR),
sugerindo que algumas das características destes animais são resultantes
sensibilidade à insulina aparentemente são mediados pela produção de
vitamina D, corroborando a hipótese da existência de uma interação entre a
regulação do metabolismo ósseo e dos níveis glicêmicos.
Em humanos, a deficiência de FGF-23 leva à hiperfosfatemia e
calcinose tumoral, condição debilitante por cursar com grandes e
deformantes calcificações ectópicas, principalmente em articulações (75).
A concentração sérica de FGF-23 se eleva precocemente com a
queda do ritmo de filtração glomerular (76, 77) e em estágios mais
avançados da DRC, atinge níveis até 1000 vezes maiores que os
encontrados em indivíduos com função renal preservada (78). As possíveis
causas de aumento do FGF-23 nesta população são o estímulo da
hiperfosfatemia, a alteração de seu metabolismo e a menor excreção renal.
O aumento do FGF-23 sérico no estágio 3 da DRC acontece antes
mesmo que sejam observadas alterações laboratoriais de cálcio, fósforo,
PTH ou calcitriol (76, 77). Em pacientes com clearance de creatinina acima
de 30 ml/min/1,73m2 o FGF-23 apresenta correlação inversa com a
1,25(OH)2vitD e reabsorção tubular máxima de fósforo, o que já não ocorre
em situações de perdas mais acentuadas de função renal (79). Estes
achados mudaram paradigmas e permitiram uma nova interpretação da
tradicional teoria do trade-off (80). Nos estágios precoces da DRC a
elevação do FGF-23 consegue aumentar a excreção de fósforo e exerce
efeito direto de inibição da produção do PTH. A produção de calcitriol, no
entanto, é reduzida, o que estimula as paratireóides. À medida que néfrons
a fosfatemia. Os níveis elevados de fósforo passam a ser mais um estímulo
ao desenvolvimento do hiperparatireoidismo secundário (81) e o efeito
inibidor sobre o PTH é perdido, pois as células paratireóides tornam-se
progressivamente menos sensíveis ao FGF-23 devido à redução na
expressão dos receptores para FGF e de klotho em glândulas hiperplásicas
(82). Podemos concluir, portanto, que durante a fase precoce de perda renal
o FGF-23 protege o organismo da hiperfosfatemia e do hiperparatireoidismo,
enquanto em estágios mais avançados de DRC passa a se comportar como
marcador de perda de massa renal e da sobrecarga de fósforo.
O FGF-23 basal medido em 103 pacientes dialíticos foi fator preditor
para o surgimento de hiperparatireoidismo secundário após dois anos de
seguimento (83) e em outro estudo os níveis de FGF-23 foram capazes de
predizer a resposta dos níveis de PTH à terapia com calcitriol (84). Além
disto, o tipo de quelante de fósforo utilizado também altera os níveis de
FGF-23. Quelantes que não contêm cálcio reduzem de maneira mais significativa
o FGF-23 sérico quando comparados aos quelantes à base de cálcio tanto
em pacientes dialíticos (60) quando naqueles em tratamento conservador
(85). O impacto de possíveis intervenções sobre o FGF-23, no entanto,
ainda é desconhecido.
Os níveis séricos do FGF-23 são mensurados por ensaios
imunoenzimáticos (ELISA) capazes de detectar apenas a molécula inteira
(FGF-23 intacto) ou a molécula inteira e o fragmento C-terminal, resultado da
quebra da molécula. A utilidade dos dois tipos de ensaio varia de acordo
hipofosfatêmicas, o ensaio que mensura apenas a molécula intacta foi mais
sensível para o diagnóstico (86). Entretanto, não existem estudos
comparando os ensaios na população geral sem disfunção renal.
1.2.1.4 Klotho
O gene klotho, cujo nome se refere a uma das deusas da mitologia
grega que controlam o fio da vida, foi identificado acidentalmente em 1997
como uma mutação presente em uma linhagem de camundongos que
apresentavam um fenótipo de envelhecimento precoce (87). Os animais com
expressão defeituosa da proteína klotho apresentavam alterações
características do processo de senescência, como tempo de vida reduzido,
atrofia muscular, osteopenia, calcificações vasculares e enfisema pulmonar
(87), que posteriormente foram reconhecidas também em animais knockout
para FGF-23 (73). Por outro lado, demonstrou-se que o aumento da
expressão do gene klotho era capaz de prolongar o tempo de vida (88), o
que sugeriu seu envolvimento direto na regulação da sobrevida e do ritmo de
envelhecimento.
O gene klotho codifica uma proteína transmembrana que possui uma
porção extracelular passível de ser secretada na circulação e é codificado
apenas em alguns tecidos, como o rim, mais especificamente nos túbulos
contorcidos distais, e no plexo coróide. Sua forma livre, no entanto, pode ser
responsável pela ação em outros tecidos. A presença do klotho na
por seus receptores, principalmente os FGF-23, FGF-21 e FGF-19/15,
responsáveis, respectivamente, pela regulação do metabolismo mineral,
metabolismo energético e produção da bile (89). Como a proteína Klotho é
essencial para a ligação do FGF-23 ao seu receptor, sua presença apenas
em algumas células confere seletividade à ação deste hormônio.
Além de alterações relacionadas ao envelhecimento, animais
deficientes em klotho desenvolvem alterações no metabolismo energético e
mineral. (90) A falta de sinalização do FGF-23 nas células tubulares renais,
por exemplo, leva à produção exagerada de calcitriol e ao aumento da
reabsorção tubular de fósforo, com conseqüente hiperfosfatemia (90).
O papel do klotho na homeostase do cálcio é mais complexo e
envolve vários mecanismos. Animais knockout para klotho cursam com
hipercalcemia secundária aos níveis séricos elevados de calcitriol e
conseqüente aumento na reabsorção intestinal de cálcio e hipercalciúria
devido à hipercalcemia (90). No entanto, estudos recentes demonstram que
o klotho aumenta a quantidade de canais TPRV5 nos túbulos distais, e sua
ausência provoca um defeito na reabsorção tubular de cálcio, adicionando
um mecanismo alternativo às alterações encontradas nestes animais (91).
As calcificações vasculares presentes neste modelo acometem a camada
média dos vasos e se assemelham à arteriosclerose de Monckeberg
presente em pacientes idosos, diabéticos e portadores de DRC (87).
Os fenótipos decorrentes da ausência dos genes responsáveis pela
produção tanto do klotho quanto do FGF-23 podem ser atribuídos
do gene responsável pelo cotransportador Na/Pi 2a corrige a fosforemia e o
envelhecimento precoce (92). Por outro lado, uma dieta rica em fósforo
resgata as alterações fenotípicas nestes animas duplo knockout para klotho
e Na/Pi 2a (92). Em outras palavras, neste modelo, o fósforo induz ao
envelhecimento.
O excesso de calcitriol também colabora para a senescência, pois a
ablação do receptor de vitamina D (VDR) (74) ou da 1α-hidroxilase (93) é
capaz de reverter as alterações encontradas nestes animais. Este efeito, no
entanto, é provavelmente indireto e mediado pela hiperfosfatemia induzida
pelo excesso de calcitriol.
Em humanos, polimorfismos do gene klotho já foram associados à
osteoartrite (94), densidade mineral óssea reduzida (95), doença arterial
coronariana (96, 97, 98) e seus fatores de risco como níveis de HDL e
pressão arterial (99) e LDL e ácido úrico (100), além de longevidade (99) na
população geral e mortalidade em pacientes em hemodiálise (101).
A Figura 2 ilustra resumidamente o controle da homeostase do
Figura 2 – Regulação da homeostase do fósforo
Pit-2: Transportador de fósforo inorgânico-2, PTHR1: receptor para PTH-1 FONTE: Adaptado de Bergwitz e cols (72).
1.2.2 Homeostase do cálcio
O cálcio é o quinto elemento mais abundante no organismo humano,
que contém em média 1000 gramas de cálcio. Deste total, mais de 99% se
encontram no osso sob a forma de cristais de cálcio, principalmente
hidroxiapatita, o que proporciona rigidez à estrutura óssea. O cálcio que não
se encontra imobilizado perfaz menos de 10 g. No entanto, é responsável
por vários processos essenciais como a sinalização intra e extracelular, a
contração muscular e a transmissão dos impulsos nervosos. Os níveis
Osso Paratireóides
P dieta
Intestino Rins
normais de cálcio total vão de 8,8 a 10,4 mg/dL e incluem íons livres (51%),
ligados a proteínas como albumina e globulina (40%) e a outros íons (9%).
Os níveis de cálcio iônico são regulados rigidamente e se mantêm entre 4,4
e 5,4 mg/dL. O balanço de cálcio depende da ingestão, da absorção
intestinal e da excreção renal de cálcio e varia de acordo com a faixa etária e
sexo do indivíduo (33, 102).
A homeostase do cálcio depende basicamente de dois hormônios
reguladores: o paratormônio (PTH) e o calcitriol. Além disso, os receptores
para estes hormônios (PTHR e VDR) e o receptor sensível a cálcio (CaR)
também são fundamentais na manutenção de níveis séricos adequados.
Quando há redução dos níveis séricos de cálcio, o CaR presente nas
glândulas paratireóides deixa de ser ativado, o que induz a produção de
PTH. O PTH secretado aumenta a reabsorção óssea e liberação de cálcio
pelo osso, além de estimular a reabsorção de cálcio e a produção de
calcitriol pelos túbulos renais. O calcitriol contribui para a normalização dos
níveis de cálcio agindo no epitélio intestinal no sentido de aumentar a
reabsorção a partir do lúmen (33, 102).
1.3 Metabolismo mineral e mortalidade/morbidade cardiovascular
1.3.1 Fósforo e doenças cardiovasculares
A associação entre fósforo e morbimortalidade foi demonstrada há
alguns anos, inicialmente na população de portadores de DRC em
confirmada em diversas outras coortes populacionais (103). Análise dos
dados de pacientes originários de 12 países participantes do DOPPS
(Dialysis Outcomes and Practice Pattern Study) mostrou que níveis séricos
de fósforo entre 6,1 e 7,0 mg/dL e acima de 7,1 mg/dL no início do estudo
conferiam um aumento no risco de morte por qualquer causa de 18% e 43%,
respectivamente, quando comparados a níveis de fósforo na faixa de
referência (entre 3,6-5,0 mg/dL). Para o risco de morte por causas
cardiovasculares o aumento de risco foi de 61% e 81%, respectivamente
(103). Níveis muito reduzidos de fósforo também foram associados à
mortalidade aumentada, refletindo talvez o pior estado nutricional deste
pacientes. Sabe-se ainda que concentrações elevadas de fósforo estão
associadas à presença de calcificações vasculares, valvares e de tecidos
moles nesta população (104) e em fases mais precoces da disfunção renal
(105).
Posteriormente, estas observações foram estendidas à população
geral e, surpreendentemente, níveis de P ainda nos limites superiores da
normalidade mostraram-se associados à maior morbimortalidade (106). O
primeiro estudo a replicar estes achados na população sem disfunção renal
foi uma análise post hoc do estudo CARE (Cholesterol And Recurrent
Event), cujo objetivo original era avaliar o benefício do uso de pravastatina
em pacientes com história prévia de infarto agudo do miocárdio (106). Tonelli
et al (106) estudaram 4.127 pacientes e após 5 anos de seguimento foi
encontrada associação positiva e gradual entre o fósforo sérico basal e
1,02-1,58; P=0,03). Pacientes com P sérico superior a 4 mg/dL
apresentaram um HR aumentado para o surgimento de insuficiência
cardíaca e eventos coronarianos não-fatais (1,43 e 1,50, respectivamente)
quando comparados àqueles com P entre 2,5 e 3,4 mg/dL. Apenas 5,2% dos
pacientes apresentavam hiperfosfatemia. As associações permaneceram
inalteradas após a exclusão de pacientes com clearance de creatinina
estimado inferior a 60 ml/min/1,73m2.
Mais recentemente, uma análise derivada da coorte de Framingham
incluindo mais de 3000 pacientes com tempo médio de seguimento de 16
anos confirmou a associação entre os quartis mais altos de P sérico basal e
mortalidade cardiovascular no período observado (4).
Os mecanismos através dos quais o fósforo aumenta a mortalidade e
a incidência de eventos cardiovasculares ainda não estão estabelecidos,
mas é provável que esta associação seja consequência de sua participação
na patogênese da calcificação vascular e do processo de aterosclerose (107,
108).
Se a relação entre P e eventos parece cada vez mais clara, pelo
menos dois estudos demonstraram associação entre P e gravidade das
lesões coronarianas à angiografia. Estudo de 1997 realizado em indivíduos
com função renal normal e suspeita de doença coronariana mostrou uma
correlação positiva entre os níveis de fósforo e o número de vasos
acometidos observados na angiografia (109). Neste estudo, um aumento de
de 3,01 para doença coronariana significativa. No entanto, não houve
correlação entre os níveis de cálcio e a gravidade das lesões. A termo de
comparação com outros fatores de risco já descritos para doença
coronariana, os autores observaram que as razões de chance obtidas para
hipercolesterolemia e tabagismo foram, respectivamente, 2,69 e 2,29 (109).
Já Rasouli et al (110), estudando 260 pacientes portadores de doença
coronariana, encontraram correlação não só entre fósforo e gravidade das
lesões coronarianas como também entre o grau de lesão arterial e as
concentrações de cálcio e produto CaxP.
O fósforo também parece ser fator de risco para o surgimento de
calcificações vasculares na população geral. Em uma coorte de 3015
pacientes adultos jovens (média de idade 25 anos) partipantes do estudo
CARDIA e seguidos durante 15 anos houve associação entre o nível sérico
de P basal e o aparecimento de calcificações coronarianas avaliadas por
tomografia computadorizada (111). Em outra coorte que incluiu pacientes
com média de idade mais elevada (48 anos) seguidos por 6 anos, o fósforo
sérico na primeira avaliação foi preditor tanto do surgimento de calcificação
coronariana quanto da sua progressão durante o seguimento (OR 1,54, p
=0,002), embora não tenha havido correlação entre os dois parâmetros
1.3.1.1 Fósforo e calcificação vascular
O fósforo participa do processo de calcificação vascular induzindo a
transformação fenotípica das células musculares lisas dos vasos em
osteoblastos. Jono et al (5), estudaram células musculares lisas humanas
extraídas da aorta expostas a diferentes concentrações de fósforo e
observaram que quando essas células eram expostas a concentrações
elevadas de fósforo no meio (>2mmol/L), ocorria uma mudança no padrão
da expressão gênica celular. Ao invés de marcadores musculares, como a
alfa-actina, estas células passavam a produzir marcadores tipicamente
osteocondrogênicos como o fator transcripcional Runx2 (também conhecido
como Cbfa). Esta mudança fenotípica conduzia à calcificação acentuada do
meio extracelular, com deposição de colágeno e cálcio. Foi demonstrado
ainda que o efeito do fósforo é dependente do co-transportador sódio-fosfato
tipo III (Pit-1) presente na membrana celular (5). A figura 3 ilustra o
processo.
Além disso, a hiperfosfatemia induz a elevação de PTH, que tem
efeito deletério sobre os cardiomiócitos (113) e participa da regulação da
calcificação vascular (114). Este mecanismo explicaria a relação entre
calcificação vascular e fósforo sérico na vigência de hiperfosfatemia, como,
por exemplo, na população portadora de disfunção renal. Entretanto, a
possui fosforemia dentro dos limites de referência implicaria no envolvimento
dos outros mecanismos de regulação e uma possível supressão dos
inibidores da calcificação vascular.
Figura 3 – Efeito da hiperfosfatemia sobre as células vasculares lisas
Alkaline phosphatase: fosfatase alcalina; calcium-binding proteins: proteínas ligadoras de cálcio; collagen-rich ECM: matriz extracelular rica em colágeno; matrix vesicles: vesículas de matriz; CML: células musculares lisas.
Adaptado de Giachelli e cols (115).
1.3.2 Vitamina D e doenças cardiovasculares
O impacto do status de vitamina D no sistema cardiovascular
começou a despertar interesse a partir de observações epidemiológicas que
relacionaram a deficiência de vitamina D ao aumento da mortalidade Hiperfosfatemia