Análise Funcional e Estrutural da Proteína
Pub1 de Saccharomyces cerevisiae
Tese apresentada ao Instituto de Química, Universidade Estadual Paulista, como parte dos requisitos para a obtenção do título de Doutor em Biotecnologia
Orientador: Prof. Dr. Sandro Roberto Valentini
Dedico este trabalho aos meus pais, que se
esforçaram para me proporcionar uma
boa educação e sempre respeitaram e
AGRADECIMENTOS
Aos meus pais e à minha irmã, pelo amor, carinho e apoio incondicionais.
Ao prof. Sandro, pela oportunidade de desenvolver este trabalho, pela orientação e pelo
estímulo de sempre ir além.
À professora Anita Corbett, pela oportunidade da realização dos estágios em seu laboratório
e pela grande colaboração com esse trabalho.
À professora Maria Célia Bertolini e ao Renato de Paula, aos quais serei eternamente grato
pelos primeiros passos em ciência.
Aos professores Carla Columbano de Oliveria, do IQ-USP, e Jörg Kobarg, do LNLS, pela
participação e contribuição na banca de qualificação deste trabalho.
À professora Leila Maria Beltramini, do IFSC - USP, pelas análises de dicroísmo circular.
Aos companheiros de laboratório, Glória, José Rodrigo, Rogério, Carlos Alberto, Wagner,
Ana Letícia, Ana Paula, Ana Beatriz, Suzana, Alan, Mariana, Cleslei, Marcus, Aline,
Marcelo, Daniella, pela experiência diária de convívio social saudável e democrático.
Às amigas Veridiana e Camila, por ultrapassarem o companheirismo profissional e terem se
tornado companheiras para vida.
À Cláudia, Maicon, Kátia e Danieli, pela grande ajuda com os coelhos e todos outros
trabalhos técnicos.
Aos companheiros do laboratório de Atlanta Michelle, Sara, Seth, Milo, Shana e Anna, pela
receptividade e essencial ajuda na adaptação a outro laboratório e outra cultura.
À FCF-UNESP, FAPESP, CAPES e CNPq, pelo apoio financeiro e institucional para a
realização desse trabalho.
Aos meus amigos de república Alessandro, Anselmo, Cícero, Jonas e Wagner, pelo convívio
diário e conversas infindáveis.
Dance, Monkeys, Dance
Ernest Cline
There are billions of galaxies in observable Universe, and each of them contains hundreds of billion of stars. In one of these galaxies, orbiting one of these stars, lies a little blue planet,
and this planet is runned by a bunch of monkeys. Now, the monkeys don't think of themselves as monkeys. They don't even think of themselves as animals. In fact, they love to list all the things
that they think separate them from the animals: opposable thumbs, self awareness.
They use words like Homo erectus
andAustralopithecus.
You say Toe-mate-o, I say Toe-motto. They're animals all right. They're monkeys.
Monkeys with high-speed digital fiber optic technology, but monkeys nevertheless.
I mean, they're clever. You've got to give them that. The Pyramids, skyscrapers,
phantom jets, the Great Wall of China.
That's some pretty impressive for a bunch of monkeys. Monkeys whose brains have evolved
to such an unmanageable size
that it's now pretty much impossible for them stay happy for any length of time.
In fact, they're the only animals that think they're supposed to be happy.
All of the other animals can just be. But it's not that simple for the monkeys.
You see, the monkeys are cursed with consciousness and so the monkeys are afraid.
So the monkeys worry.
The monkeys worry about everything,
but mostly about what all the other monkeys think. Because the monkeys desperately want to fit in with the other monkeys.
Which is pretty hard to do,
because a lot of the monkeys hate each other.
This is what really separates them from the other animals. These monkeys hate.
They hate monkeys that are different. Monkeys from different places, monkeys who are a different color... You see, the monkeys feel alone. All six billion of them.
The monkeys want answers
and the monkeys know they're going to die.
So the monkeys make up gods and then they worship them.
Then the monkeys start to argue over whose made-up god is better.
Then the monkeys get really pissed off and this is usually when the monkeys decide that it's a good time to start killing each other. So the monkeys wage war.
The monkeys make hydrogen bombs.
The monkeys got their entire planet wired up to explode. The monkeys just can't help it.
The monkeys make trophies
and then they give them to each other. Like it means something…
Some of the monkeys think that they have it all figured out. Some of the monkeys read Nietzsche.
The monkeys argue about Nietzsche
without giving any consideration to the fact that Nietzsche was just another monkey.
The monkeys make plans.
The monkeys fall in love. The monkeys have sex and then they make more monkeys.
The monkeys make music and then the monkeys dance: dance, monkeys, dance.
The monkeys make a hell of a lot of noise. The monkeys have so much potential. If they only apply it to themselves...
The monkeys shave the hair off their bodies in blatant denial of their true monkey nature.
The monkeys build giant monkey hives that they call cities. The monkeys draw a lot of imaginary lines in the earth. The monkeys are running out of oil
which is what fuels their precarious civilization. The monkeys are polluting and rapping their planet like there's no tomorrow...
The monkeys like to pretend that everything is just fine!!! Some of the monkeys actually believe
that the entire Universe was created to their benefit. As you can see... these are some messed up monkeys!!! These monkeys are at once
the ugliest and most beautiful creatures on the planet. And the monkeys don't want to be monkeys.
RESUMO
A expressão gênica pode ser regulada em eucariotos em diversas etapas do metabolismo de mRNA, como transcrição, processamento, tradução e degradação. A estabilidade de mRNA é modulada por elementos presentes no transcrito e por
proteínas ligantes de RNA associadas a esses elementos. Pub1 de S. cerevisiae é
uma proteína citoplasmática capaz de estabilizar transcritos contendo elementos ricos em AU (ARE e ARE-like) ou elementos estabilizadores (STE). O presente trabalho identificou num rastreamento de duplo-híbrido a proteína Nab2 como ligante de Pub1. Nab2 é uma proteína nucleocitoplasmática essencial que regula o comprimento da cauda poli(A) e a exportação nuclear de mRNA. A interação entre
Pub1 e Nab2 foi confirmada por co-purificação e ensaio de interação in vitro. Foi
demonstrado também que essa interação é mediada pelo domínio de dedos de zinco presente na região C-terminal de Nab2. A análise da relação funcional entre essas duas proteínas revelou que Nab2, assim como Pub1, é capaz de modular a
estabilidade de mRNA. A estabilidade do transcrito de RPS16B, mensageiro
contendo sequência ARE-like e regulado por Pub1, é diminuída nos mutantes
nab2-1 e nab2-67. No entanto, a estabilidade do transcrito de GCN4, mensageiro
contendo STE e também regulado por Pub1, não é afetada nos mesmos mutantes. Resultados semelhantes foram observados para outros transcritos contendo sequências ARE-like ou STE. Ainda, dados obtidos com um mutante da via NMD
(
'
upf1) mostraram que esta via de decaimento não está envolvida com omecanismo de estabilização de RPS16B mediada por Pub1 e Nab2. Uma análise
mais profunda mostrou que a sequência ARE-like presente no mensageiro de
RPS16B é necessária para a estabilização mediada por Nab2. A proteína Pub1 e
ABSTRACT
Regulation of gene expression can occur at different levels of mRNA life cycle, including transcription, processing, translation and degradation. mRNA stability is modulated by elements in the mRNA transcript and their cognate RNA-binding
proteins. Poly(U)-binding protein 1 (Pub1) is a cytoplasmic S. cerevisiae mRNA
binding protein that stabilizes transcripts containing AU-Rich Elements (ARE and ARE-like) or Stabilizer Elements (STE). In a yeast two-hybrid screen, we identified Nuclear poly(A)-binding protein 2 (Nab2) as a Pub1-interacting protein. Nab2 is an essential nucleocytoplasmic shuttling mRNA binding protein that regulates poly(A) tail length and mRNA export. The interaction between Pub1 and Nab2 was
confirmed by co-purification and in vitro binding assays. The interaction is mediated
by the Nab2 zinc finger domain. Analysis of the functional link between these proteins reveals that Nab2, like Pub1, can modulate the stability of specific target
mRNA transcripts. We find that the half-life of the RPS16B transcript, an
ARE-like-containing Pub1 target, is decreased in both nab2-1 and nab2-67 mutants. In
contrast,GCN4, an STE-containing Pub1 target, is not affected. Similar results were
obtained with other ARE- and STE-containing Pub1 target transcripts. Additionally,
results obtained with a mutant of the NMD pathway (
'
upf1) showed that this pathwayis not involved in the mechanism of RPS16B stabilization mediated by Pub1 and
LISTA DE FIGURAS
Figura 1. Mecanismos de degradação de mRNAs. 18
Figura 2. Esquemas da estrutura primária de Pub1 e dos transcritos de mRNA regulados por Pub1.
23
Figura 3. Esquema do sistema de duplo-híbrido utilizado para análise de interação proteína-proteína.
24
Figura 4. Análise da produção e purificação de His-Pub1. 49
Figura 5. Produção de anticorpo policlonal anti-Pub1 em coelho. 50
Figura 6. Análise da produção e teste de auto-ativação da proteína isca lexA-Pub1.
54
Figura 7. Ativação dos genes repórteres pelos clones obtidos no rastreamento por duplo-híbrido de ligantes de Pub1.
55
Figura 8. Confirmação da interação entre Pub1 e Nab2 por co-purificação. 60
Figura 9. Análise da produção e purificação de GST-Nab2. 61
Figura 10. Confirmação da interação direta entre Pub1 e Nab2 por ensaio de interaçãoin vitro.
62
Figura 11. Mapeamento por duplo-híbrido da interação entre Pub1 e Nab2. 63
Figura 12. Nocauteamento do gene PUB1emS. cerevisiae. 66
Figura 13. Análise de interação genética entre PUB1e mutantes de NAB2. 67
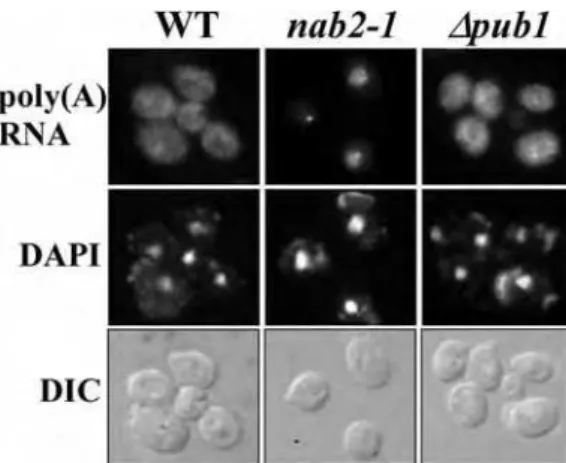
Figura 14. Localização de mRNA na linhagem 'pub1. 69
Figura 15. Padronização da reação de qRT-PCR. 74
Figura 16. Análise da estabilidade de RPS16B e GCN4 no mutante nab2-1. 75
Figura 17. Análise do decaimento de RPS16B no mutante nab2-1 por northern blot. 77
Figura 18. Geração de mutações no domínio CCCH de Nab2. 81
Figura 19. Avaliação da interação entre Pub1 e Nab2-67. 83
Figura 20. Análise da funcionalidade do alelo nab2-67. 84
Figura 21. Análise da estabilidade de RPS16B e GCN4 no mutante nab2-67. 85
Figura 22. Análise da estabilidade de RPL17A, RPS24B e WSC3 nos mutantes nab2-1 e nab2-67.
Figura 23. Análise do efeito da deleção do elemento ARE na estabilidade de
RPS16Bem mutantes de NAB2.
90
Figura 24. Análise do efeito do bloqueio da via NMD na estabilidade de RPS16B. 92
Figura 25. Análise da estrutura secundária de Pub1 por dicroísmo circular. 97
Figura 26. Esquema da estrutura primária de Pub1 e das formas truncadas em estudo.
98
Figura 27. Análise da produção e purificação da proteína Pub1 truncada His-' C-Pub1.
99
Figura 28. Análise da produção e purificação dos domínios de Pub1 His-RRM1 e His-RRM3.
100
LISTA DE TABELAS
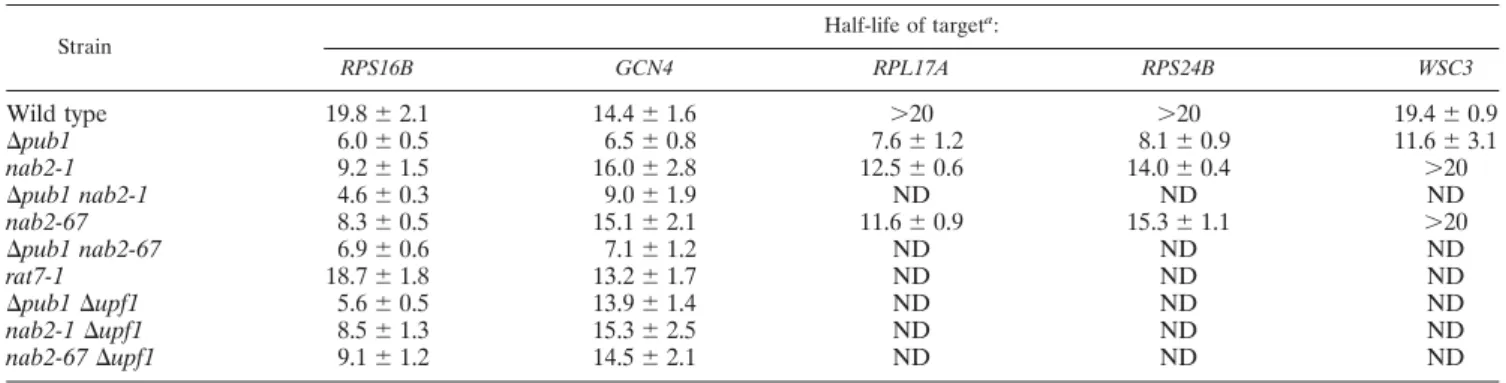
Tabela 1. Meia-vida dos transcritos regulados por Pub1 no mutantes de NAB2. 76
Tabela 2. Substituições de aminoácidos no domínio CCCH de Nab2. 82
Tabela 3. Meia-vida dos transcritos RPS16B e RPS16B-'ARE nos mutantes de
NAB2.
ABREVIATURAS
°C graus Celcius
Pg micrograma
PF microFaraday
PL microlitro
: Ohm
ARE “AU-rich element” (elemento rico em AU)
ARE-like elemento semelhante a ARE
BSA soroalbumina bovina
cDNA DNA complementar
cpm contagem por minuto
C-terminal carboxi-terminal
DAPI 4'-6-diamino-2-fenilindol
DEPC dietil pirocarbonato
DMSO dimetilssulfóxido
DNA ácido desoxirribonucléico
dNTPs mistura de desoxirribonucleotídeos trifosfatados (dATP, dCTP, dGTP
e dTTP)
D.O. densidade ótica
DTT ditiotreitol
EDTA ácido etilenodiaminotetracético
FITC isotiocianato de fluoresceína
5-FOA ácido 5-flúor-orótico
IPTG isopropil-E-D-tiogalactopiranosídeo
kb quilobase
kDa quilodalton
kV quilovolt
LB meio Luria-Bertani
M molar
mL mililitro
mM milimolar
mRNA RNA mensageiro ng nanograma
nm nanômetro
N-terminal amino-terminal
ONPG O-nitrofenil-E-D-galactopiranosídeo
ORF “Open Reading Frame” (janela aberta de leitura)
PAGE eletroforese em gel de poliacrilamida
PAP anticorpo anti-peroxidase conjugado com peroxidase
pb pares de bases
PBS solução salina tamponada com fosfato
PBST PBS com Tween 20
PCI fenol:clorofórmio:álcool isoamílico (24:25:1)
PCR “Polymerase Chain Reaction” (reação em cadeia de polimerase)
PEG polietilenoglicol
pH potencial hidrogeniônico
Pipes N,N’-bis(2-etanossulfonato) de piperazina
PMSF fluoreto de fenilmetilsulfonila
PLAC solução de inibidores de proteases (pepstatina, leupeptina, aprotinina e
quimostatina)
q.s.p. quantidade suficiente para
RNA ácido ribonucléico
RNase ribonuclease
qRT-PCR “quantitative real-time PCR” (PCR em tempo real quantitativa)
SC meio sintético completo para levedura
SDS dodecil sulfato de sódio
SSC solução salina/citrato de sódio
SSPE solução salina/fosfato de sódio/EDTA
S-SPO “super-sporulation” (meio para esporulação de levedura)
STE “stabilizer element” (elemento estabilizador)
TAE Tris-acetato EDTA
Tris tris-hidroximetilaminometano
Triton X-100 polietilenoglicol-terc-octilfenil éter
U unidade enzimática
uORF “upstream”-ORF (ORF anterior à ORF principal)
UTR “untranslated region” (região não traduzida de mRNA)
V volt
YNB “yeast nitrogen base” (base nitrogenada para levedura)
YPD “yeast extract, peptone, dextrose” (meio rico para levedura)
xg aceleração gravitacional
SUMÁRIO
1. INTRODUÇÃO ...15
1.1. Decaimento de mRNA e seu envolvimento na regulação da expressão gênica... 15
1.2. Pub1 e o controle da estabilidade de mRNAs... 19
1.3. O sistema de duplo-híbrido como ferramenta de identificação de interações proteína-proteína ... 21
2. OBJETIVOS...25
2.1. Objetivo geral... 25
2.2. Objetivos específicos ... 25
3. MATERIAIS E MÉTODOS ...26
3.1. Linhagens, plasmídeos e oligonucleotídeos... 26
3.1.1. Linhagens... 26
3.1.2. Plasmídeos. ... 27
3.1.3. Oligonucleotídeos. ... 28
3.2. Meios de cultura ... 30
3.3. Manipulação de bactérias ... 30
3.3.1. Transformação química de E. coli... 30
3.3.2. Eletroporação de E. coli... 31
3.4. Manipulação de leveduras ... 31
3.4.1. Transformação rápida de S. cerevisiae ... 31
3.4.2. Transformação de alta eficiência de S. cerevisiae ... 32
3.4.3. Cruzamento de leveduras ... 32
3.4.4. Esporulação e dissecção de leveduras ... 33
3.5. Manipulação de DNA... 33
3.5.1. Isolamento de DNA plasmidial de bactéria (mini preparação)... 33
3.5.2. Isolamento de DNA plasmidial de levedura... 34
3.5.3. Isolamento de DNA genômico de levedura ... 34
3.5.4. Eletroforese de DNA em gel de agarose ... 35
3.5.5. Isolamento de fragmentos de DNA de gel de agarose ... 35
3.5.6. Amplificação de DNA por PCR... 35
3.5.7. Manipulação enzimática de DNA ... 35
3.5.8. Sequenciamento de DNA ... 36
3.5.9. Mutação sítio-dirigida de DNA... 36
3.6. Análise de proteínas ... 36
3.6.1. Quantificação de proteínas... 36
3.6.2. Separação de proteínas por SDS-PAGE... 36
3.6.3. Coloração de proteínas por “Coomassie Blue” ... 37
3.6.4. Western blot ... 37
3.7. Produção e purificação de proteína recombinante ... 37
3.7.1. Teste de indução da produção de proteína recombinante... 37
3.7.2. Produção de proteína recombinante em larga escala... 38
3.7.3. Purificação de proteína em fusão com cauda de histidinas... 38
3.7.4. Purificação de proteína em fusão com GST (Glutationa S-transferase) ... 38
3.9. Rastreamento de ligantes físicos por duplo-híbrido... 39
3.10. Ensaio de co-purificação utilizando a fusão TAP... 41
3.11. Ensaio in vitro de interação proteína-proteína... 41
3.12. Nocauteamento genético em S. cerevisiae ... 42
3.13. Ensaio de localização de mRNA por FISH (Fluorescence In Situ Hybridization)... 42
3.14. Extração de RNA total de S. cerevisiae... 43
3.15. PCR em tempo real quantitativo (qRT-PCR)... 43
3.16. Northern blot ... 44
3.16.1. Eletroforese de RNA... 44
3.16.2. Hibridização da sonda ... 44
3.16.3. Marcação radioativa de sondas ... 44
3.17. Ensaio de estabilidade de mRNA... 45
3.18. Teste de determinação in vitro da atividade de E-galactosidase ... 45
3.19. Testes de varredura de condições de cristalização de proteína... 46
4. RESULTADOS...47
4.1. Produção e purificação de His-Pub1 ... 47
4.2. Produção de anticorpo policlonal anti-Pub1 em coelho ... 47
4.3. Rastreamento de ligantes físicos de Pub1 utilizando o sistema de duplo-híbrido ... 51
4.4. Estudo da interação física entre Pub1 e Nab2 ... 56
4.4.1. Ensaios de co-purificação entre Pub1 e Nab2 ... 56
4.4.2. Ensaios de interação in vitro entre Pub1 e Nab2 ... 57
4.4.3. Determinação do domínio de Nab2 que interage com Pub1 ... 58
4.5. Avaliação de interação genética entre PUB1 e NAB2 ... 64
4.6. Avaliação da localização de mRNA no nocaute de Pub1 ... 68
4.7. Determinação da estabilidade de mRNA nos mutantes de NAB2... 70
4.7.1. Obtenção de linhagens contendo o alelo rpb1-1... 70
4.7.2. Padronização da reação de PCR em tempo real quantitativo ... 71
4.7.3. Determinação da estabilidade de mRNA... 72
4.8. Identificação e caracterização de um novo mutante de NAB2 ... 78
4.8.1. Mutagênese sítio-dirigida do domínio CCCH de NAB2 ... 78
4.8.2. Avaliação da funcionalidade do mutante nab2-67 ... 79
4.8.3. Análise da estabilidade de mRNA no mutante nab2-67 ... 80
4.9. Análise da dependência da sequência ARE-like na estabilização de RPS16B por Nab2 ... 87
4.10. Análise da dependência da via de NMD no decaimento de RPS16B... 88
4.11. Análise estrutural da proteína Pub1 ... 93
4.11.1. Produção, purificação e varredura de condições de cristalização de Pub1... 93
4.11.2. Produção, purificação e varredura de condições de cristalização de domínios de Pub1... 94
5. DISCUSSÃO ...101
6. CONCLUSÕES ...107
1. INTRODUÇÃO
1.1. Decaimento de mRNA e seu envolvimento na regulação da expressão gênica
A expressão gênica em eucariotos é um mecanismo altamente diversificado, envolvendo diversas etapas do metabolismo de RNA mensageiro (mRNA), como transcrição, processamento, tradução e decaimento (1,2). Apesar de a regulação da expressão gênica ter sido historicamente mais extensivamente estudada em seu nível transcricional, recentes estudos têm demonstrado com sucesso que a expressão gênica pode ocorrer nas diversas etapas do ciclo de vida do mRNA (3).
O processo de decaimento de mRNA é um importante ponto de controle pós-transcricional que auxilia na modulação da quantidade de um transcrito. A meia-vida de um mensageiro pode ser alterada em resposta a uma variedade de estímulos, como fatores ambientais, mitógenos, fatores de crescimento e hormônios (4). Interessantemente, transcritos codificadores de proteínas com funções de manutenção celular são tipicamente caracterizados por meias-vidas longas, enquanto transcritos codificadores de proteínas que são requeridas transientemente na célula, como durante o ciclo celular ou diferenciação, frequentemente têm meias-vidas curtas (4). Por exemplo, em humanos, o mensageiro codificador da enzima gliceraldeído 3-fosfato desidrogenase (GAPDH), uma proteína de manutenção
celular, tem meia-vida de mais de 24 horas (5), enquanto o transcrito de c-myc, um
fator de transcrição transiente, possui meia-vida de apenas 10 minutos (6). Um grande número de transcritos com relevância clínica exibe um decaimento regulado em resposta a sinais celulares e a desregulação da sua taxa de decaimento está diretamente relacionada a estados patológicos (2,7,8).
Um considerável número de estudos que analisaram o decaimento de mRNA como mecanismo de regulação da expressão gênica utilizaram a levedura
Saccharomyces cerevisiae como modelo, e esses estudos revelaram componentes
de transcritos de levedura que codificam proteínas funcionalmente relacionadas exibem perfis de degradação similares em resposta a estímulos ambientais (11), sugerindo que o decaimento de mRNA é coordenado aos requisitos biológicos da célula. Essas observações enfatizam a contribuição significante do decaimento de mRNA na regulação da expressão gênica.
Durante sua biogênese, o mRNA é complexado a várias proteínas ligantes de RNA (“RNA-binding proteins”, RBPs), as quais se associam e dissociam em determinadas etapas da maturação do transcrito (13), e essa associação e dissociação entre RBPs e deferentes elementos do transcrito regula o ciclo de vida de um mRNA (13). Evidências relevantes sugerem que os diferentes passos do metabolismo de mRNA são coordenados e mecanisticamente acoplados ao invés de serem sequenciais (14). Já foram caracterizados mecanismos que acoplam a síntese de mRNA à exportação nuclear (14) e também que acoplam a tradução à degradação do transcrito (15,16). No entanto, o mecanismo que acopla processamento/exportação à degradação de mRNA ainda não é bem compreendido. Recentemente têm sido demonstrados alguns exemplos de como eventos nucleares relativos ao processamento de mRNA podem influenciar o destino desse mensageiro no citoplasma. A proteína Pab1, por exemplo, apresenta localização primordialmente citoplasmática (17) e é um fator crucial no controle da iniciação da tradução estimulada por cauda de poliadenosina (poli(A)) e na modulação da estabilidade de mRNA no citoplasma (18). No entanto, Pab1 pode ser translocada entre o núcleo e o citoplasma (19,20) e, no núcleo, é capaz de regular o processamento da extremidade 3´ do mRNA (21) e o tamanho da cauda poli(A) (22). Contudo, ainda não é compreendido como essas funções nucleares de Pab1 influencia suas funções citoplasmática já bem caracterizadas. Pab1 é apenas uma de várias proteínas ligantes de RNA que se complexam a transcritos no núcleo e exercem efeitos também no citoplasma.
deve ser comprometida ou o mRNA deve sofrer clivagem por ataque endonucleásico (23).
Em eucariotos, a principal via de degradação de mRNA, chamada de decaimento de mRNA dependente de desadenilação, é iniciada com o encurtamento da cauda poli(A) por ação de desadenilases (Figura 1A). Esta etapa do decaimento de mRNA é a única etapa que é reversível - transcritos contendo certos determinantes podem ser readenilados e retornarem aos polissomos (23). No entanto, uma vez determinado que o mRNA deve ser destruído, uma das duas rotas de degradação é seguida (Figura 1A): o “cap” é removido da estremidade 5’ pelo processo conhecido por “decapping”, permitindo que o mRNA seja degradado na
direção 5’o3’ ou a extremidade 3’ desprotegida é atacada por ação exonucleásica
3’o5’ (23). Na via de “decapping”, o complexo Lsm se associa à extremidade 3’ do
transcrito e induz a remoção do “cap” de 7-metilguanosina pela ação do complexo de
Dcp1 e 2, deixando o mRNA suscetível à degradação 5’o3’ por ação da
exorribonuclease Xrn1. Alternativamente, o mRNA desadenilado por ser degradado
na direção 3’o5’ por ação do complexo de exorribonucleases conhecido como
exossomo, seguido da hidrólise do “cap” pela enzima DcpS (Figura 1A) (23).
No entanto, apesar de o decaimento da maioria dos transcritos ser dependente de desadenilação, existem várias exceções a esta via, o que permite que mRNAs específicos possam transpor algumas etapas da via “padrão” de degradação, oferecendo um mecanismo de regulação própria da estabilidade (23). Na via de decaimento independente de desadenilação, elementos específicos na região 3’UTR podem desencadear a degradação do transcrito independentemente da sua desadenilação (23). Como exemplificado na Figura 1B, o fator Edc3, juntamente com Rps28B, é capaz de se ligar à região 3’UTR de mRNAs específicos e recrutar a maquinaria de “decapping”, deixando o mRNA suscetível à degradação
5’o3’ por ação da exorribonuclease Xrn1. Na via de decaimento mediado por
endonucleases, a degradação do mRNA é iniciada por ação de enzimas endonucleolíticas (em mamíferos, por exemplo, PMR1, IRE1 e RNAse MRP), gerando dois fragmentos com uma das extremidades desprotegida, sujeitas a
AA
Desadenilase
Decaimento 3' 5' Decaimento 5' 3'
Decaimento 5' 3'
Dcp2
Dcp1
DcpS
Exossomo
Xrn1
AAAAA AAAAA
Dcp2
Dcp1
Xrn1
Rps28b Edc3
Rps28b Edc3
AAAAA AAAAA
Exossomo
Decaimento 5' 3' Decaimento 3' 5'
Xrn1
Endonucleases
B
C
1.2. Pub1 e o controle da estabilidade de mRNAs
Análises por microarranjos de DNA (“microarrays”) têm revelado que 40-50% das mudanças na expressão gênica em resposta a sinais celulares ocorrem ao nível da estabilidade de mRNA (24,25). Essas mudanças são usualmente induzidas por alterações na composição de fatores que interagem com diferentes sequências localizadas mRNA (2), inibindo ou facilitando a sua degradação. Em leveduras e mamíferos existe um grande número de proteínas ligantes de RNA que agem tanto como ativadores quanto inibidores da estabilidade e eficiência de tradução de mRNA (2). Muitas dessas proteínas medeiam seu efeito por se ligarem seletivamente às regiões 3’UTR ou 5’UTR de alvos específicos.
A proteína Pub1 ("poli-U binding protein 1") de levedura, por exemplo, é a
principal proteína que se liga a RNA poliadenilado nuclear e citoplasmático de S.
cerevisiae (26) e tem sido implicada na regulação do decaimento de mRNA. É uma
proteína com 453 aminoácidos, com ponto isoelétrico teórico 4,76 e massa molecular estimada em 50kDa e observada de aproximadamente 60kDa (26,27). Esta proteína pertence a uma grande família de proteínas que se ligam a RNA por conservar domínios de reconhecimento de RNA (“RNA recognition motifs” – RRMs), como as hnRNPs e as snRNPs. Pub1 contém três RRMs, um domínio rico em metionina e asparagina entre o
2o e o 3o RRM e um domínio rico em glutamina na região C-terminal (Figura 2A) (26).
Várias proteínas ortólogas a Pub1 (Slx, TIA-1 e HuR) possuem uma organização estrutural muito semelhante e desempenham um importante papel na diferenciação e proliferação celular, compondo uma família de proteínas semelhantes a ELAV, altamente conservada em vertebrados (28). Especiamente a proteína HuR, com 30% de identidade, regula a estabilidade de mRNAs envolvidos com diferenciação celular (29) e resposta a várias condições de estresse celular (30,31).
Estudos relacionam o envolvimento de Pub1 com a estabilização de mRNA contendo ARE (“AU-rich elements”) (10). O controle da estabilidade de mRNA contendo ARE é um importante mecanismo de controle da expressão gênica, sendo observado em vários transcritos envolvidos em proliferação e diferenciação celular em mamíferos, como proto-oncogenes, fatores de transcrição, citocinas e linfocinas (8). A alteração no controle da degradação de mRNA mediado por ARE resulta em expressão gênica anormal, resultando, por exemplo, em oncogênese devido à estabilização do transcrito
Em alguns casos, as sequências ARE são capazes, por si próprias, de recrutar a maquinaria de degradação de mRNA - os exossomos, por exemplo, mostram afinidade por esses elementos (34). Foi mostrado também que várias proteínas de mamíferos, como AUF1, TTP, RHAU e KSRP, se ligam às sequências e interagem, direta ou indiretamente com fatores de decaimento de mRNA (23). Ainda não é claro o mecanismo de degradação desses transcritos, mas existem evidências que mostram o aumento da atividade de desadenilases, do exossomo e da enzima de “decapping” (23). Por outro lado, algumas proteínas podem estabilizar esse tipo de transcrito, simplesmente por remover esse mRNA dos sítios de degradação ou por competir com fatores de desestabilização pela ligação às sequências ARE (23). Alternativamente, essas proteínas podem interagir diretamente e inibir a maquinaria de degradação de mRNA ou de alguma forma aumentar a interação entre Pab1 e a cauda poli(A), prevenindo assim a desadenilação. A proteína HuR por exemplo, ortólogo de Pub1 em mamíferos, compete com as proteínas AUF1, KSRP e TTP na ligação a ARE, fazendo com que, sob condições nas quais a interação entre HuR e ARE é favorecida, o transcrito contendo a ARE seja estabilizado (35,36). Análogo ao decaimento de mRNA mediado por ARE em mamíferos, foi mostrado que AREs regulam o decaimento de transcritos de levedura, e que Pub1 é capaz de estabilizar transcritos sujeitos a essa via de regulação (Figura 2B) (10).
de desadenilação (Figura 1B) (23). No entanto, a via de NMD não é restrita ao decaimento de transcritos aberrantes, tendo também um importante papel na regulação da expressão gênica de transcritos normais. Devido à similaridade da organização cistrônica entre mRNAs contendo mutação “nonsense” e mRNAs contendo naturalmente uORFs, a regulação da estabilidade de mRNA contendo uORFs também é mediada por NMD (43). Pub1 impediria a ação da via NMD estabilizando esses transcritos por se ligar a elementos de estabilização (“Stabilizer Elements”, STEs) presentes em transcritos contendo uORFs (Figura 2B) (44).
No entanto, ainda não está claro como Pub1 se relaciona funcionalmente com outras RNPs para estabilizar transcritos de mRNA específicos. A descoberta de parceiros físicos de Pub1 com função conhecida pode contribuir para a melhor caracterização do seu mecanismo de ação.
1.3. O sistema de duplo-híbrido como ferramenta de identificação de interações proteína-proteína
O sistema de duplo-híbrido desenvolvido por Fields e Song (45) é capaz de
identificar interações proteína-proteína in vivo através da reconstituição da atividade
de um fator de transcrição em levedura. Muitos ativadores transcricionais eucarióticos possuem pelo menos dois domínios funcionais distintos. A proteína Gal4, por exemplo, contém na porção amino um domínio de ligação a sequências específicas de DNA (BD, “binding domain”) e na porção carboxila um domínio acídico que é necessário para a ativação da transcrição (AD, “activation domain”). Dessa forma, conforme esquema mostrado na Figura 3, estes domínios independentes podem ser fusionados separadamente a outras proteínas, as quais, quando interagem entre si, reconstituem artificialmente a proximidade natural destes
domínios, promovendo a transcrição de genes repórteres, como HIS3 e lacZ,
contendo na sua região ativadora de transcrição (UAS, “uptream activating
sequence) a sequência de ligação de Gal4 (UASGal4). Então, ao ser utilizada uma
Existem ainda outras variações do sistema original de duplo-híbrido, desenvolvidos com os objetivos de aumentar a sensibilidade do método e diminuir a detecção de interações falso-positivas (46). Foi demonstrado que o domínio de
ativação da proteína Gal4 de S. cerevisiae pode ser fusionado à proteína ligante de
DNA de Escherichia coli lexA, criando um ativador transcricional funcional em
levedura (47). A substituição do domínio de ligação de DNA de Gal4 pela proteína lexA leva a uma maior sensibilidade do sistema, principalmente devido ao fato de lexA, por ser uma proteína de bactéria, não possuir nenhum sítio de ligação ao DNA de levedura, diminuindo a obtenção de interações falso-positivas (48). Nesse sitema, também é possível variar a sensibilidade do gene repórter através do
número de operadores de lexA (lexAop) utilizadas como promotor (48). A utilização
de outros domínios de ativação de transcrição diferentes de Gal4, como o domínio
acídico da proteína B42 de E. coli (48), também oferece vantagens na diminuição da
detecção de clones falso-positivos. Também com o objetivo de prevenir a obtenção de clones falso-positivos, podem ser utilizados mútiplos genes repórteres, inclusive
URA3, que permite seleção negativa em meio 5-FOA (46).
Os diferentes sistemas de duplo-híbrido têm sido extensamente aplicados na detecção de interações proteína-proteína (46), inclusive na determinação de redes de interações (interactomas) de diversos organismos (49). Nosso grupo de pesquisa especificamente já utilizou o sistema de duplo-híbrido na busca de fatores celulares
que interagem com o possível fator de início de tradução 5A (eIF5A) de S. cerevisiae,
o que permitiu identificar a proteína Lia1 (“Ligand of eIF5A”) (50), naquele momento com função desconhecida, mas identificada recentemente como uma das enzimas envolvidas na maturação pós-traducional de eIF5A. Atualmente outras proteínas de levedura e mamíferos têm sido utilizadas como isca na busca de ligantes físicos.
pQ - C
N -
RRM1
RRM2
RRM3
Pub1
B
AAAAA
m7G
uORF STE ORF
Pub1
AAAAA
m7G
ORF ARE
his+
azul
XY
Interação entre proteínas X eY
Gal4 AD
X
Y
HIS3
LacZ
UASGal4
Gal4 BD
GAL4 BD
2. OBJETIVOS
2.1. Objetivo geral
Este trabalho teve como objetivo geral o entendimento dos mecanismos de regulação pós-transcricionais exercidos por Pub1 na última etapa do metabolismo de mRNA, ou seja, na sua degradação citoplasmática.
2.2. Objetivos específicos
Identificação de parceiros físicos de Pub1 e confirmação das possíveis
interações.
Estudo da relação funcional das interações observadas.
3. MATERIAIS E MÉTODOS
3.1. Linhagens, plasmídeos e oligonucleotídeos
As linhagens de E. coli e S. cerevisiae, assim como os plasmídeos e os
oligonucleotídeos utilizados no desenvolvimento deste trabalho, estão relacionados abaixo.
3.1.1. Linhagens.
Genótipo Origem
E. coli
SVB2 (BL21 DE) F-ompThsdSB(rB-mB-)gal dcm (DE3) pLysS (CamR) Invitrogen SVB3 (DH5D) F'/endA1 hsdR17(rK-mK+) supE44 thi-1 recA1 gyrA (Na1r) relA1
'(lacZYA-argF)U169(I80lacZ'M15)
Life Technologies SVB8 (M15[pREP4]) NaIS, StrS, RifS, Thi–, Lac–, Ara+, Gal+, Mtl–, F–, RecA+, Uvr+, Lon+. Qiagen SVB28 (HB101) '(gpt-proA)62 leuB6 thi-1 lacY1 hsdSB20 recA rpsL20 (Strr) ara-14
galK2 xyl-5 mtl-1 supE44 mcrBB Carla Oliveira
S. cerevisiae
SVL55 MATa ura3 leu2 trp1 Pamela Silver
SVL56 MATD ura3 leu2 his3 Pamela Silver
SVL86 (L40) MATa his3 trp1 leu2 ade2 LYS::(lexAop)4-HIS3 URA3::(lexA op)8-lacZ Clontech
SVL129 MATa his3 trp1 leu2 ade2 LYS::(lexAop)4-HIS3 URA3::(lexA op)8-lacZ
[pBTM-NIP7] [pACT-NOP8] (51)
SVL130 (L40) MATa his3 trp1 leu2 ade2 LYS::(lexAop)4-HIS3 URA3::(lexA op)8-lacZ
[pBTM-NIP7] [pACT-RRP43] (51)
SVL296 MATD ura3 his4 rpb1-1 (52)
SVL316 MATDtrp1 ura3 his3 leu2::lexA-LEU2 [URA3 lacZ] Invitrogen
SVL371 MATa ura3 leu2 his3 met15 pub1::KAN MX4 Open Biosystems
SVL377 MATD ura3 his3 leu2 rat7-1 Anita Corbett
SVL538 MATa ura3 met15 leu2 NAB2-TAP::HIS3 Open Biosystems
SVL544 MATa ura3 trp1 leu2 nab2::HIS3 [pSV877] (53)
SVL549 MATa ura3 trp1 leu2 pub1::KAN MX4 nab2::HIS3 [pSV877] Este estudo
SVL565 MATa ura3 met15 leu2 SEC27-TAP::HIS3 Open Biosystems
SVL577 MATa ura3 trp1 leu2 his3 rpb1-1 pub1::KAN MX4 nab2::HIS3 [pSV877] Este estudo
SVL677 MATa ura3 trp1 leu2 his3 rpb1-1 Este estudo
SVL678 MATa ura3 trp1 leu2 his3 rpb1-1 pub1::KAN MX4 Este estudo
SVL679 MATa ura3 trp1 leu2 his3 rpb1-1 nab2::KAN MX4 [pSV877] Este estudo
SVL680 MATa ura3 trp1 leu2 rpb1-1 pub1::KAN MX4 upf1::HIS3 Este estudo
SVL682 MATa ura3 tr1 leu2 rpb1-1 nab2::KAN MX4 upf1::HIS3 [pSV877] Este estudo
SVL688 MATa ura3 trp1 leu2 his3 rpb1-1 rat7-1 Este estudo
SVL690 MATa ura3 leu2 his3 rpb1-1 pub1::KAN MX4 RPS16B'ARE-TRP1 Este estudo
3.1.2. Plasmídeos.
Plasmídeos Descrição Origem
pSV59 (pRS315) CEN6,LEU2 (54)
pSV70 (pRS304) TRP1integrativo (54)
pSV148 (pBTM116) lexA,2P,TRP1 Clontech
pSV149 (pACT) GAL4 AD,2P,LEU2 Clontech
pSV255 pQE30 Qiagen
pSV408 pQE30-PUB1 Este estudo
pSV424 pBTM116-PUB1, 2P, TRP1 Este estudo
pSV440 pJG4-5 Origene Technologies
pSV446 pEG202 Origene Technologies
pSV491 pGEX-4T-NAB2 Este estudo
pSV507 pQE-'C-PUB1 Este estudo
pSV545 pJG4-5-PUB1, 2P, TRP1 Este estudo
pSV567 pEG202-NAB2, 2P, HIS3 (53)
pSV568 pEG202-nab2-1, 2P, HIS3 Anita H. Corbett
pSV569 pEG202-'RGG NAB2, 2P, HIS3 Anita H. Corbett
pSV570 pEG202-'QQQP NAB2, 2P, HIS3 Anita H. Corbett
pSV571 pEG202-'CCCH NAB2, 2P, HIS3 Anita H. Corbett
pSV572 NAB2,CEN6,LEU2 (55)
pSV573 'RGG-nab2,CEN6,LEU2 (55)
pSV575 nab2-1,CEN6,LEU2 (55)
pSV587 pQE-RRM1 Este estudo
pSV597 pEG202-CCCH NAB2, 2P, HIS3 Anita H. Corbett
pSV605 pQE-RRM3 Este estudo
pSV607 pGEX-4T-'CCCH NAB2 Este estudo
pSV609 pGEX-4T-CCCH NAB2 Este estudo
pSV645 pEG202-'C3 NAB2, 2P, HIS3 Este estudo
pSV646 pEG202-'CT NAB2, 2P, HIS3 Este estudo
pSV661 'QQQP-nab2, CEN6,LEU2 (55)
pSV690 nab2-C437S, CEN6,LEU2 Anita H. Corbett
pSV741 pEG202-'C4-NAB2, 2P, HIS3 Este estudo
pSV859 pGEX-4T-nab2-67 Este estudo
pSV864 pEG202-ccch-67 NAB2, 2P, HIS3 Este estudo
pSV870 pSV70-RPS16B-'ARE,TRP1 Este estudo
pSV876 nab2-67, CEN6, LEU2 Este estudo
pSV878 pEG202-ccch-14 NAB2, 2P, HIS3 Este estudo
pSV879 pEG202-ccch-57 NAB2, 2P, HIS3 Este estudo
pSV880 pEG202-ccch-17 NAB2, 2P, HIS3 Este estudo
pSV881 pEG202-ccch-5 NAB2, 2P, HIS3 Este estudo
pSV882 pEG202-ccch-6 NAB2, 2P, HIS3 Este estudo
pSV883 pEG202-ccch-7 NAB2, 2P, HIS3 Este estudo
pSV884 pEG202-ccch-56 NAB2, 2P, HIS3 Este estudo
pAC1112 'CT-nab2, CEN6,LEU2 (55)
pAC1113 'C3-nab2, CEN6,LEU2 (55)
3.1.3. Oligonucleotídeos.
Oligonucleotídeos Sequências Sítio ou
Mutações
SVO135 5’-TACCACTACAATGGATG-3’
-SVO145 5’-CGCGGATCCATGTCTGAAAATAACGAAG-3’ BamHI
SVO146 5’-CCGCTCGAGTGTGCCTGGACTCAAGACTG-3’ XhoI
SVO171 5’-CGCGGATCCCAATGTCTGAAAATAACGAAG-3’ BamHI
SVO183 5’-CCGGAATTCATGTCTGAAAATAACGAAG-3’ EcoRI
SVO199 5’-CACGATGCACAGTTG-3’
-SVO225 5’-CTGCAGCGAGGAGCCGTAAT-3’
-SVO246 5’-CGGGATCCATGTCTCAAGAACAGTAC-3’ BamHI
SVO247 5’-CCGGAATTCAGTGCCCTTATGATCAGG-3’ EcoRI
SVO256 5’-CGCGGATCCTTAGGATTGGAAGGCCC-3’ BamHI
SVO258 5’-CGCGGATCCTTAGTGGTCAACAGCTTG-3’ BamHI
SVO260 5’-CGCGGATCCTTATGATCTTTCCTTACC-3’ BamHI
SVO270 5’-CGCGGATCCATGTCCCAACAGAGTTC-3’ BamHI
SVO295 5'-CGCGAATCCATGCAGCGTCGTAACTACG-3' BamHI
SVO325 5'-GCTTACCCTTCCCACTGCCGTTACAAGGGAGG-3'
-SVO326 5'-GTCGCTGGAGGCTGGACTCAAGACTGACATGTCTCC-3'
-SVO345 5'-CTTGAACTTACGCCCCTCTTTCTTGGTTGGAGTGAAGTTC-3'
-SVO346 5'-GAGGGGCGTAAGATCAAGGAAGTAAAACCAATAAGC-3'
-SVO351 5'-CCGGAATTCATGTCTCAAGAACAGTAC-3' EcoRI
SVO352 5'-CCGCTCGAGAGTGCCCTTATGATCAGG-3' XhoI
SVO403 5'-CTGCTTTGCCAACCATCAAGT-3'
-SVO404 5'-GCAACTGGAGCCAAAGAGTATTTT-3'
-SVO469 5'-TAAGTAAAAAGTTTTTTAATACAATAGATGTGC-3'
-SVO470 5'-GATTCTAGAAGACCAGAACCAAAGAAAT-3'
-SVO476 5'-CCCTCGAGATAGTAGACATACCC-3' XhoI
SVO495 5'-AGAGATTTCTGGAAAAAATCTTGAAATTTTCTTAAC-3'
-SVO496 5'-TTTTTTCCAGAAATCTCTATCTATTGTATTAAAAAAC-3'
-SVO497 5'-GTAAAAAGTTTTTTAATACAATAGATAGAGATTTCTGGA-3
-AC2277 5'-TGTCTGCCGTCCCAAGTGTC-3'
-AC2278 5'-AGAACCGTTGACCTTAATCAAACC-3'
-AC2281 5'-ACTGGATCATCTAGGTGTTGTTGCT-3'
-AC2282 5'-ATTCGGGCACAATTGGAGAA-3'
-AC2285 5'-GCACACCCAACTAAGGTACGTAATGAATATCCAAATTGTCC-3' C283R
AC2286 5'-CAATTTGGATATTCATTACGTACCTTAGTTGGGTGTGCATGTG-3' C283R
AC2287 5'-ACTGGTATCGTTCTGCGTAAATTTGGGGCTCTGTG-3' C304R
AC2288 5'-CAGAGCCCCAAATTTACGCAGAACGATACCAGTTTG-3' C304R
AC2289 5'-AAAGTCATTGATCTAATGTGGCGTGACAAGAATTTGACATGTG-3' C371R
AC2290 5'-TATCACATGTCAAATTCTTGTCACGCCACATTAGATCAATGAC-3' C371R
AC2291 5'-AAGTCCTTAGAACAACGTAAGTTCGGTACGCACTGC-3' C415R
AC2292 5'-CAGTGCGTACCGAACTTACGTTGTTCTAAGGACTTTTC-3' C415R
AC2293 5'-CGTTCTCATATTATGCGCCGTGAAGGAGCAAACTG-3' C437R
AC2294 5'-TTGCTCCTTCACGGCGCATAATATGAGAACGAGC-3' C437R
AC2295 5'-TTGGCCATCCAATTAATGAAGATCGTAGATTTGGTGTCAATTG-3' C458R
AC2296 5'-TACAATTGACACCAAATCTACGATCTTCATTAATTGGATGGCC-3' C458R
AC2297 5'-GACCGAATATACTTTTTATATTACATCAATCATTGTCATTATCAAG GCCTCCTCTAGTAC-3' -AC2298 5'-ATCACAAGCCAAGTTTAACATTTTATTTTAACAGGGTTCACCGAA GCGCGCCTCGTTCAG-3'
-AC2327 5'-GGCTGGAATTCCGTCGCAGATTGTTTCCTCAC-3' C262R
AC2328 5'-GAGGAAACAATCTGCGACGGAATTGCCAGCCAG-3' C262R
AC2366 5'-TCTGATCATAAGGAAGTGGAAGTACATCAGAAAGGCCTCCTCTA GTACACTC-3' -AC2367 5'-TGTACTTAAACGTTCCGTGATTTTAATAGTAAGCGCGCCTCGTTC AGAATG-3' -AC2370 5'-TCTGATCATAAGGAAGTGGAAGTACATCAGAAACGTACGCTGCA GGTCGAC-3' -AC2371 5'-TGTACTTAAACGTTCCGTGATTTTAATAGTAAATCGATGAATTCG AGCTCG-3'
-AC2450 5'-AAGTTCGGTACGCACCGCACCAATAAACGTGTC-3' C412R
AC2451 5'-GACACGTTTATTGGTGCGGTGCGTACCGAACTT-3' C412R
AC2452 5'-GCCGTGAAGGAGCAAACCGTACTAGAATTGATTG-3' C443R
AC2453 5'-CAATCAATTCTAGTACGGTTTGCTCCTTCACGGC-3' C443R
AC2454 5'-GTAGATTTGGTGTCAATCGTAAGAATATTTACTGTCTATTCAG-3' C464R
AC2455 5'-CTGAATAGACAGTAAATATTCTTACGATTGACACCAAATCTAC-3' C464R
AC2468 5'-CCTGAGTCAGCAGTAGTAGCATTATTTAAC-3'
-AC2469 5'-AGCCAGAGCATTTAGTTCCACAG-3'
-AC2472 5'-AAGGTTATCTCCAACCCATTGTTG-3'
-AC2473 5'-TTTTCAGCCTTGTAGACTTCAGCTAA-3'
-AC2474 5'-ACTAACCCTGCTAAATCCGCTTC-3'
-AC2475 5'-ACTTGTTCCAAGTATTTTTGAGCCTT-3'
3.2. Meios de cultura
- Meio LB - triptona 1%; NaCl 0,5%; extrato de levedura 1% (ágar 2% para meio
sólido).
- Meio SOB - triptona 2%; extrato de levedura 0,5%; NaCl 10mM; KCl 2,5mM;
MgCl2 10mM; MgSO4 10mM.
- M9 - Na2HPO4 46mM; KH2PO4 22mM; NaCl 8,5mM; NH4Cl 18mM; glicose 0,4%;
prolina 20Pg/mL (ágar 1,8% para meio sólido).
- Meio completo (YPD) - extrato de levedura 1%; peptona 2%; glicose 2% (ágar 2%
para meio sólido).
- Meio sintético (SC) - YNB 0,67% (sem aminoácidos); glicose 2%; suplementado
com aminoácidos, uracila ou adenina quando necessário (ágar 2% para meio sólido).
- Meio SC / X-Gal - Na2HPO4 50mM; NaH2PO4 25mM; YNB 0,67% (sem
aminoácidos); galactose 2%; rafinose 1%; ágar 2%; suplementado com aminoácidos, uracila ou adenina quando necessário.
- Meio S-SPO - extrato de levedura 0,25%; acetato de potássio 1,5%; 40mg/L de
adenina, uracil e tirosina; 20mg/L de histidina, leucina, lisina, triptofano, metionina e arginina; 100mg/L de fenilalanina e 350mg/L de treonina.
3.3. Manipulação de bactérias
3.3.1. Transformação química de E. coli
Uma cultura estacionária de E. coli foi diluída em 250mL de meio SOB líquido
e incubada a 37qC até atingir D.O.600nm 0,6. A cultura bacteriana foi resfriada em
banho de gelo por 10 minutos e centrifugada a 3.200xg por 10 minutos a 4qC.
Posteriormente, o precipitado celular foi suspenso em 100mL de solução TB gelada
(Pipes 10mM pH7,0; MnCl2 55mM; CaCl2 15mM; KCl 250mM). As células foram
incubadas por 10 minutos em banho de gelo e centrifugadas a 3.200xg por 10
minutos a 4qC. O sobrenadante foi desprezado e as células foram suspensas em
20mL de solução TB gelada contendo 1,5mL de DMSO. Para armazenamento,
Células de E. coli competentes foram descongeladas em banho de gelo.
Foram adicionados 50–250ng de DNA plasmidial ou 10PL de reação de ligação em
um tubo de microcentrífuga contendo 100PL de células competentes. Os tubos
foram mantidos em banho de gelo por 30 minutos e, então, em banho-maria a 42qC
por 2 minutos. A seguir, 1mL de meio LB líquido foi adicionado ao tubo, o qual foi
incubado por 1 hora a 37qC. Decorrido o período de incubação, a cultura foi
centrifugada, o sobrenadante descartado e as células suspensas em 200PL de meio
LB. A suspensão foi distribuída em meio LB sólido contendo o antibiótico requerido
para seleção de transformantes e as placas foram incubadas por uma noite a 37qC.
3.3.2. Eletroporação de E. coli
Uma cultura estacionária de E. coli foi diluída 1:100 em 1L de meio LB
líquido e incubada a 37qC, sob agitação até atingir D.O.600nm 1,0-1,2. As células
foram coletadas por centrifugação a 3.200xg por 20 minutos e lavadas uma vez com 500mL de água milli-Q estéril gelada e duas vezes com 250mL de água gelada. Foi realizada uma nova lavagem com 30mL de glicerol 20% gelado. As células foram suspensas em 3mL de glicerol 20% gelado e armazenadas em alíquotas a -80ºC.
O volume de 2PL da preparação de DNA plasmidial de levedura (item 3.5.2)
foi adicionado a 40PL de células eletrocompetentes. Estas foram transferidas para
uma cubeta de eletroporação (0,1cm) e submetidas a um pulso elétrico com as
seguintes constantes elétricas: voltagem 1,7kV, resistência 200: e capacitância
25PF. Em seguida, as células foram removidas da cubeta com 1mL de meio LB e
incubadas a 37qC por 1 hora sob agitação. Finalmente, as células foram plaqueadas
em meio seletivo M9 contendo o antibiótico requerido para seleção dos transformantes. A seguir, as placas foram incubadas a 37ºC até a obtenção de colônias.
3.4. Manipulação de leveduras
3.4.1. Transformação rápida de S. cerevisiae
lítio 100mM e incubadas a 30qC por 15 minutos. Após incubação, a suspensão foi centrifugada e o sobrenadante removido. Ao precipitado obtido foram então
adicionados 240PL de 40% PEG 4000, 36PL 1M acetato de lítio, 50Pg de DNA de
esperma de salmão, 0,1-5Pg de DNA plasmidial e H2O milli-Q estéril q.s.p 350PL. A
suspensão foi agitada até completa homogeneização e incubada a 42qC por 20
minutos. Após incubação, as células foram coletadas por centrifugação, suspensas
em 200PL de H2O milli-Q estéril e plaqueadas em meio seletivo adequado.
3.4.2. Transformação de alta eficiência de S. cerevisiae
A cultura de interesse em fase estacionária foi diluída a 5,0x106 células/mL
em 10mL de meio SC líquido pré-aquecido e incubada a 30qC até a concentração de
2,0x107 células/mL. As células foram coletadas por centrifugação a 3.000xg por 5
minutos e lavadas com água milli-Q estéril. O precipitado foi suspenso em 600PL de
solução de acetato de lítio 100mM e incubado por 15 minutos a 30˚C. As células foram novamente coletadas por centrifugação e ao precipitado foram adicionados,
na seguinte ordem, 480PL de 50% PEG5000, 72PL de 1M acetato de lítio, 50PL de
2mg/mL DNA de esperma de salmão, 10PL da biblioteca de cDNA e o volume de
reação completado com 108PL de água milli-Q estéril. O tubo foi então agitado
vigorosamente por 1 minuto e incubado a 30˚C por 30 minutos. A suspensão foi incubada a 42˚C por 20 minutos, tomando-se cuidado de inverter o tubo a cada 5 minutos para equilíbrio da temperatura. Após nova centrifugação, as células foram suspensas em 2mL de água milli-Q estéril, plaqueadas em meio seletivo e incubadas a 30˚C até o aparecimento das colônias.
3.4.3. Cruzamento de leveduras
As linhagens haplóides de S. cerevisiae desejadas foram cruzadas na
3.4.4. Esporulação e dissecção de leveduras
Os diplóides foram crescidos em meio S-SPO líquido a 30ºC, sob agitação até
a formação de ascos (3-5 dias). Foram então centrifugados 200PL da cultura e, após
remoção do sobrenadante, as células foram suspensas em 100PL de sorbitol 20%.
Foram então adicionados 3PL de zimoliase (10mg/mL) e a reação foi incubada a
temperatura ambiente por 15 minutos. A seguir, foi adicionado 1mL de sorbitol 20% e as células foram mantidas em gelo até a dissecção. Os ascos foram dissecados em microscópio de dissecção, utilizando placa de meio YPD, a qual foi incubada a 30ºC até a obtenção de colônias. O genótipo das células haplóides obtidas foi caracterizado utilizando diferentes meios de cultura seletivos. O fator de acasalamento foi definido através do cruzamento com cepas de fator de acasalamento conhecido e seleção dos diplóides em meio seletivo.
3.5. Manipulação de DNA
3.5.1. Isolamento de DNA plasmidial de bactéria (mini preparação)
Uma colônia da bactéria de interesse foi inoculada em 3mL de meio LB
líquido contendo antibiótico para seleção. O inóculo foi incubado a 37qC sob
agitação por uma noite. Após crescimento, 1mL da cultura bacteriana foi centrifugada a 16.000xg por 2 minutos, o sobrenadante descartado e as bactérias
suspensas em 200PL de TE pH8,0 (Tris-HCl 10mM pH8,0; EDTA 1mM pH8,0). À
suspensão foram adicionados 200PL de solução SDS/NaOH (SDS 1%; NaOH 0,2M),
o conteúdo homogeneizado por inversão e incubado a 37qC por 5 minutos. Em
seguida, foram adicionados 150PL de acetato de sódio 3M pH4,8 e a solução foi
misturada várias vezes por inversão. Os tubos foram então centrifugados a 16000xg por 6 minutos, o sobrenadante foi transferido para novos tubos e 1mL de isopropanol foi adicionado. Após serem misturados por inversão, os tubos foram incubados a
37qC por 5 minutos e centrifugados a 16.000xg por 10 minutos. O sobrenadante foi
descartado, o precipitado lavado duas vezes com 1mL de etanol 75% gelado e, após
3.5.2. Isolamento de DNA plasmidial de levedura
O volume de 1,5mL da cultura de interesse em fase estacionária foi centrifugado a 16.000xg por 1 minuto. A seguir, o sobrenadante foi desprezado, as
células lavadas com 1mL de água milli-Q estéril e suspensas em 200PL de tampão
de lise (Tris-HCl 10mM pH8,0; Triton X-100 2%; SDS 1%; NaCl 100mM; 1mM
EDTA). Foram adicionados 200PL de PCI e aproximandamente 300mg de pérolas
de vidro. O tubo foi então agitado vigorosamente por 5 minutos e centrifugado a 16.000xg por 1 minuto. A fase aquosa foi transferida para um novo tubo e adicionada de igual volume de isopropanol. Após centrifugação por 10 minutos a
16.000xg o sobrenadante foi removido e o precipitado lavado com 500PL de etanol
75% gelado. O precipitado, depois de seco, foi suspenso em 50PL de água milli-Q.
3.5.3. Isolamento de DNA genômico de levedura
O volume de 10mL de uma cultura em fase estacionária foi centrifugado a 3.000xg por 15 minutos e as células lavadas em 1mL de água milli-Q. Foram
adicionados ao precipitado de células 200PL de tampão de lise (Tris-HCl 10mM
pH8,0; Triton X-100 2%; SDS 1%; NaCl 100mM; 1mM EDTA), 200PL de PCI e
aproximadamente 300mg de contas de vidro. O tubo foi então agitado
vigorosamente por 5 minutos e posteriormente foram adicionados 200PL de TE
pH8,0. Após nova agitação por 1 minuto, a mistura foi centrifugada a 16.000xg por 5 minutos. A fase aquosa foi transferida para um tubo de microcentrífuga ao qual
foram adicionados 200 PL de PCI. A mistura foi agitada novamente e submetida a
centrifugação 16.000xg por 5 minutos e a fase aquosa transferida para um novo tubo de microcentrífuga. Esse procedimento de extração foi repetido mais duas vezes. Após extração foi adicionado 1mL de etanol absoluto gelado à fase aquosa e a
solução foi centrifugada a 16.000xg por 2 minutos a 4qC. O precipitado obtido foi
suspenso em 400PL de TE pH 8,0 contendo 30Pg de RNAse A e a solução foi
incubada por 30 minutos a 37oC. Foi realizada então uma nova extração com PCI
seguida de precipitação com etanol absoluto. O precipitado foi lavado com etanol
3.5.4. Eletroforese de DNA em gel de agarose
A eletroforese de DNA foi realizada em gel de agarose 0,8% em TAE 1X (Tris-acetato 40mM pH8,5; EDTA 2mM). À amostra de DNA foi adicionado tampão de amostra 5X (Tris-HCl 10mM pH8,0; EDTA 1mM pH8,0; azul de bromofenol 0,125%; xilenocianol 0,125%; glicerol 30%; EDTA 1mM), e aplicada no gel contendo
0,5Pg/mL de brometo de etídeo. O gel foi submetido à voltagem de 100V em
tampão TAE 1X.
3.5.5. Isolamento de fragmentos de DNA de gel de agarose
O fragmento de DNA separado em gel de agarose 0,8% foi visualizado por coloração com brometo de etídeo e iluminação ultravioleta. O fragmento de interesse foi cortado do gel e purificado utilizando o kit “QIAquick Gel Extraction” (Qiagen) conforme recomendado pelo fabricante.
3.5.6. Amplificação de DNA por PCR
As reações foram realizadas em tubos de microcentrífuga de 0,5mL contendo
aproximadamente 50ng de DNA molde, 1PM de cada oligonucleotídeo, 2U de Taq
HiFi DNA polimerase (Invitrogen) e 200PM de dNTPs em tampão adequado num
volume final de reação de 100PL. A solução foi incubada por 3 minutos a 94qC,
seguida de 20 ciclos de incubações a 94qC por 30 segundos (desnaturação), a 58qC
por 40 segundos (hibridização) e a 72qC por 1 minuto (extensão). Por último,
seguiu-se incubação a 72qC por 10 minutos. Quando necessário, as condições de
hibridização e extensão foram variadas de acordo com os oligonucleotídeos utilizados e o fragmento de DNA a ser amplificado. Os produtos da reação foram analisados por eletroforese em gel de agarose 0,8% e purificados utilizando o kit “QIAquick PCR Purification” (QIAGEN).
3.5.7. Manipulação enzimática de DNA
3.5.8. Sequenciamento de DNA
O sequenciamento de DNA foi realizado utilizando o kit “Big Dye Terminator” e o sequenciador semi-automático ABI377 (Applied Biosystem), conforme protocolo recomendado pelo fabricante.
3.5.9. Mutação sítio-dirigida de DNA
Para a obtenção de mutantes através de mutação sítio-dirigida foi utilizado o kit “QuickChange Site-Directed Mutagenesis” (Stratagene), de acordo com o protocolo fornecido pelo fabricante. Esse método baseia-se na amplificação do plasmídeo contendo o gene de interesse por PCR utilizando a enzima de alta
processividade Pfx turbo e oligonucleotídeos contendo a mutação deseja, seguida
da remoção do DNA molde pela ação endonucleásica de DpnI e transformação de E.
colicompetente.
3.6. Análise de proteínas
3.6.1. Quantificação de proteínas
Para a determinação da concentração protéica de extratos celulares e de proteínas purificadas foi utilizado o kit “BioRad Protein Assay” (BioRad), baseado no método de Bradford.
3.6.2. Separação de proteínas por SDS-PAGE
Proteínas foram separadas em gel de poliacrilamida descontínuo. O gel de empacotamento continha acrilamida:bis-acrilamida (29:1) 5%; Tris-HCl 380mM
pH6,8; SDS 0,1%; persulfato de amônio 0,1%; 1PL/mL de TEMED e o gel de
separação acrilamida:bis-acrilamida (29:1) 10% ou 12%; Tris-HCl 380mM pH8,8;
SDS 0,1%; persulfato de amônio 0,1%; 0,4PL/mL de TEMED. As amostras em
tampão de amostra para proteína (Tris-HCl 40mM pH6,8; glicerol 8%; SDS 2%; DTT
100mM; azul de bromofenol 0,1%) foram incubadas a 96qC por 5 minutos e
3.6.3. Coloração de proteínas por “Coomassie Blue”
O gel de acrilamida foi incubado em solução de Coomassie Blue R 0,25% em metanol 45% e ácido acético 10% por 30 minutos e descorado em solução de etanol 50% e ácido acético 10%.
3.6.4. Western blot
Após separação por SDS-PAGE, as proteínas foram transferidas para membrana de nitrocelulose (Hybond-ECL GE Healthcare) por 1 hora a 100V em tampão de transferência (Tris-HCl 12mM; glicina 125mM) em sistema de transferência (BioRad). A transferência foi avaliada por coloração com Ponceau S (Pounceau S 1mg/mL; ácido acético 5%) e a membrana foi incubada com tampão PBST (PBS pH7,4; 0,25% Tween-20) contendo 5% de leite desnatado a temperatura ambiente por ao menos 30 minutos. Após bloqueio, a membrana foi incubada em PBST contendo 5% de leite desnatado e o anticorpo primário de interesse por 2 horas a temperatura ambiente sob agitação branda. Posteriormente, a membrana foi lavada três vezes por 5 minutos com PBST, e incubada por 1 hora com anti-IgG de coelho conjugado com peroxidase (Sigma) na diluição de 1:5.000 em PBST contendo 5% de leite desnatado. Após três lavagens de 5 minutos com PBST, a membrana foi tratada com reagentes quimioluminescentes (ECL-GE Healthcare) e utilizada na exposição de filme autorradiográfico.
3.7. Produção e purificação de proteína recombinante
3.7.1. Teste de indução da produção de proteína recombinante
Uma cultura estacionária de E. coli transformada com plasmídeo de
expressão de interesse foi diluída 1:100 em 25mL de meio LB líquido contendo o
antibiótico adequado e crescida a 37qC sob agitação até D.O.600nm 0,6. Após
crescimento, uma amostra de 400PL foi retirada da cultura, as células coletadas por
centrifugação 16.000xg por 1 minuto e suspensas em 40PL de tampão de amostra
para proteína. No restante da cultura foi induzida a expressão do gene recombinante
com 0,4mM de IPTG e 400PL de amostra foram retirados após 1, 2 e 3 horas de
crescimento. Da mesma forma, as células foram coletadas por centrifugação e
respectivamente. As amostras foram analisadas por SDS-PAGE e coloração por “Coomassie Blue”.
3.7.2. Produção de proteína recombinante em larga escala
Excetuando-se a adequação volumétrica para o aumento de escala, foi utilizado a mesmo procedimento que para o teste de indução da produção de proteína recombinante. Foram utilizadas culturas de 1L de LB líquido contendo antibiótico adequado. Após 3 horas de indução com 0,4mM de IPTG, as células foram coletadas por centrifugação a 5.000xg por 20 minutos, lavadas com tampão
PBS pH7,4 e congeladas a -80qC.
3.7.3. Purificação de proteína em fusão com cauda de histidinas
As células foram descongeladas em gelo e lisadas em tampão de lise (tampão fosfato 50mM pH8,0; NaCl 300mM; imidazol 10mM; DTT 2mM; PMSF 2mM; 1X PLAC). Para a lise celular, a suspensão foi submetida à sonicação, em gelo, a ciclos de 4 segundos de sonicação e 10 segundos de repouso, durante 2 minutos, numa amplitude de 60%. O lisado foi então centrifugado a 20.000xg por 20 minutos
e o sobrenadante filtrado em filtro de 0,45Pm. O lisado clarificado e filtrado foi
submetido à cromatografia de afinidade utilizando-se uma coluna de Ni-NTA de 5mL e o sistema cromatográfico ÄKTA FPLC (GE Healthcare). O procedimento para purificação foi realizado como descrito pelo fabricante. Resumidamente, após a aplicação da amostra na coluna, esta foi lavada com tampão de lavagem (tampão fosfato 50mM pH8,0; NaCl 300mM; imidazol 20mM). Em seguida, foi feita a eluição da proteína de fusão com um gradiente de tampão de lavagem e tampão de eluição (tampão fosfato 50mM pH8,0; NaCl 300mM; imidazol 250mM). As frações obtidas da cromatografia foram analisadas por SDS-PAGE e coloração por “Coomassie Blue”. As frações contendo a proteína de interesse foram reunidas e submetidas à diálise em membrana SpectraPor (Spectrum) com poro de seleção de 12-14.000Da
em 2L de tampão 10mM Tris-HCL pH7,5 por uma noite a 4qC.
3.7.4. Purificação de proteína em fusão com GST (Glutationa S-transferase)
submetida à sonicação, clarificada por centrifugação e filtrada como descrito anteriormente. O lisado clarificado e filtrado foi submetido à cromatografia de afinidade utilizando-se uma coluna de glutationa-Sepharose GSTrap 5mL e o sistema cromatográfico ÄKTA FPLC (GE Healthcare). O procedimento para purificação foi realizado como descrito pelo fabricante. Resumidamente, após a aplicação da amostra na coluna, esta foi lavada com tampão PBS pH7,4 e a proteína de fusão foi eluída em tampão de eluição (Tris-HCl 100mM pH8,0; glutationa reduzida 10mM). As frações obtidas da cromatografia foram analisadas por SDS-PAGE e coloração por “Coomassie Blue”. As frações obtidas da cromatografia contendo a proteína foram reunidas e submetidas à diálise como descrito anteriormente. Quando necessária a remoção de GST, a proteína purificada foi submetida à clivagem por trombina. Para isso, foi adicionada 0,5U de trombina por mg de proteína e a reação incubada a temperatura ambiente por 2 horas de clivagem. A amostra foi submetida então a uma nova cromatografia de afinidade para a remoção de GST e purificação da proteína livre.
3.8. Produção de anticorpo policlonal anti-Pub1 em coelhos
A proteína de fusão His-Pub1 purificada foi utilizada na imunização de
coelhos. Para isso, 300Pg de proteína foram homogeneizados com o mesmo
volume de adjuvante completo de Freund e a emulsão obtida foi inoculada na região
dorsal do coelho por via intradérmica. Após 21 dias da primeira inoculação, 300Pg
de proteína foram homogeneizados com o mesmo volume de adjuvante incompleto de Freund e inoculado na região dorsal do coelho por via subcutânea. Outras duas inoculações foram realizadas da mesma forma com intervalos de 21 dias. Decorridos 11 dias das inoculações, a partir da segunda inoculação, foram coletados 15mL de sangue periférico do coelho e o título do anticorpo obtido foi determinado por western blot utilizando extrato total de levedura selvagem.
3.9. Rastreamento de ligantes físicos por duplo-híbrido
transcrição da proteína Gal4 de levedura e da proteína lexA de procariotos, capaz de se ligar a sequências específicas de DNA (48). A linhagem SVL86 (L40) contém os
genes repórteres do sistema de duplo-híbrido lexAop-HIS3 e lexAop-lacZ. A
proteína isca lexA-Pub1 foi utilizada para rastrear uma biblioteca de cDNA clonada em fusão com o domínio de ativação de Gal4 no vetor pACT. Este sistema de rastreamento por duplo-híbrido foi cedido pela Profa. Dra. Carla C. Oliveira, do Instituto de Química – USP.
O rastreamento foi procedido da seguinte maneira:
- Clonagem de PUB1 por PCR no vetor pBTM116, que possibilita a produção
da proteína isca em fusão com lexA.
- Análise da expressão da proteína isca por western blot.
- Análise da autoativação dos genes repórteres HIS3 e lacZ pela proteína
isca:
Teste de crescimento em meio deficiente em histidina - as células foram
crescidas em meio SC Leu Trp e, após diluição seriada, plaqueadas em meio SC -Leu -Trp -His.
Teste de atividade de
E
-galactosidase - as células foram transferidas paramembrana de nitrocelulose e permeabilizadas em nitrogênio líquido. A membrana foi colocada sobre papel de filtro previamente umedecido em tampão Z (tampão
fosfato 100mM pH7,0; MgCl2 10mM; E-mercaptoetanol 50mM) contendo 1mg/mL de
X-Gal e incubada a 30qC por 30 minutos para o desenvolvimento de coloração azul
pelos clones positivos.
- Transformação da linhagem contendo o gene PUB1 em vetor plasmidial com
a biblioteca de cDNA de S. cerevisiae construída no vetor pACT.
- Seleção dos clones transformantes His+ em meio seletivo.
- Teste de ativação do gene repórter Lac Z por ensaio da atividade de E
-galactosidase.
- Recuperação dos plasmídeos dos clones positivos e nova transformação da linhagem repórter ("plasmid linkage") após amplificação do plasmídeo da biblioteca emE. coli HB101.
3.10. Ensaio de co-purificação utilizando a fusão TAP
A linhagem contendo o alelo integrado no locus do gene de interesse em fusão
com a região codificadora de TAP (“Tandem Affinity Protein”, Open Biosystems, (56)) foi utilizada para os ensaios de co-purificação. Esta linhagem produz a proteína de interesse em fusão C-terminal com a proteína TAP, a qual consiste de um peptídeo de ligação à calmodulina, um sítio de clivagem por TEV (subunidade catalítica da proteína NIa do vírus “etch” do tabaco) e dois domínios de ligação a IgG de proteína A de Staphylococcus aureus. Dessa forma, a proteína de fusão produzida pode ser
purificada por afinidade utilizando resina de IgG-Sepharose.
A linhagem de interesse foi crescida em meio YPD a 30qC sob agitação até
D.O.600nm 1,5. As células coletadas por centrifugação foram lisadas em tampão
IPP150 (Tris-HCl 20mM pH8,0; NaCl 150mM; Igepal CA-630 0,1%; PMSF 2mM; 1X PLAC) com contas de vitro por 5 ciclos de 1 minuto de agitação vigorosa, com intervalos de 1 minuto em gelo. Após centrifugação 16.000xg por 10 minutos, uma alíquota do extrato total contendo 3mg de proteína foi incubada em 1mL de tampão
IPP150 com 50PL de IgG-Sepharose (GE Healthcare) por 30 minutos a temperatura
ambiente na presença ou não de 50Pg de RNase A e então por 2 horas a 4qC sob
agitação branda. A resina foi lavada três vezes com 1mL de tampão IPP150 e
incubada com 10Pg de protease TEV por 2h a 16qC em tampão IPP150 contendo
DTT 1mM e EDTA 0,5mM. Após centrifugação, o sobrenadante foi coletado e submetido a SDS-PAGE e western blot.
3.11. Ensaio in vitro de interação proteína-proteína
Uma alíquota contendo 10Pg da proteína em fusão com GST foi incubada com
20PL de glutationa-Sepharose (GE Healthcare) em 500PL de tampão PBS pH7,4 por
30 minutos a temperatura ambiente sob agitação branda. A resina foi lavada duas
vezes com 1mL de tampão PBS pH7,4 e incubada com 1Pg da proteína em fusão
com cauda de histidina em 500PL de tampão A (Tris-HCl 20mM pH8,0; Triton X-100